合成生物学 ›› 2023, Vol. 4 ›› Issue (3): 464-487.DOI: 10.12211/2096-8280.2023-008
人工智能蛋白质结构设计算法研究进展
陈志航, 季梦麟, 戚逸飞
- 复旦大学药学院,上海 201203
-
收稿日期:2023-01-13修回日期:2023-03-15出版日期:2023-06-30发布日期:2023-07-05 -
通讯作者:戚逸飞 -
作者简介:陈志航 (1998—),男,硕士研究生。研究方向为人工智能蛋白质设计。 E-mail:zhihangchen21@m.fudan.edu.cn季梦麟 (2000—),男,硕士研究生。研究方向为人工智能蛋白质设计。 E-mail:22211030067@m.fudan.edu.cn戚逸飞 (1983—),男,副研究员,硕士生导师。研究方向为生物大分子结构和功能模拟以及人工智能药物设计。 E-mail:yfqi@fudan.edu.cn -
基金资助:国家自然科学基金(22033001)
Research progress of artificial intelligence in desiging protein structures
CHEN Zhihang, JI Menglin, QI Yifei
- School of Pharmacy,Fudan University,Shanghai 201203,China
-
Received:2023-01-13Revised:2023-03-15Online:2023-06-30Published:2023-07-05 -
Contact:QI Yifei
摘要:
蛋白质是各类生命活动不可缺少的承担者,其序列决定了折叠后的三维结构和功能。这些具有特定功能的蛋白质在生物医学等多个领域具有重要的应用价值。计算蛋白质设计可以根据所需的蛋白功能和结构设计氨基酸序列,生成自然界中不存在的蛋白质。传统计算蛋白质设计通常采用能量函数和特定的搜索优化算法获得设计的序列。近年来,随着先进算法的发展、大数据的积累和计算机硬件算力的增长,人工智能技术得到了蓬勃发展,并逐渐应用于蛋白质设计领域。本文综述了近年人工智能在蛋白质结构设计中的进展,侧重于各类算法的介绍,从固定骨架设计、可变骨架设计和序列结构生成三个方面回顾了最新的蛋白质结构设计算法,并阐明了其相对于传统计算方法的新颖性和创新性。在人工智能技术的赋能下,蛋白质设计的成功率和合理性获得大幅提高,按需功能蛋白设计的时代即将到来。
中图分类号:
引用本文
陈志航, 季梦麟, 戚逸飞. 人工智能蛋白质结构设计算法研究进展[J]. 合成生物学, 2023, 4(3): 464-487.
CHEN Zhihang, JI Menglin, QI Yifei. Research progress of artificial intelligence in desiging protein structures[J]. Synthetic Biology Journal, 2023, 4(3): 464-487.
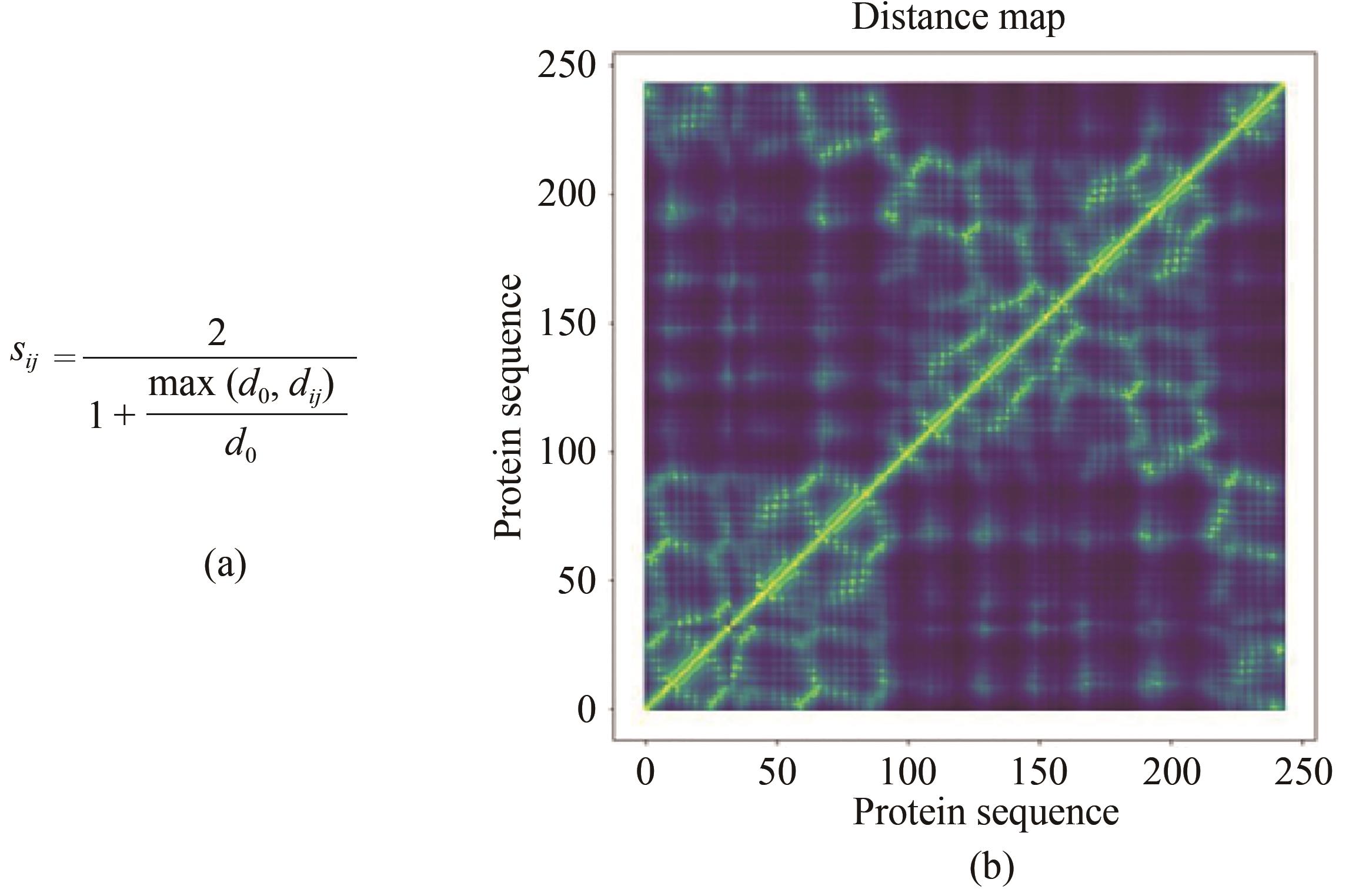
图1 SPROF中残基距离计算方法(a)dij 为残基i和j的Cα原子之间的距离,d0=0.4 nm;(b)蛋白质残基-残基距离矩阵
Fig. 1 Calculating the distance of residues in SPROF(a) dij is the distance between the Cα atoms of residues i and j, d0=0.4 nm, and (b) matrix for residue-residue distance of a protein structure.
