CRISPR-Cas9 assisted gene disruption in the higher fungus Ganoderma species
1
2017
... 2015年之前,CRISPR-Cas9系统尚未应用于真菌,研究者一直使用经典的基因工程手段来研究丝状真菌体系,尽管传统手段编辑丝状真菌基因可以达到多种研究目的,但仅限于一些模式真菌,其效率较低,编辑位点较少,且花费大量的时间和劳动力.CRISPR-Cas系统作为新一代基因组编辑技术,自出现以来,经过多种巧妙的改造,在多个物种中的基因编辑能力被相继证实,得到迅速而广泛的应用,为生命科学和医学研究带来前所未有的革新.自2015年起,CRISPR-Cas9系统被国内外研究学者陆续在丝状真菌体系中进行研发和应用,在短短几年内飞速发展,在不同的丝状真菌中各种高效的、CRISPR-Cas体系已经被陆续建立起来[45-50].近三年来,一些大型真菌的CRISPR-Cas9编辑系统也开始发展并建立,例如灵芝[88]、灰盖鬼伞[89]、蛹虫草[58, 90]和竹黄菌[91]等.CRISPR基因组编辑技术能对真菌基因组特定基因位点进行精准、高效的改造,在真菌细胞工厂构建和优化改造中展示了强大的功能,对真菌功能基因组研究和合成生物学的发展起着非常重要的作用. ...
Genome editing in the mushroom-forming basidiomycete Coprinopsis cinerea, optimized by a high-throughput transformation system
1
2017
... 2015年之前,CRISPR-Cas9系统尚未应用于真菌,研究者一直使用经典的基因工程手段来研究丝状真菌体系,尽管传统手段编辑丝状真菌基因可以达到多种研究目的,但仅限于一些模式真菌,其效率较低,编辑位点较少,且花费大量的时间和劳动力.CRISPR-Cas系统作为新一代基因组编辑技术,自出现以来,经过多种巧妙的改造,在多个物种中的基因编辑能力被相继证实,得到迅速而广泛的应用,为生命科学和医学研究带来前所未有的革新.自2015年起,CRISPR-Cas9系统被国内外研究学者陆续在丝状真菌体系中进行研发和应用,在短短几年内飞速发展,在不同的丝状真菌中各种高效的、CRISPR-Cas体系已经被陆续建立起来[45-50].近三年来,一些大型真菌的CRISPR-Cas9编辑系统也开始发展并建立,例如灵芝[88]、灰盖鬼伞[89]、蛹虫草[58, 90]和竹黄菌[91]等.CRISPR基因组编辑技术能对真菌基因组特定基因位点进行精准、高效的改造,在真菌细胞工厂构建和优化改造中展示了强大的功能,对真菌功能基因组研究和合成生物学的发展起着非常重要的作用. ...
Efficient CRISPR-Cas9 gene disruption system in edible-medicinal mushroom Cordyceps militaris
1
2018
... 2015年之前,CRISPR-Cas9系统尚未应用于真菌,研究者一直使用经典的基因工程手段来研究丝状真菌体系,尽管传统手段编辑丝状真菌基因可以达到多种研究目的,但仅限于一些模式真菌,其效率较低,编辑位点较少,且花费大量的时间和劳动力.CRISPR-Cas系统作为新一代基因组编辑技术,自出现以来,经过多种巧妙的改造,在多个物种中的基因编辑能力被相继证实,得到迅速而广泛的应用,为生命科学和医学研究带来前所未有的革新.自2015年起,CRISPR-Cas9系统被国内外研究学者陆续在丝状真菌体系中进行研发和应用,在短短几年内飞速发展,在不同的丝状真菌中各种高效的、CRISPR-Cas体系已经被陆续建立起来[45-50].近三年来,一些大型真菌的CRISPR-Cas9编辑系统也开始发展并建立,例如灵芝[88]、灰盖鬼伞[89]、蛹虫草[58, 90]和竹黄菌[91]等.CRISPR基因组编辑技术能对真菌基因组特定基因位点进行精准、高效的改造,在真菌细胞工厂构建和优化改造中展示了强大的功能,对真菌功能基因组研究和合成生物学的发展起着非常重要的作用. ...
