|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
CRISPR/Cas9编辑系统在微生物天然产物研究中的应用
合成生物学
2024, 5 (3):
658-671.
DOI:10.12211/2096-8280.2023-110
微生物作为天然产物的巨大宝库,一直以来都是研究人员挖掘和开发新的活性化合物的重要来源。目前,利用基因编辑工具发现、生物合成和代谢调控天然产物的研究方法受到该领域研究者的广泛关注。CRISPR/Cas9遗传编辑系统以其独特的灵活靶向优势克服了其他遗传编辑方法常见的对序列同源或位点限制,简化了实验步骤,提高了实验效率,促进了天然产物研究领域的发展。本文主要介绍CRISPR/Cas9系统在微生物天然产物发现、生物合成和工程改造方面的应用,分别从CRISPR/Cas9系统的发展、天然产物生物合成基因簇的克隆和遗传编辑、天然产物结构衍生化和代谢调节、沉默天然产物基因簇的激活这几个方面阐述CRISPR/Cas9系统在微生物天然产物研究领域的优势。最后,针对CRISPR/Cas9系统无法克服的重组效率和宿主适应性问题提供了可行的解决思路。相信随着合成生物学和信息技术的发展,越来越多的与CRISPR/Cas9系统相关的遗传操作工具和方法会被开发,将不断推动天然产物领域的发展进步。
表1
CRISPR/Cas相关的生物合成基因簇编辑策略
正文中引用本图/表的段落
然而,这些传统的克隆技术严重依赖于对生物合成基因簇序列两端的恰当切割,以获得同源末端序列供连接重组使用。CRISPR/Cas9系统的靶向切割能力完美解决了该限制,通过gRNA引导的靶向剪切,可显著提高传统方法的克隆效率。根据该设计思路,许多团队通过结合CRISPR/Cas9系统开发了多种克隆技术,实现了生物合成基因簇的灵活高效克隆(表1)。
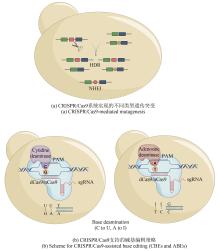
与上面这些较为复杂的基因组编辑手段相比,现在广泛使用的CRISPR/Cas9系统可通过靶向编辑技术直接完成上述复杂重组过程。此外,该系统可实现多位点同时编辑,Cas9蛋白的靶向剪切又能帮助阳性克隆的筛选(表1)。武汉大学孙宇辉团队[34]开发了高效体外ICE(In vitro CRISPR/Cas9-mediated editing)编辑系统,通过额外的T4 DNA连接酶修复体外Cas9/sgRNA反应带来的黏性末端,进而实现了精准无缝的靶向编辑。随后,该团队借助ICE系统分别成功地敲除了来自tetronate RK-682和 dithiolopyrrolone holomycin生物合成基因簇的rkD和homE基因。美国伊利诺伊大学香槟分校(UIUC)赵惠民团队[35]使用温敏型质粒表达来自酿脓链球菌的CRISPR/Cas9系统,并在链霉菌属中实现了多个基因组的靶向编辑。该团队构建的pCRISPomyces质粒可通过Golden Gate组装和等温组装(或传统的消化连接)插入spacer和修复模板,满足快速灵活的基因组编辑。而后通过多重靶向能力成功实现了Streptomyces lividans内31 kb red基因簇的删除。此外,他们还证实该系统适用于多种链霉菌菌株的基因编辑。类似的CRISPR/Cas9介导的基因编辑也被多个团队报道,其中丹麦技术大学Sang Yup Lee团队还证实了在没有引入修复模板时,NHEJ修复通路会在靶向位点周围引入不同大小的缺失突变[36-37,61][图2(a)]。
CRISPR/Cas9在基因编辑方面展现出强大的可拓展的能力,因此很多研究人员开始借助该系统来产生新颖天然产物衍生物、调控天然产物产量和激活沉默的天然产物生物合成基因簇(表1)。天然产物药物的衍生化是优化先导化合物的主要和关键步骤。将生物合成基因簇作为天然产物的遗传单元进行修改,为研究天然产物药物衍生物提供了一个易于操作的平台。日本AIST研究所的Kazuo Shin-ya团队[41]通过体外Cas9反应和Gibson组装在rapamycin生物合成基因簇上成功地实现了不同AT结构域(acyltransferase domain)的替换并对应获得了多个不同的rapamycin衍生物[图3(a)]。此外,该团队进一步通过模块删除插入、结构域的编辑和交换也获得了对应的rapamycin衍生物。该研究表明CRISPR/Cas9可帮助实现模块化天然产物生物合成基因簇的编辑,并实现目的天然产物的衍生化。基于蛋白结构活性位点的突变策略,针对合成脂肽类抗生素enduracidin的生物合成基因簇,Jason Micklefield团队[42]通过替换存在于A结构域(adenylation domain)中的高度保守的类黄素氧还蛋白单元实现了A结构域底物选择性靶向突变,从而合成了多个新的脂肽类抗生素。