图2 三维卷积神经网络提取网格中的蛋白质空间结构信息
Fig. 2 Extracting spatial information of a protein structure from 3D convolutional neural network

图3 trDesign模型架构图
Fig. 3 Architecture for the trDesign model

图 4 GraphTrans编码器与解码器示意图
Fig. 4 Architecture for the GraphTrans encoder and decoder
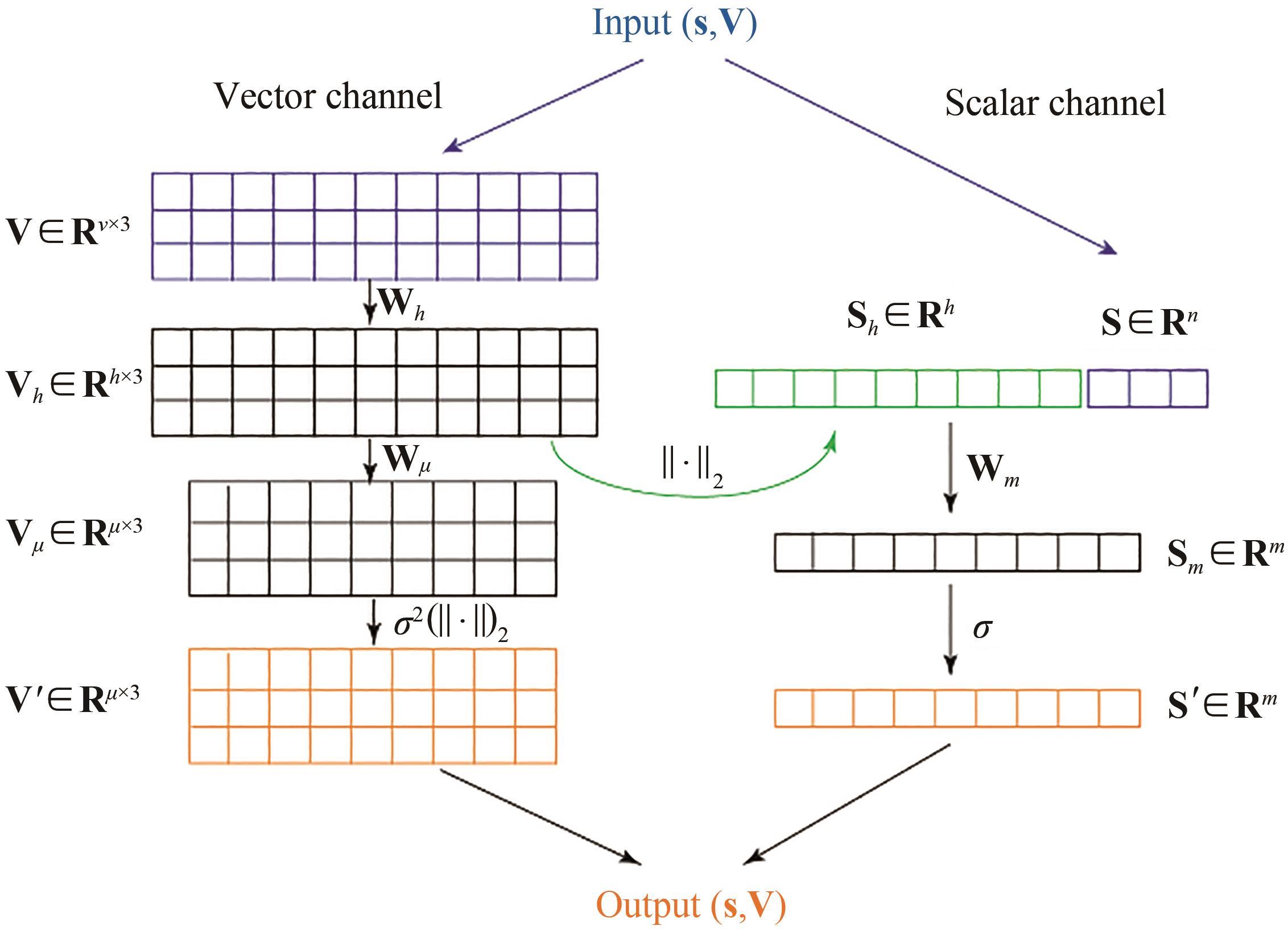
图5 GVP原理示意图
Fig. 5 Architecture for GVP

图 6 ABACUS-R模型架构示意图
Fig. 6 Architecture for the ABACUS-R model
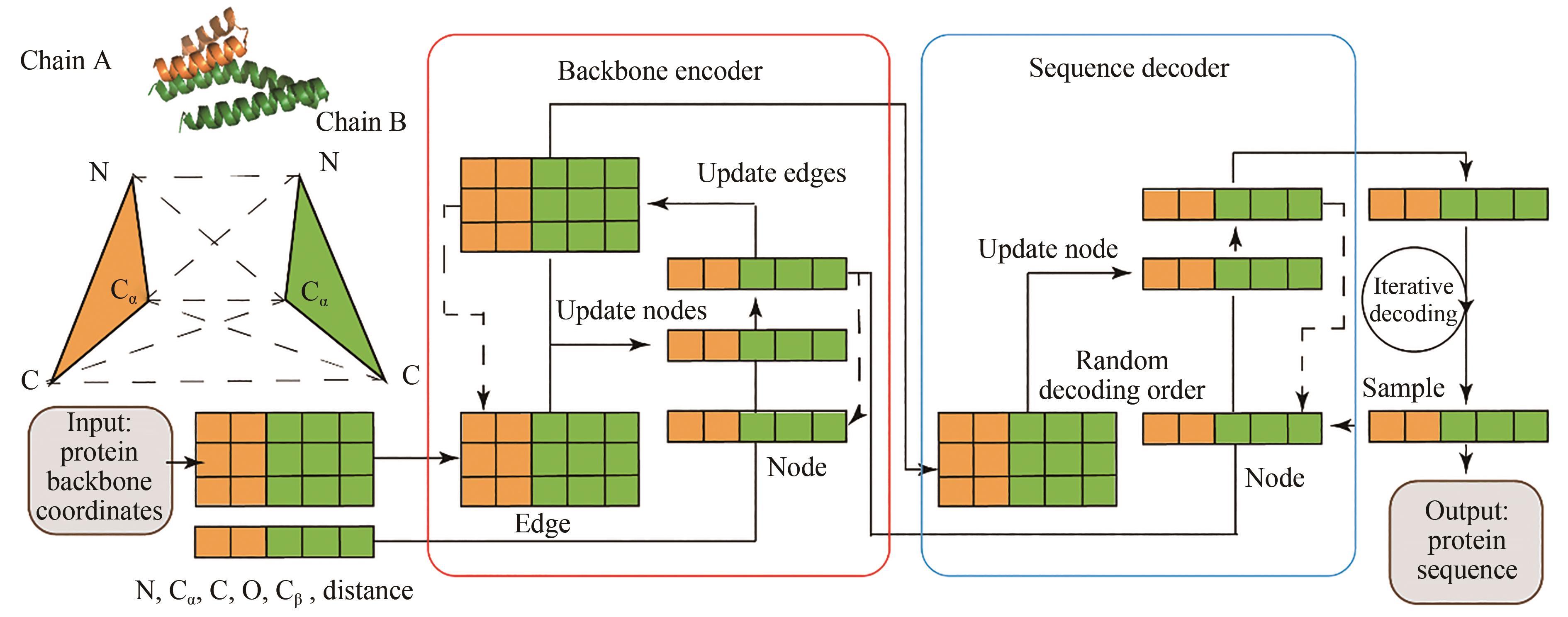
图 7 ProteinMPNN模型的整体结构
Fig. 7 Architecture for the ProteinMPNN model
模型 Models | 恢复率/%(↑) Recovery/% (↑) | 困惑度(↓) Perplexity (↓) |
|---|---|---|
| GraphTrans | 35.82 | 6.63 |
| StructGNN[ | 37.1 | 6.49 |
| GVP-GNN-large | 39.20 | 6.17 |
| GVP-GNN-Transformer | 38.30 | 6.44 |
| GVP-GNN-Transformer+AF2 | 51.60 | 4.01 |
| ProteinMPNN | 45.96 | 4.61 |
| ProDesign | 50.22 | 4.69 |
| PiFold[ | 50.22 | 4.62 |
| LM-DESIGN[ | 55.65 | 4.52 |
表1 固定骨架序列设计模型在CATH 4.2测试集上的序列恢复率和困惑度
Table 1 Sequence recovery rate and perplexity of the fixed-backbone sequence design model on CATH 4.2 test set
模型 Models | 恢复率/%(↑) Recovery/% (↑) | 困惑度(↓) Perplexity (↓) |
|---|---|---|
| GraphTrans | 35.82 | 6.63 |
| StructGNN[ | 37.1 | 6.49 |
| GVP-GNN-large | 39.20 | 6.17 |
| GVP-GNN-Transformer | 38.30 | 6.44 |
| GVP-GNN-Transformer+AF2 | 51.60 | 4.01 |
| ProteinMPNN | 45.96 | 4.61 |
| ProDesign | 50.22 | 4.69 |
| PiFold[ | 50.22 | 4.62 |
| LM-DESIGN[ | 55.65 | 4.52 |
模型类别 Group | 模型 Models | TS50 | TS500 | ||
|---|---|---|---|---|---|
恢复率/%(↑) Recovery/%(↑) | 困惑度(↓) Perplexity(↓) | 恢复率/%(↑) Recovery/%(↑) | 困惑度(↓) Perplexity (↓) | ||
| MLP | SPIN | 30.00 | — | — | — |
| SPIN2 | 34.00 | — | — | — | |
| Wang’s model | 33.00 | — | — | — | |
| CNN | SPROF | 39.80 | — | — | — |