Genome editing in Shiraia bambusicola using CRISPR-Cas9 system
1
2017
... 2015年之前,CRISPR-Cas9系统尚未应用于真菌,研究者一直使用经典的基因工程手段来研究丝状真菌体系,尽管传统手段编辑丝状真菌基因可以达到多种研究目的,但仅限于一些模式真菌,其效率较低,编辑位点较少,且花费大量的时间和劳动力.CRISPR-Cas系统作为新一代基因组编辑技术,自出现以来,经过多种巧妙的改造,在多个物种中的基因编辑能力被相继证实,得到迅速而广泛的应用,为生命科学和医学研究带来前所未有的革新.自2015年起,CRISPR-Cas9系统被国内外研究学者陆续在丝状真菌体系中进行研发和应用,在短短几年内飞速发展,在不同的丝状真菌中各种高效的、CRISPR-Cas体系已经被陆续建立起来[45-50].近三年来,一些大型真菌的CRISPR-Cas9编辑系统也开始发展并建立,例如灵芝[88]、灰盖鬼伞[89]、蛹虫草[58, 90]和竹黄菌[91]等.CRISPR基因组编辑技术能对真菌基因组特定基因位点进行精准、高效的改造,在真菌细胞工厂构建和优化改造中展示了强大的功能,对真菌功能基因组研究和合成生物学的发展起着非常重要的作用. ...
Evolved Cas9 variants with broad PAM compatibility and high DNA specificity
1
2018
... 随着CRISPR技术的发展,相信CRISPR-Cas9系统在诸多方面可以进一步优化改进,包括降低脱靶效应、解除Cas蛋白的PAM序列的限制性、提升精确定点编辑的效率等.例如近期研究表明可以通过人工改造Cas9蛋白(xCas9、SpCas9-non-G PAMs等)[92-95]或者是寻找并鉴定更多的Cas同源蛋白(Cas12b、Cas14等)[96-98],使其识别更多的PAM位点.与此同时,基于dCas9或者nCas9的CRISPR相关衍生技术在工业真菌中有待开发,例如CRISPRa和CRISPRi介导的基因表达调控技术[99-103]在丝状真菌中尚未报道,多功能CRISPR介导的组合调控技术还没有在丝状真菌中研发和建立.未来建立不局限于某个真菌物种的通用编辑技术体系,以及将CRISPR系统与高通量技术相结合进行高通量编辑改造等都值得期待.除前文提到的在黑曲霉中建立了胞嘧啶碱基编辑器(cytosine base editor,CBE)以外,在其他的丝状真菌中还没有构建起来,并且腺嘌呤碱基编辑器(adenine base editor,ABE)也尚未在丝状真菌中研发报道.随着CRISPR基因组编辑及相关技术的创新发展,我们相信CRISPR系统将在真菌基因功能研究、代谢途径重构、精确表达调控、蛋白定向进化以及高性能底盘构建等方面得到快速发展和技术革新,从而促进真菌合成生物学的快速发展. ...
Engineered CRISPR-Cas9 nuclease with expanded targeting space
0
2018
Continuous evolution of SpCas9 variants compatible with non-G PAMs
0
2020
Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants
1
2020
... 随着CRISPR技术的发展,相信CRISPR-Cas9系统在诸多方面可以进一步优化改进,包括降低脱靶效应、解除Cas蛋白的PAM序列的限制性、提升精确定点编辑的效率等.例如近期研究表明可以通过人工改造Cas9蛋白(xCas9、SpCas9-non-G PAMs等)[92-95]或者是寻找并鉴定更多的Cas同源蛋白(Cas12b、Cas14等)[96-98],使其识别更多的PAM位点.与此同时,基于dCas9或者nCas9的CRISPR相关衍生技术在工业真菌中有待开发,例如CRISPRa和CRISPRi介导的基因表达调控技术[99-103]在丝状真菌中尚未报道,多功能CRISPR介导的组合调控技术还没有在丝状真菌中研发和建立.未来建立不局限于某个真菌物种的通用编辑技术体系,以及将CRISPR系统与高通量技术相结合进行高通量编辑改造等都值得期待.除前文提到的在黑曲霉中建立了胞嘧啶碱基编辑器(cytosine base editor,CBE)以外,在其他的丝状真菌中还没有构建起来,并且腺嘌呤碱基编辑器(adenine base editor,ABE)也尚未在丝状真菌中研发报道.随着CRISPR基因组编辑及相关技术的创新发展,我们相信CRISPR系统将在真菌基因功能研究、代谢途径重构、精确表达调控、蛋白定向进化以及高性能底盘构建等方面得到快速发展和技术革新,从而促进真菌合成生物学的快速发展. ...