由于CRISPR/Cas9系统带来的特异性的靶向编辑和高效的同源重组,很多借助CRISPR/Cas9系统的策略被开发用于沉默生物合成基因簇的激活(表1)。赵惠民团队[47]通过CRISPR/Cas9介导的敲入技术简单地在沉默生物合成基因簇的第一个基因上游引入单向或双向启动子,进而激活了该沉默生物合成基因簇。经过kasO*p和双向启动子P8-kasO*p的敲入,位于S. roseosporus的24号基因簇被成功激活并合成了2个polycyclic tetramate macrolactam类的化合物,同时10号基因簇被激活合成了化合物FR-900098。此外,该团队又在不同的链霉菌的不同生物合成基因簇上验证了该策略,成功获得了新的天然产物。洛克菲勒大学的Sean F. Brady团队[48]开发了包含多重CRISPR/Cas9编辑和TAR克隆的启动子工程系统(multiplexed CRISPR/Cas9- and TAR-mediated promoter engineering method,mCRISTAR)。该系统包含两个质粒——pCRCT:Tam(包含了CRISPR/Cas9系统)和pTARa:Tam[包含Tam(tetarimycin)基因簇的大肠杆菌:酵母:链霉菌穿梭质粒],最后同携带Tam特异性启动子的基因一同转入酵母细胞完成启动子替换。在单筛选标志物存在的情况下,该系统通过一次反应同时完成了4个启动子的替换。此外,经过2轮mCRISTAR反应步骤,该团队成功实现了8个启动子的替换,证实了该系统的灵活和可拓展性。韩国建国大学的Hahk-Soo Kang团队[49]进一步优化了mCRISTAR系统,通过多个质粒分别表达gRNA提高了反应效率。利用优化后的mpCRISTAR(multiple plasmied-based CRISPR/Cas9- and TAR-mediated promoter engineering method)系统,该团队凭借4个质粒单次反应成功完成了acinorhodin生物合成基因簇8个启动子的同时替换。随后,在mCRISTAR系统的基础上,Sean F. Brady团队[50]又开发了可拓展的生物合成基因簇激活方法,miCASTAR(multiplexed CRISPR TAR)。该方法将CRISPR/Cas9系统拆分得到的基因簇和PCR扩增得到的合成启动子一起转入酵母细胞完成TAR克隆。凭借该方法,该团队成功激活了atolypene生物合成基因簇并得到两个新的细菌环状二倍半萜天然产物。
大量的研究已经证实,CRISPR/Cas9系统具有强大的遗传编辑能力以及高度的特异性,可以通过基因编辑和重构基因序列实现天然产物生物合成基因簇的调控。然而,非特异性重组和非同源末端修复往往会带来许多副产物,尤其当靶向序列高度同源时,细胞内反应效率取决了宿主的同源重组能力。实际应用中,研究人员可以通过选择非同源的gRNA、更长的同源修复模板,或将生物合成基因簇转移到更适宜遗传编辑的宿主内来克服以上不足。众所周知,广泛成熟应用的来自酿脓链球菌的CRISPR/Cas9系统识别靶向序列时需要提前满足合适的PAM位点(5′-NGG-3′)。该条件限制了它在富含AT序列上的应用。此外,早期研究报告显示,基于SpCas9的编辑质粒无法在几种工业链霉菌中使用。CRISPR/Cpf1系统又称CRISPR/Cas12a系统,和CRISRP/Cas9系统类似,但CRISPR/Cpf1系统使用更小的Cpf1内切酶在富含T的PAM位点下游裂解gRNA靶向位置并得到黏性末端,同时Cpf1内切酶自身足以实现前体CRISPR RNA的成熟加工,该系统还可以对多个靶点同时编辑[67-69]。中国科学院芦银华团队[70]借助CRISPR/Cpf1系统在链霉菌中同样实现了同源重组突变、非同源末端突变和CRISPRi实验,进而证实了CRISPR/Cpf1系统是一种可替代CRISPR/Cas9的强大基因编辑工具,尤其是当CRISPR/Cas9系统无法很好地发挥作用时。Jochen Schmid团队[71]也开发了基于CRISPR/dCas12a的转录激活和转录抑制系统,并在Paenibacillus polymyxa和Escherichia coli上得以验证。同CRISPR/Cas9系统,多个借助CRISPR/Cas12a系统的基因簇克隆技术相继被开发。赵惠民团队[32]联合Cas12a的靶向剪切、DNA聚合酶体外连接和Cre-lox重组系统体内环化,开发了CAPTURE(Cas12a-assisted precise targeted cloning using in vivo Cre-lox recombination)策略,凭借该策略成功地从放线菌和芽孢杆菌中克隆了43个基因簇,并得到了7个成功异源表达基因簇。华东理工大学谭高翼团队[33]同样借助CRISPR/Cas12a的高效靶向剪切能力,结合细菌人工染色体文库构建,开发了CAT-FISHING技术,成功地从多个放线菌基因组DNA样本中克隆得到多个生物合成基因簇,并通过异源表达获得一个新的大环内酰胺类化合物Marinolactam A。由于不同长度的gRNA序列将指导Cpf1切割得到不同长度的黏性末端[46],上海师范大学王金团队[72]根据这一特征,先后利用不同长度的gRNA序列开发了C-Brick DNA组装标准元件,借助4个标准接口序列实现表达模块的灵活替换和CCTL(Cpf1-assisted Cutting and Taq DNA ligase-mediated Ligation)方法,联合Taq DNA连接酶实现了act基因簇启动子的高效替换。
本文的其它图/表
|