| ProDCoNN | 46.50 | — | — | — | |
| DenseCPD | 50.71 | — | 55.53 | — | |
| GNN | StructGNN | 43.89 | 5.40 | 45.69 | 4.98 |
| GraphTrans | 42.20 | 5.60 | 44.66 | 5.16 | |
| GVP-GNN | 44.14 | 4.71 | 49.14 | 4.20 | |
| GCA[ | 47.02 | 5.09 | 47.74 | 4.72 | |
| ADesign[ | 48.36 | 5.25 | 49.23 | 4.93 | |
| ProteinMPNN | 54.43 | 3.93 | 58.08 | 3.53 | |
| PiFold | 58.72 | 3.86 | 60.42 | 3.44 | |
| LM-DESIGN(PiFold) | 57.89 | 3.50 | 67.78 | 3.19 | |
表2 固定骨架序列设计模型在TS50 &TS500测试集上的序列恢复率和困惑度
Table 2 Sequence recovery rate and perplexity of the fixed-backbone sequence design model on TS50 &TS500 test sets
模型类别 Group | 模型 Models | TS50 | TS500 | ||
|---|---|---|---|---|---|
恢复率/%(↑) Recovery/%(↑) | 困惑度(↓) Perplexity(↓) | 恢复率/%(↑) Recovery/%(↑) | 困惑度(↓) Perplexity (↓) | ||
| MLP | SPIN | 30.00 | — | — | — |
| SPIN2 | 34.00 | — | — | — | |
| Wang’s model | 33.00 | — | — | — | |
| CNN | SPROF | 39.80 | — | — | — |
| ProDCoNN | 46.50 | — | — | — | |
| DenseCPD | 50.71 | — | 55.53 | — | |
| GNN | StructGNN | 43.89 | 5.40 | 45.69 | 4.98 |
| GraphTrans | 42.20 | 5.60 | 44.66 | 5.16 | |
| GVP-GNN | 44.14 | 4.71 | 49.14 | 4.20 | |
| GCA[ | 47.02 | 5.09 | 47.74 | 4.72 | |
| ADesign[ | 48.36 | 5.25 | 49.23 | 4.93 | |
| ProteinMPNN | 54.43 | 3.93 | 58.08 | 3.53 | |
| PiFold | 58.72 | 3.86 | 60.42 | 3.44 | |
| LM-DESIGN(PiFold) | 57.89 | 3.50 | 67.78 | 3.19 | |
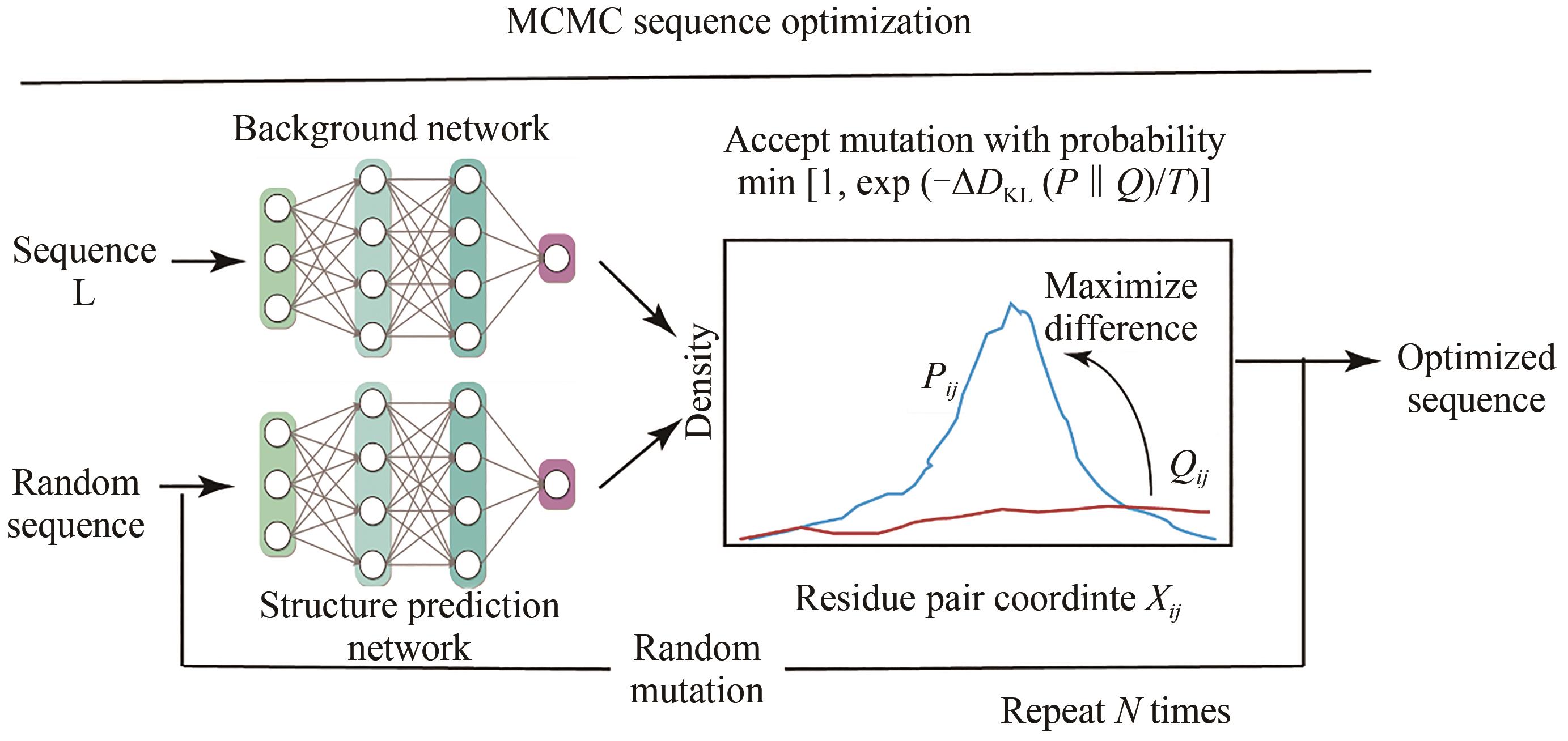
图8 hallucination模型原理示意图
Fig. 8 Architecture for the hallucination model

图9 Constrained hallucination模型原理示意图
Fig. 9 Architecture for the Constrained hallucination model

图10 生成对抗模型用于蛋白二维接触图和三维骨架的生成
Fig. 10 Generative adversarial network for generating contact map and 3D backbone structure.