Repurposing CRISPR-Cas12b for mammalian genome engineering
1
2018
... 随着CRISPR技术的发展,相信CRISPR-Cas9系统在诸多方面可以进一步优化改进,包括降低脱靶效应、解除Cas蛋白的PAM序列的限制性、提升精确定点编辑的效率等.例如近期研究表明可以通过人工改造Cas9蛋白(xCas9、SpCas9-non-G PAMs等)[92-95]或者是寻找并鉴定更多的Cas同源蛋白(Cas12b、Cas14等)[96-98],使其识别更多的PAM位点.与此同时,基于dCas9或者nCas9的CRISPR相关衍生技术在工业真菌中有待开发,例如CRISPRa和CRISPRi介导的基因表达调控技术[99-103]在丝状真菌中尚未报道,多功能CRISPR介导的组合调控技术还没有在丝状真菌中研发和建立.未来建立不局限于某个真菌物种的通用编辑技术体系,以及将CRISPR系统与高通量技术相结合进行高通量编辑改造等都值得期待.除前文提到的在黑曲霉中建立了胞嘧啶碱基编辑器(cytosine base editor,CBE)以外,在其他的丝状真菌中还没有构建起来,并且腺嘌呤碱基编辑器(adenine base editor,ABE)也尚未在丝状真菌中研发报道.随着CRISPR基因组编辑及相关技术的创新发展,我们相信CRISPR系统将在真菌基因功能研究、代谢途径重构、精确表达调控、蛋白定向进化以及高性能底盘构建等方面得到快速发展和技术革新,从而促进真菌合成生物学的快速发展. ...
Engineering of CRISPR-Cas12b for human genome editing
0
2019
Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants
1
2020
... 随着CRISPR技术的发展,相信CRISPR-Cas9系统在诸多方面可以进一步优化改进,包括降低脱靶效应、解除Cas蛋白的PAM序列的限制性、提升精确定点编辑的效率等.例如近期研究表明可以通过人工改造Cas9蛋白(xCas9、SpCas9-non-G PAMs等)[92-95]或者是寻找并鉴定更多的Cas同源蛋白(Cas12b、Cas14等)[96-98],使其识别更多的PAM位点.与此同时,基于dCas9或者nCas9的CRISPR相关衍生技术在工业真菌中有待开发,例如CRISPRa和CRISPRi介导的基因表达调控技术[99-103]在丝状真菌中尚未报道,多功能CRISPR介导的组合调控技术还没有在丝状真菌中研发和建立.未来建立不局限于某个真菌物种的通用编辑技术体系,以及将CRISPR系统与高通量技术相结合进行高通量编辑改造等都值得期待.除前文提到的在黑曲霉中建立了胞嘧啶碱基编辑器(cytosine base editor,CBE)以外,在其他的丝状真菌中还没有构建起来,并且腺嘌呤碱基编辑器(adenine base editor,ABE)也尚未在丝状真菌中研发报道.随着CRISPR基因组编辑及相关技术的创新发展,我们相信CRISPR系统将在真菌基因功能研究、代谢途径重构、精确表达调控、蛋白定向进化以及高性能底盘构建等方面得到快速发展和技术革新,从而促进真菌合成生物学的快速发展. ...
Beyond editing: repurposing CRISPR-Cas9 for precision genome regulation and interrogation
1
2016
... 随着CRISPR技术的发展,相信CRISPR-Cas9系统在诸多方面可以进一步优化改进,包括降低脱靶效应、解除Cas蛋白的PAM序列的限制性、提升精确定点编辑的效率等.例如近期研究表明可以通过人工改造Cas9蛋白(xCas9、SpCas9-non-G PAMs等)[92-95]或者是寻找并鉴定更多的Cas同源蛋白(Cas12b、Cas14等)[96-98],使其识别更多的PAM位点.与此同时,基于dCas9或者nCas9的CRISPR相关衍生技术在工业真菌中有待开发,例如CRISPRa和CRISPRi介导的基因表达调控技术[99-103]在丝状真菌中尚未报道,多功能CRISPR介导的组合调控技术还没有在丝状真菌中研发和建立.未来建立不局限于某个真菌物种的通用编辑技术体系,以及将CRISPR系统与高通量技术相结合进行高通量编辑改造等都值得期待.除前文提到的在黑曲霉中建立了胞嘧啶碱基编辑器(cytosine base editor,CBE)以外,在其他的丝状真菌中还没有构建起来,并且腺嘌呤碱基编辑器(adenine base editor,ABE)也尚未在丝状真菌中研发报道.随着CRISPR基因组编辑及相关技术的创新发展,我们相信CRISPR系统将在真菌基因功能研究、代谢途径重构、精确表达调控、蛋白定向进化以及高性能底盘构建等方面得到快速发展和技术革新,从而促进真菌合成生物学的快速发展. ...
Filamentous fungi
1
2016
... 丝状真菌(filamentous fungi)作为一类重要的真核微生物,在自然界中分布广泛,在生物技术领域和基础生物学研究中扮演着重要的角色[1-3].例如,粗糙脉孢菌(Neurospora crassa)和构巢曲霉(Aspergillus nidulans)等被作为模式真菌用于研究遗传、发育和细胞生物学基础问题.球孢白僵菌(Beauveria bassiana)、稻瘟病菌(Magnaporthe oryzae)、玉米黑粉菌(Ustilago maydis)和烟曲霉(Aspergillus fumigatus)等,分别是昆虫、植物和人类的致病型真菌,深入研究其致病机理对解决真菌病害问题意义重大.里氏木霉(Trichoderma reesei)、黑曲霉(Aspergillus niger)、米曲霉(Aspergillus oryzae)、青霉菌(Penicillium)和嗜热毁丝霉(Myceliophthora thermophila)等工业丝状真菌因其代谢多样性、蛋白分泌效率高、翻译后修饰能力强等特点,被广泛用作生产有机酸、工业酶制剂、抗生素等大宗发酵产品的“细胞工厂”[3-5]. ...
The importance of fungal pathogens and antifungal coatings in medical device infections
0
2018
Production of recombinant proteins by filamentous fungi
2
2012
... 丝状真菌(filamentous fungi)作为一类重要的真核微生物,在自然界中分布广泛,在生物技术领域和基础生物学研究中扮演着重要的角色[1-3].例如,粗糙脉孢菌(Neurospora crassa)和构巢曲霉(Aspergillus nidulans)等被作为模式真菌用于研究遗传、发育和细胞生物学基础问题.球孢白僵菌(Beauveria bassiana)、稻瘟病菌(Magnaporthe oryzae)、玉米黑粉菌(Ustilago maydis)和烟曲霉(Aspergillus fumigatus)等,分别是昆虫、植物和人类的致病型真菌,深入研究其致病机理对解决真菌病害问题意义重大.里氏木霉(Trichoderma reesei)、黑曲霉(Aspergillus niger)、米曲霉(Aspergillus oryzae)、青霉菌(Penicillium)和嗜热毁丝霉(Myceliophthora thermophila)等工业丝状真菌因其代谢多样性、蛋白分泌效率高、翻译后修饰能力强等特点,被广泛用作生产有机酸、工业酶制剂、抗生素等大宗发酵产品的“细胞工厂”[3-5]. ...