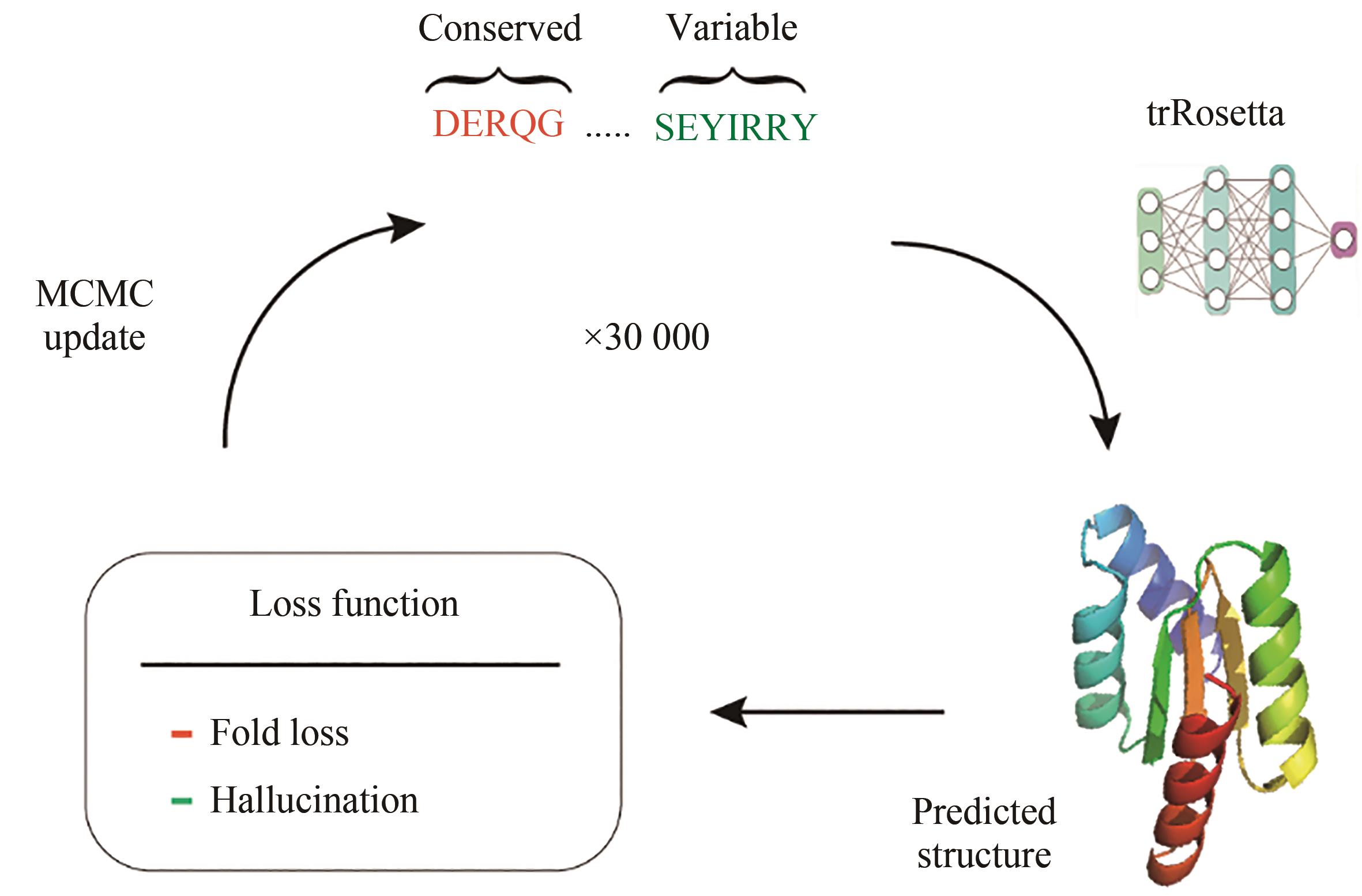
图11 Famliy-wide幻想蛋白质结构生成模型架构图
Fig. 11 Architectuer for the famliy-wide hallucination protein structure generation model

图12 Ig-VAE模型架构
Fig. 12 Framwork for Ig-VAE
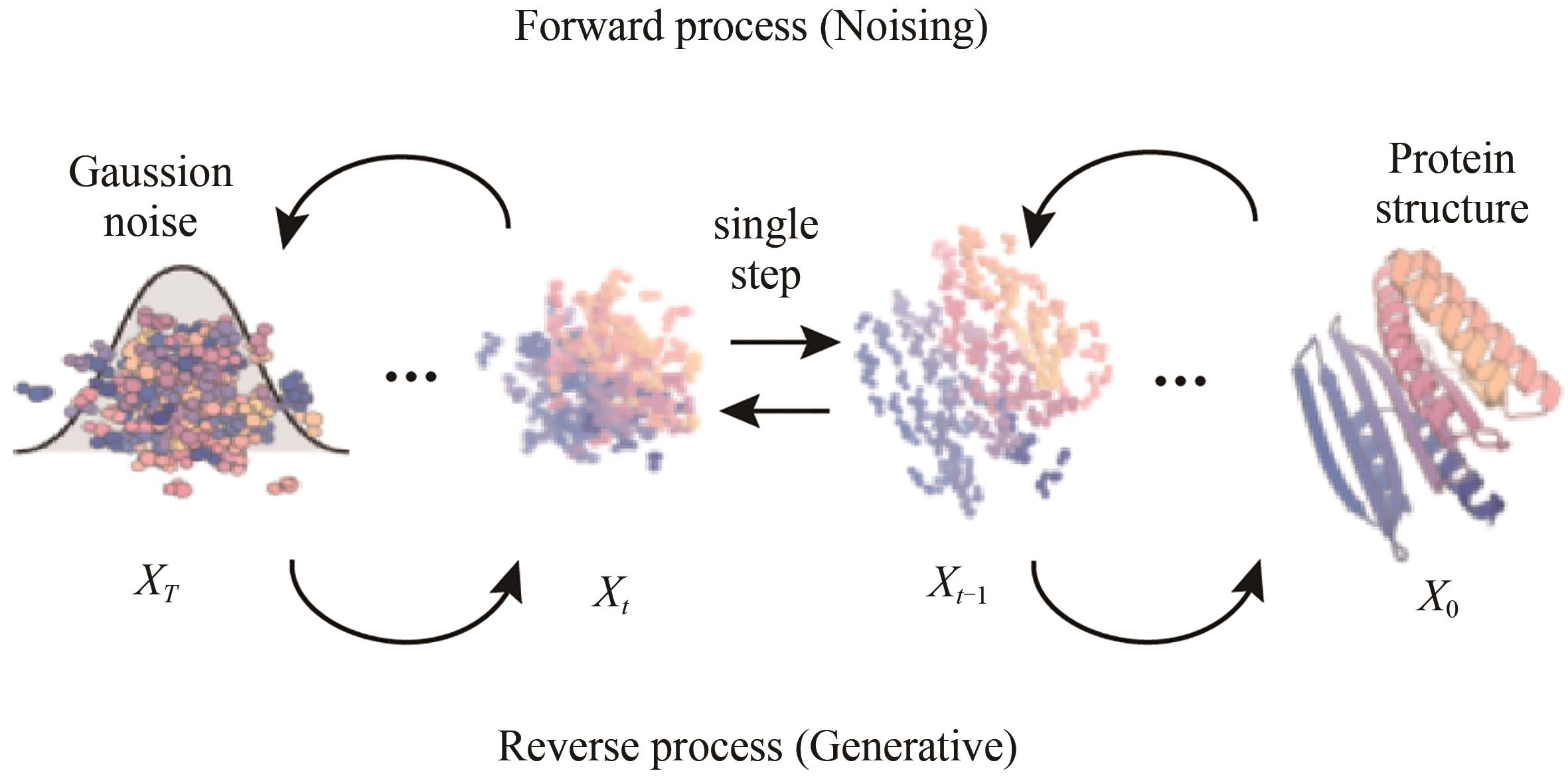
图 13 蛋白质结构生成扩散模型的原理示意图[100]
Fig. 13 Schematic diagram of the diffusion model for protein structure generation[100]

图14 FoldingDiff训练流程
Fig. 14 Training flow of the FoldingDiff model
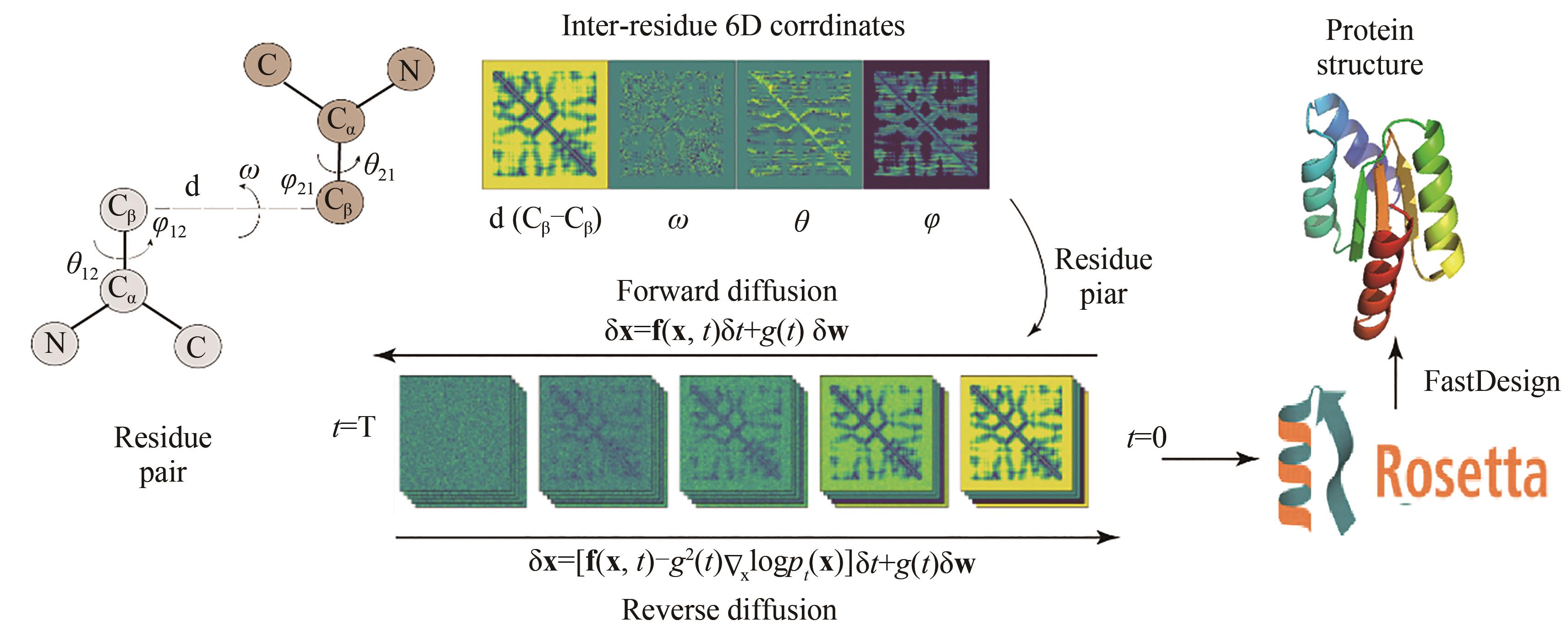
图15 ProteinSGM蛋白编码和模型架构图
Fig. 15 Protein structure encoding and model architecture of ProteinSGM

图 16 RFdiffusion模型原理示意图
Fig. 16 Schematic diagram of the RFdiffusion model
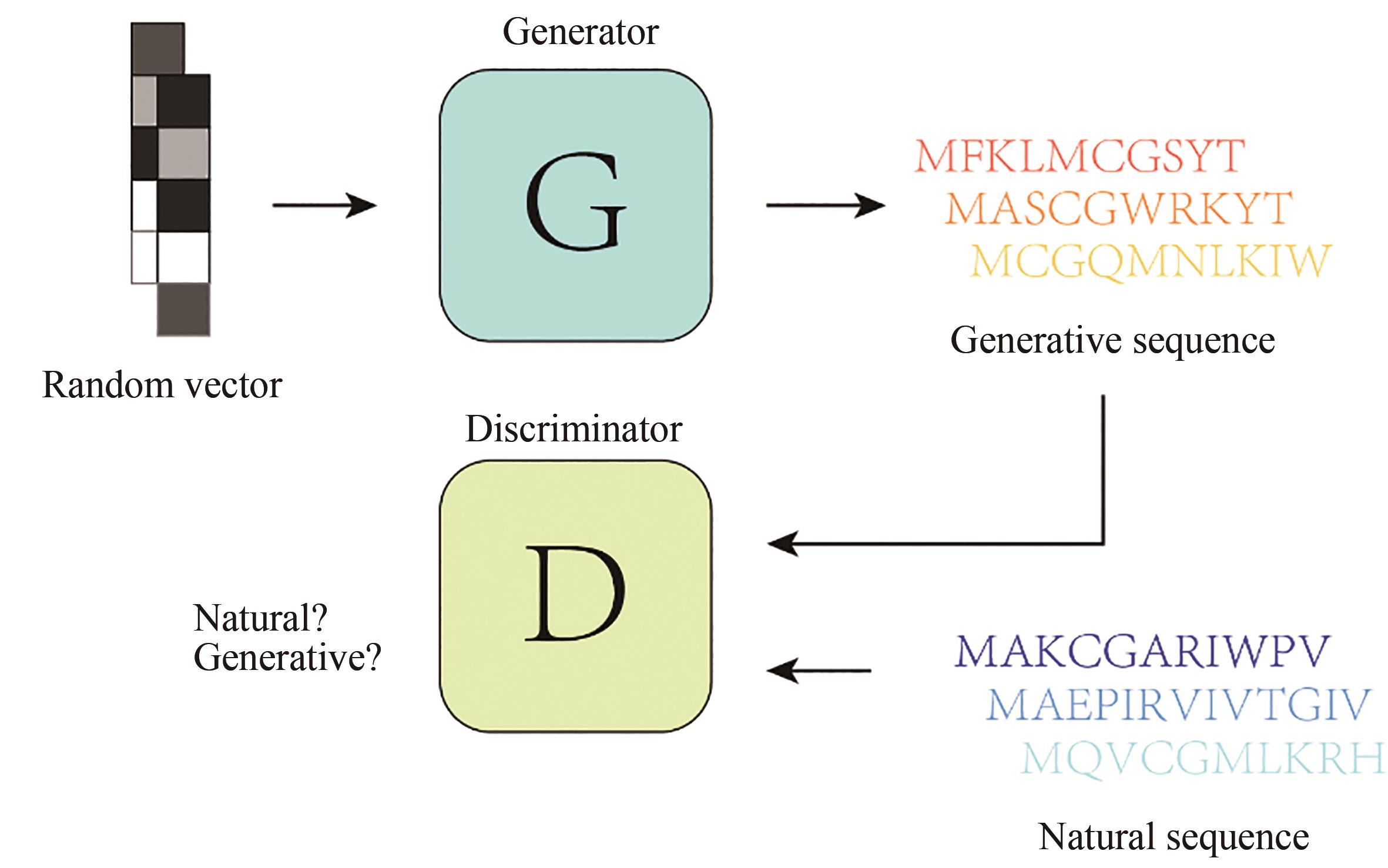
图17 ProteinGAN基本架构
Fig. 17 Architecture for ProteinGAN
| 1 | HUANG P S, BOYKEN S E, BAKER D. The coming of age of de novo protein design[J]. Nature, 2016, 537(7620): 320-327. |
| 2 | KHERSONSKY O, LIPSH R, AVIZEMER Z, et al. Automated design of efficient and functionally diverse enzyme repertoires[J]. Molecular Cell, 2018, 72(1): 178-186.e5. |
| 3 | GLASGOW A A, HUANG Y M, MANDELL D J, et al. Computational design of a modular protein sense-response system[J]. Science, 2019, 366(6468): 1024-1028. |
| 4 | ANFINSEN C B. Principles that govern the folding of protein chains[J]. Science, 1973, 181(4096): 223-230. |
| 5 | LEAVER-FAY A, O'MEARA M J, TYKA M, et al. Scientific benchmarks for guiding macromolecular energy function improvement[J]. Methods in Enzymology, 2013, 523: 109-143. |
| 6 | LEMAN J K, WEITZNER B D, LEWIS S M, et al. Macromolecular modeling and design in Rosetta: recent methods and frameworks[J]. Nature Methods, 2020, 17(7): 665-680. |
| 7 | NADRA A D, SERRANO L, ALIBÉS A. Chapter one-DNA-binding specificity prediction with FoldX[M]//Methods in enzymology. New York: Academic Press. 2011, 498: 3-18. |
| 8 | HUANG X Q, PEARCE R, ZHANG Y. EvoEF2: accurate and fast energy function for computational protein design[J]. Bioinformatics, 2020, 36(4): 1135-1142. |
| 9 | ALFORD R F, LEAVER-FAY A, JELIAZKOV J R, et al. The Rosetta all-atom energy function for macromolecular modeling and design[J]. Journal of Chemical Theory and Computation, 2017, 13(6): 3031-3048. |
| 10 | KUHLMAN B, DANTAS G, IRETON G C, et al. Design of a novel globular protein fold with atomic-level accuracy[J]. Science, 2003, 302(5649): 1364-1368. |
| 11 | SIEGEL J B, ZANGHELLINI A, LOVICK H M, et al. Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction[J]. Science, 2010, 329(5989): 309-313. |
| 12 | SILVA D A, YU S, ULGE U Y, et al. De novo design of potent and selective mimics of IL-2 and IL-15[J]. Nature, 2019, 565(7738): 186-191. |
| 13 | MOHAN K, UEDA G, KIM A R, et al. Topological control of cytokine receptor signaling induces differential effects in hematopoiesis[J]. Science, 2019, 364(6442): eaav7532. |
| 14 | CHEVALIER A, SILVA D A, ROCKLIN G J, et al. Massively parallel de novo protein design for targeted therapeutics[J]. Nature, 2017, 550(7674): 74-79. |
| 15 | CAO L X, GORESHNIK I, COVENTRY B, et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors[J]. Science, 2020, 370(6515): 426-431. |
| 16 | LANGAN R A, BOYKEN S E, NG A H, et al. De novo design of bioactive protein switches[J]. Nature, 2019, 572(7768): 205-210. |
| 17 | DAWSON W M, LANG E J M, RHYS G G, et al. Structural resolution of switchable states of a de novo peptide assembly[J]. Nature Communications, 2021, 12: 1530. |
| 18 | SHEN H, FALLAS J A, LYNCH E, et al. De novo design of self-assembling helical protein filaments[J]. Science, 2018, 362(6415): 705-709. |
| 19 | HSIA Y, BALE J B, GONEN S, et al. Design of a hyperstable 60-subunit protein icosahedron[J]. Nature, 2016, 535(7610): 136-139. |
| 20 | ROCKLIN G J, CHIDYAUSIKU T M, GORESHNIK I, et al. Global analysis of protein folding using massively parallel design, synthesis, and testing[J]. Science, 2017, 357(6347): 168-175. |
| 21 | BERMAN H M, WESTBROOK J, FENG Z K, et al. The protein data bank[J]. Nucleic Acids Research, 2000, 28(1): 235-242. |
| 22 | FOX N K, BRENNER S E, CHANDONIA J M. SCOPe: structural classification of proteins—extended, integrating SCOP and ASTRAL data and classification of new structures[J]. Nucleic Acids Research, 2014, 42(D1): D304-D309. |
| 23 | CONSORTIUM T U, BATEMAN A, MARTIN M J, et al. UniProt: the universal protein knowledgebase[J]. Nucleic Acids Research, 2017, 45(D1): D158-D169. |
| 24 | MISTRY J, CHUGURANSKY S, WILLIAMS L, et al. Pfam: the protein families database in 2021[J]. Nucleic Acids Research, 2021, 49(D1): D412-D419. |
| 25 | FRAPPIER V, KEATING A E. Data-driven computational protein design[J]. Current Opinion in Structural Biology, 2021, 69: 63-69. |
| 26 | KWON Y, SHIN W H, KO J, et al. AK-score: accurate protein-ligand binding affinity prediction using an ensemble of 3D-convolutional neural networks[J]. International Journal of Molecular Sciences, 2020, 21(22): 8424. |
| 27 | JIANG D J, HSIEH C Y, WU Z X, et al. InteractionGraphNet: a novel and efficient deep graph representation learning framework for accurate protein-ligand interaction predictions[J]. Journal of Medicinal Chemistry, 2021, 64(24): 18209-18232. |
| 28 | JONES D, KIM H, ZHANG X H, et al. Improved protein-ligand binding affinity prediction with structure-based deep fusion inference[J]. Journal of Chemical Information and Modeling, 2021, 61(4): 1583-1592. |
| 29 | JIMÉNEZ J, ŠKALIČ M, MARTÍNEZ-ROSELL G, et al. KDEEP: protein-ligand absolute binding affinity prediction via 3D-convolutional neural networks[J]. Journal of Chemical Information and Modeling, 2018, 58(2): 287-296. |
| 30 | SLEDZIESKI S, SINGH R, COWEN L, et al. D-SCRIPT translates genome to phenome with sequence-based, structure-aware, genome-scale predictions of protein-protein interactions[J]. Cell Systems, 2021, 12(10): 969-982.e6. |
| 31 | BARANWAL M, MAGNER A, SALDINGER J, et al. Struct2Graph: a graph attention network for structure based predictions of protein-protein interactions[J]. BMC Bioinformatics, 2022, 23(1): 370. |
| 32 | WANG S, CHEN W Q, HAN P F, et al. RGN: residue-based graph attention and convolutional network for protein-protein interaction site prediction[J]. Journal of Chemical Information and Modeling, 2022, 62(23): 5961-5974. |
| 33 | SHEN W X, ZENG X, ZHU F, et al. Out-of-the-box deep learning prediction of pharmaceutical properties by broadly learned knowledge-based molecular representations[J]. Nature Machine Intelligence, 2021, 3(4): 334-343. |
| 34 | BUTTON A, MERK D, HISS J A, et al. Automated de novo molecular design by hybrid machine intelligence and rule-driven chemical synthesis[J]. Nature Machine Intelligence, 2019, 1(7): 307-315. |
| 35 | DE CAO N, KIPF T. MolGAN: an implicit generative model for small molecular graphs[EB/OL]. arXiv, 2018: 1805.11973[2023-10-01]. |
| 36 | WINTER R, MONTANARI F, STEFFEN A, et al. Efficient multi-objective molecular optimization in a continuous latent space[J]. Chemical Science, 2019, 10(34): 8016-8024. |
| 37 | DING W Z, NAKAI K T, GONG H P. Protein design via deep learning[J]. Briefings in Bioinformatics, 2022, 23(3): bbac102. |
| 38 | JUMPER J, EVANS R, PRITZEL A, et al. Highly accurate protein structure prediction with AlphaFold[J]. Nature, 2021, 596(7873): 583-589. |
| 39 | BAEK M, DIMAIO F, ANISHCHENKO I, et al. Accurate prediction of protein structures and interactions using a three-track neural network[J]. Science, 2021, 373(6557): 871-876. |
| 40 | DAHIYAT B I, MAYO S L. Protein design automation[J]. Protein Science, 1996, 5(5): 895-903. |
| 41 | DAHIYAT B I, MAYO S L. De novo protein design: fully automated sequence selection[J]. Science, 1997, 278(5335): 82-87. |
| 42 | LI Z X, YANG Y D, FARAGGI E, et al. Direct prediction of profiles of sequences compatible with a protein structure by neural networks with fragment-based local and energy-based nonlocal profiles[J]. Proteins: Structure, Function, and Bioinformatics, 2014, 82(10): 2565-2573. |
| 43 | DAI L, YANG Y D, KIM H R, et al. Improving computational protein design by using structure-derived sequence profile[J]. Proteins: Structure, Function, and Bioinformatics, 2010, 78(10): 2338-2348. |
| 44 | YANG Y D, ZHOU Y Q. Ab initio folding of terminal segments with secondary structures reveals the fine difference between two closely related all-atom statistical energy functions[J]. Protein Science, 2008, 17(7): 1212-1219. |
| 45 | WANG J X, CAO H L, ZHANG J Z H, et al. Computational protein design with deep learning neural networks[J]. Scientific Reports, 2018, 8: 6349. |
| 46 | O'CONNELL J, LI Z X, HANSON J, et al. SPIN2: predicting sequence profiles from protein structures using deep neural networks[J]. Proteins: Structure, Function, and Bioinformatics, 2018, 86(6): 629-633. |
| 47 | CHEN S, SUN Z, LIN L H, et al. To improve protein sequence profile prediction through image captioning on pairwise residue distance map[J]. Journal of Chemical Information and Modeling, 2020, 60(1): 391-399. |
| 48 | KRIZHEVSKY A, SUTSKEVER I, HINTON G E. ImageNet classification with deep convolutional neural networks[J]. Communications of the ACM, 2017, 60(6): 84-90. |
| 49 | ZHANG Y, CHEN Y, WANG C R, et al. ProDCoNN: protein design using a convolutional neural network[J]. Proteins: Structure, Function, and Bioinformatics, 2020, 88(7): 819-829. |
| 50 | ANAND N, EGUCHI R, MATHEWS I I, et al. Protein sequence design with a learned potential[J]. Nature Communications, 2022, 13: 746. |
| 51 | QI Y F, ZHANG J Z H. DenseCPD: improving the accuracy of neural-network-based computational protein sequence design with DenseNet[J]. Journal of Chemical Information and Modeling, 2020, 60(3): 1245-1252. |
| 52 | HUANG G, LIU Z, VAN DER MAATEN L, et al. Densely connected convolutional networks[C]//2017 IEEE Conference on Computer Vision and Pattern Recognition (CVPR). July 21-26, 2017, Honolulu, HI, USA. IEEE, 2017: 2261-2269. |
| 53 | SHROFF R, COLE A W, DIAZ D J, et al. Discovery of novel gain-of-function mutations guided by structure-based deep learning[J]. ACS Synthetic Biology, 2020, 9(11): 2927-2935. |
| 54 | LU H Y, DIAZ D J, CZARNECKI N J, et al. Machine learning-aided engineering of hydrolases for PET depolymerization[J]. Nature, 2022, 604(7907): 662-667. |
| 55 | NORN C, WICKY B I M, JUERGENS D, et al. Protein sequence design by conformational landscape optimization[J]. Proceedings of the National Academy of Sciences of the United States of America, 2021, 118(11): e2017228118. |
| 56 | YANG J Y, ANISHCHENKO I, PARK H, et al. Improved protein structure prediction using predicted interresidue orientations[J]. Proceedings of the National Academy of Sciences of the United States of America, 2020, 117(3): 1496-1503. |
| 57 | WANG X, FLANNERY S T, KIHARA D. Protein docking model evaluation by graph neural networks[J]. Frontiers in Molecular Biosciences, 2021, 8: 647915. |
| 58 | KIPF T N, WELLING M. Semi-supervised classification with graph convolutional networks[EB/OL]. arXiv, 2016: 1609.02907[2023-01-10]. |
| 59 | INGRAHAM J, GARG V K, BARZILAY R, et al. Generative models for graph-based protein design[C/OL]// Advances in Neural Information Processing Systems 32 (NeurIPS 2019), December 2019, Vancouver, Canada, Neural Information Processing Systems Foundation, 2019[2023-01-10]. . |
| 60 | VASWANI A, SHAZEER N, PARMAR N, et al. Attention is all you need[C]//Proceedings of the 31st International Conference on Neural Information Processing Systems. December 4-9, 2017, Long Beach, California, USA. New York: ACM, 2017: 6000-6010. |
| 61 | STROKACH A, BECERRA D, CORBI-VERGE C, et al. Fast and flexible protein design using deep graph neural networks[J]. Cell Systems, 2020, 11(4): 402-411.e4. |
| 62 | JING B, EISMANN S, SURIANA P, et al. Learning from protein structure with geometric vector perceptrons[EB/OL]. arXiv, 2020: 2009.01411[2023-01-10]. . |
| 63 | ORELLANA G A, CACERES-DELPIANO J, IBAÑEZ R, et al. Protein sequence sampling and prediction from structural data[EB/OL]. bioRxiv, 2021[2023-01-10] . |
| 64 | LI A J, LU M, DESTA I, et al. Neural network-derived Potts models for structure-based protein design using backbone atomic coordinates and tertiary motifs[J]. Protein Science, 2023, 32(2): e4554. |
| 65 | ZHENG F, ZHANG J, GRIGORYAN G. Tertiary structural propensities reveal fundamental sequence/structure relationships[J]. Structure, 2015, 23(5): 961-971. |
| 66 | HSU C, VERKUIL R, LIU J, et al. Learning inverse folding from millions of predicted structures[C/OL]//Proceedings of the 39th International Conference on Machine Learning. Proceedings of Machine Learning Research, PMLR. 2022: 8946-8970 [2023-01-10]. . |
| 67 | MCPARTLON M, LAI B, XU J B. A deep SE(3)-equivariant model for learning inverse protein folding[EB/OL]. bioXiv, 202[2023-01-10]. . |
| 68 | ZADEH A, CHEN M, PORIA S, et al. Tensor fusion network for multimodal sentiment analysis[EB/OL]. arXiv, 2017: 1707.07250[2023-01-10]. . |
| 69 | XIONG P, HU X H, HUANG B, et al. Increasing the efficiency and accuracy of the ABACUS protein sequence design method[J]. Bioinformatics, 2020, 36(1): 136-144. |
| 70 | LIU Y F, ZHANG L, WANG W L, et al. Rotamer-free protein sequence design based on deep learning and self-consistency[J]. Nature Computational Science, 2022, 2(7): 451-462. |
| 71 | XIONG P, WANG M, ZHOU X Q, et al. Protein design with a comprehensive statistical energy function and boosted by experimental selection for foldability[J]. Nature Communications, 2014, 5: 5330. |
| 72 | RONEY J P, OVCHINNIKOV S. State-of-the-art estimation of protein model accuracy using AlphaFold[J]. Physical Review Letters, 2022, 129(23): 238101. |
| 73 | DAUPARAS J, ANISHCHENKO I, BENNETT N, et al. Robust deep learning-based protein sequence design using ProteinMPNN[J]. Science, 2022, 378(6615): 49-56. |
| 74 | HUANG B, FAN T W, WANG K Y, et al. Accurate and efficient protein sequence design through learning concise local environment of residues[J]. Bioinformatics, 2023: btad122. |
| 75 | ZHENG Z, DENG Y, XUE D, et al. Structure-informed language models are protein designers[EB/OL]. arXiv, 2023: 2302.01649[2023-02-10]. . |
| 76 | INGRAHAM J, GARG V K, BARZILAY R, et al. Generative models for graph-based protein design[C]// Proceedings of the 33rd International Conference on Neural Information Processing Systems, 8-14 December 2019, Vancouver, Canada, Curran Associates Inc, 2019:1417[2023-01-10]. . |
| 77 | GAO Z Y, TAN C, LI S Z. ProDesign: toward effective and efficient protein design[EB/OL]. arXiv, 2022[2023-01-10]. . |
| 78 | TAN C, GAO Z Y, XIA J, et al. Generative de novo protein design with global context[EB/OL]. arXiv, 2022[2023-01-10]. . |
| 79 | GAO Z Y, TAN C, LI S Z. AlphaDesign: a graph protein design method and benchmark on AlphaFoldDB[EB/OL]. arXiv, 2022[2023-01-10]. . |
| 80 | ANISHCHENKO I, PELLOCK S J, CHIDYAUSIKU T M, et al. De novo protein design by deep network hallucination[J]. Nature, 2021, 600(7889): 547-552. |
| 81 | TISCHER D, LISANZA S, WANG J, et al. Design of proteins presenting discontinuous functional sites using deep learning[EB/OL]. bioXiv, 2020[2023-01-10]. . |
| 82 | WANG J, LISANZA S, JUERGENS D, et al. Scaffolding protein functional sites using deep learning[J]. Science, 2022, 377(6604): 387-394. |
| 83 | ZHANG S H, XU Y J, PEI J F, et al. AutoFoldFinder: an automated adaptive optimization toolkit for de novo protein fold design[EB/OL]. 2021[2023-01-10]. . |
| 84 | YEH A H W, NORN C, KIPNIS Y, et al. De novo design of luciferases using deep learning[J]. Nature, 2023, 614(7949): 774-780. |
| 85 | DOU J Y, VOROBIEVA A A, SHEFFLER W, et al. De novo design of a fluorescence-activating β-barrel[J]. Nature, 2018, 561(7724): 485-491. |
| 86 | CAO L X, COVENTRY B, GORESHNIK I, et al. Design of protein-binding proteins from the target structure alone[J]. Nature, 2022, 605(7910): 551-560. |
| 87 | HUANG B, XU Y, HU X H, et al. A backbone-centred energy function of neural networks for protein design[J]. Nature, 2022, 602(7897): 523-528. |
| 88 | LIANG S D, LI Z X, ZHAN J, et al. De novo protein design by an energy function based on series expansion in distance and orientation dependence[J]. Bioinformatics, 2021, 38(1): 86-93. |
| 89 | LIANG S D, ZHENG D D, ZHANG C, et al. Fast and accurate prediction of protein side-chain conformations[J]. Bioinformatics, 2011, 27(20): 2913-2914. |
| 90 | LIANG S D, ZHOU Y Q, GRISHIN N, et al. Protein side chain modeling with orientation-dependent atomic force fields derived by series expansions[J]. Journal of Computational Chemistry, 2011, 32(8): 1680-1686. |
| 91 | LIANG S D, ZHANG C, ZHOU Y Q. LEAP: highly accurate prediction of protein loop conformations by integrating coarse-grained sampling and optimized energy scores with all-atom refinement of backbone and side chains[J]. Journal of Computational Chemistry, 2014, 35(4): 335-341. |
| 92 | ANAND N, HUANG P S. Generative modeling for protein structures[C/OL]// 6th International Conference on Learning Representations, Vancouver, BC, Canada, April 30-May 3, 2018[2023-01-10]. . |
| 93 | ANAND N, EGUCHI R, HUANG P S. Fully differentiable full-atom protein backbone generation[EB/OL]. ICLR 2019 Workshop on Deep Generative Models for Highly Structured Data, 2019[2023-01-10]. . |
| 94 | EGUCHI R R, CHOE C A, HUANG P S. Ig-VAE: generative modeling of protein structure by direct 3D coordinate generation[J]. PLoS Computational Biology, 2022, 18(6): e1010271. |
| 95 | LAI B Q, MCPARTLON M, XU J B. End-to-End deep structure generative model for protein design[EB/OL]. bioRxiv, 2022[2023-01-10]. . |
| 96 | GUO X J, DU Y Q, TADEPALLI S, et al. Generating tertiary protein structures via interpretable graph variational autoencoders[J]. Bioinformatics Advances, 2021, 1(1): vbab036. |
| 97 | HARTEVELD Z, SOUTHERN J, LOUKAS A, et al. Deep sharpening of topological features for de novo protein design[EB/OL]. ICLR 2022 Machine Learning for Drug Discovery, 2022 [2023-01-10]. . |
| 98 | HO J, JAIN A, ABBEEL P. Denoising diffusion probabilistic models[EB/OL]. arXiv, 2020: 2006.11239. . |
| 99 | SOHL-DICKSTEIN J, WEISS E A, MAHESWARANATHAN N, et al. Deep unsupervised learning using nonequilibrium thermodynamics[C]//Proceedings of the 32nd International Conference on International Conference on Machine Learning-Volume 37. July 6-11, 2015, Lille, France. New York: ACM, 2015: 2256-2265. |
| 100 | WATSON J L, JUERGENS D, BENNETT N R, et al. Broadly applicable and accurate protein design by integrating structure prediction networks and diffusion generative models[EB/OL]. bioXiv, 2022[2023-01-10]. . |
| 101 | TRIPPE B L, YIM J, TISCHER D, et al. Diffusion probabilistic modeling of protein backbones in 3D for the motif-scaffolding problem[EB/OL]. arXiv, 2022: 2206.04119[2023-01-10]. . |
| 102 | WU K E, YANG K K, BERG R V D, et al. Protein structure generation via folding diffusion[EB/OL]. arXiv, 2022: 2209.15611[2023-01-10]. . |
| 103 | LEE J S, KIM P. ProteinSGM: score-based generative modeling for de novo protein design[EB/OL]. 2022[2023-01-10]. . |
| 104 | LEAVER-FAY A, TYKA M, LEWIS S M, et al. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules[J]. Methods in Enzymology, 2011, 487: 545-574. |
| 105 | INGRAHAM J, BARANOV M, COSTELLO Z, et al. Illuminating protein space with a programmable generative model[EB/OL]. bioXiv, 2022[2023-01-10]. . |
| 106 | ANAND N, ACHIM T. Protein structure and sequence generation with equivariant denoising diffusion probabilistic models[EB/OL]. arXiv, 2022: 2205.15019[2023-01-10]. . |
| 107 | DEVLIN J, CHANG M, LEE K, et al. BERT: Pre-training of deep bidirectional transformers for language understanding[EB/OL]. arXiv, 2018: 1810.04805[2023-01-10]. . |
| 108 | DE BORTOLI V, MATHIEU E, HUTCHINSON M, et al. Riemannian score-based generative modelling[EB/OL]. arXiv, 2022: 2202.02763[2023-01-10]. . |
| 109 | LEACH A, SCHMON S M, DEGIACOMI M T, et al. Denoising diffusion probabilistic models on SO(3) for rotational alignment[EB/OL]. ICLR 2022 Workshop on Geometrical and Topological Representation Learning, 2022[2023-01-10]. . |
| 110 | LIU Y F, CHEN L H, LIU H Y. De novo protein backbone generation based on diffusion with structured priors and adversarial training[EB/OL]. bioRxiv, 2022[2023-01-10]. . |
| 111 | RIVES A, MEIER J, SERCU T, et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences[J]. Proceedings of the National Academy of Sciences of the United States of America, 2021, 118(15): e2016239118. |
| 112 | LUO S T, SU Y F, PENG X G, et al. Antigen-specific antibody design and optimization with diffusion-based generative models for protein structures[EB/OL]. bioXiv, 2022[2023-01-10]. . |
| 113 | REPECKA D, JAUNISKIS V, KARPUS L, et al. Expanding functional protein sequence spaces using generative adversarial networks[J]. Nature Machine Intelligence, 2021, 3(4): 324-333. |
| 114 | MADANI A, MCCANN B, NAIK N, et al. ProGen: Language modeling for protein generation[EB/OL]. arXiv, 2020: 2004.03497[2023-01-10]. . |
| 115 | ELNAGGAR A, HEINZINGER M, DALLAGO C, et al. ProtTrans: towards cracking the language of lifes code through self-supervised deep learning and high performance computing[J]. IEEE Transactions on Pattern Analysis and Machine Intelligence, 2021, 44(10), 7112-7127. |
| 116 | GLIGORIJEVIĆ V, BERENBERG D, RA S, et al. Function-guided protein design by deep manifold sampling[EB/OL]. bioRxiv, 2021[2023-01-10]. . |
| 117 | MOFFAT L, KANDATHIL S M, JONES D T. Design in the DARK: learning deep generative models for de novo protein design[EB/OL]. bioRxiv, 2022[2023-01-10]. . |
| 118 | FERRUZ N, SCHMIDT S, HÖCKER B. ProtGPT2 is a deep unsurprised language model for protein design[J]. Nature Communications, 2022,13(1): 4348. |
| 119 | HESSLOW D, ZANICHELLI N, NOTIN P, et al. RITA: a study on scaling up generative protein sequence models[EB/OL]. arXiv, 2022: 2205.05789[2023-01-10]. . |
| 120 | NIJKAMP E, RUFFOLO J, WEINSTEIN E N, et al. ProGen2: exploring the boundaries of protein language models[EB/OL]. arXiv, 2022[2023-01-10]. . |
| 121 | LI Z X, YANG Y D, ZHAN J, et al. Energy functions in de novo protein design: current challenges and future prospects[J]. Annual Review of Biophysics, 2013, 42: 315-335. |
| [1] | 温艳华, 刘合栋, 曹春来, 巫瑞波. 蛋白质工程在医药产业中的应用[J]. 合成生物学, 2025, 6(1): 65-86. |
| [2] | 王子渊, 杨立荣, 吴坚平, 郑文隆. 酶促合成手性氨基酸的研究进展[J]. 合成生物学, 2024, 5(6): 1319-1349. |
| [3] | 奚萌宇, 胡逸灵, 顾玉诚, 戈惠明. 基因组挖掘指导天然药物分子的发现[J]. 合成生物学, 2024, 5(3): 447-473. |
| [4] | 雷茹, 陶慧, 刘天罡. 基因组深度挖掘驱动微生物萜类化合物高效发现[J]. 合成生物学, 2024, 5(3): 507-526. |
| [5] | 朱景勇, 李钧翔, 李旭辉, 张瑾, 毋文静. 深度学习在基于序列的蛋白质互作预测中的应用进展[J]. 合成生物学, 2024, 5(1): 88-106. |
| [6] | 王晟, 王泽琛, 陈威华, 陈珂, 彭向达, 欧发芬, 郑良振, 孙瑨原, 沈涛, 赵国屏. 基于人工智能和计算生物学的合成生物学元件设计[J]. 合成生物学, 2023, 4(3): 422-443. |
| [7] | 康里奇, 谈攀, 洪亮. 人工智能时代下的酶工程[J]. 合成生物学, 2023, 4(3): 524-534. |
| [8] | 孟巧珍, 郭菲. “可折叠性”在酶智能设计改造中的应用研究——以AlphaFold2为例[J]. 合成生物学, 2023, 4(3): 571-589. |
| [9] | 赖奇龙, 姚帅, 查毓国, 白虹, 宁康. 微生物组生物合成基因簇发掘方法及应用前景[J]. 合成生物学, 2023, 4(3): 611-627. |
| [10] | 宋益东, 袁乾沐, 杨跃东. 深度学习在蛋白质功能预测中的应用[J]. 合成生物学, 2023, 4(3): 488-506. |
| [11] | 梁丽亚, 刘嵘明. 靶向DNA的Ⅱ类CRISPR/Cas系统的蛋白工程化改造[J]. 合成生物学, 2023, 4(1): 86-101. |
| [12] | 吕靖伟, 邓子新, 张琪, 丁伟. 基于深度学习识别RiPPs前体肽及裂解位点[J]. 合成生物学, 2022, 3(6): 1262-1276. |
| [13] | 祁延萍, 朱晋, 张凯, 刘彤, 王雅婕. 定向进化在蛋白质工程中的应用研究进展[J]. 合成生物学, 2022, 3(6): 1081-1108. |
| [14] | 涂涛, 罗会颖, 姚斌. 蛋白质工程在饲料用酶研发中的应用研究进展[J]. 合成生物学, 2022, 3(3): 487-499. |
| [15] | 王汇滨, 车昌丽, 游松. Fe/α-酮戊二酸依赖型卤化酶在绿色卤化反应中的研究进展[J]. 合成生物学, 2022, 3(3): 545-566. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