... [3-5]. ...
Aspergillus as a versatile cell factory for organic acid production
0
2017
Developments and opportunities in fungal strain engineering for the production of novel enzymes and enzyme cocktails for plant biomass degradation
1
2019
... 丝状真菌(filamentous fungi)作为一类重要的真核微生物,在自然界中分布广泛,在生物技术领域和基础生物学研究中扮演着重要的角色[1-3].例如,粗糙脉孢菌(Neurospora crassa)和构巢曲霉(Aspergillus nidulans)等被作为模式真菌用于研究遗传、发育和细胞生物学基础问题.球孢白僵菌(Beauveria bassiana)、稻瘟病菌(Magnaporthe oryzae)、玉米黑粉菌(Ustilago maydis)和烟曲霉(Aspergillus fumigatus)等,分别是昆虫、植物和人类的致病型真菌,深入研究其致病机理对解决真菌病害问题意义重大.里氏木霉(Trichoderma reesei)、黑曲霉(Aspergillus niger)、米曲霉(Aspergillus oryzae)、青霉菌(Penicillium)和嗜热毁丝霉(Myceliophthora thermophila)等工业丝状真菌因其代谢多样性、蛋白分泌效率高、翻译后修饰能力强等特点,被广泛用作生产有机酸、工业酶制剂、抗生素等大宗发酵产品的“细胞工厂”[3-5]. ...
The post-genomic era of Trichoderma reesei: what's Next?
1
2016
... 近年来,大量不同种属的丝状真菌基因组测序工作已经完成.目前,科研工作者可以从JGI和FungiDB等数据库中获取超过1000种丝状真菌基因组序列和相关注释信息,丝状真菌研究和利用进入“后基因组时代”,以基因组水平遗传改造为核心的真菌合成生物学也已成为当代生物技术中具有新兴活力的研究方向之一[6-7].尽管丝状真菌后基因组时代已经开启,但丝状真菌具有多细胞的结构特点,与酵母等这类单细胞的真菌相比较,其生长发育相对复杂,分子遗传操作较为困难,因此丝状真菌合成生物学的研究尚处在方兴未艾的起步阶段. ...
How a fungus shapes biotechnology: 100 years of Aspergillus niger research
1
2018
... 近年来,大量不同种属的丝状真菌基因组测序工作已经完成.目前,科研工作者可以从JGI和FungiDB等数据库中获取超过1000种丝状真菌基因组序列和相关注释信息,丝状真菌研究和利用进入“后基因组时代”,以基因组水平遗传改造为核心的真菌合成生物学也已成为当代生物技术中具有新兴活力的研究方向之一[6-7].尽管丝状真菌后基因组时代已经开启,但丝状真菌具有多细胞的结构特点,与酵母等这类单细胞的真菌相比较,其生长发育相对复杂,分子遗传操作较为困难,因此丝状真菌合成生物学的研究尚处在方兴未艾的起步阶段. ...
Development and applications of CRISPR-Cas9 for genome engineering
4
2014
... 基因组编辑(genome editing)是基于位点特异性的核酸酶对基因组双链DNA进行切割,从而激活特定位点的插入、删除或者替换的技术.目前应用最广泛的核酸酶主要有三种,包括锌指核酸酶技术(zinc-finger nucleases,ZFNs)、转录激活因子类似的核酸酶(transcription activator-like effector nuclease proteins,TALENs)以及成簇的规律间隔的短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR),如图1所示.其中CRISPR/Cas9系统是目前使用最为广泛的技术[8-12].基因组编辑技术作为关键的反向遗传学研究方法,是功能基因组研究与遗传改造重要的手段,显著促进了合成生物学研究的迅猛发展,在真菌遗传育种方面具有重要应用前景,是当前真菌合成生物学的研究热点[13-14].本文将对基因组编辑技术的类型和作用机理进行简要综述,重点介绍工业丝状真菌(industrial filamentous fungi)中CRISPR系统介导的基因组编辑技术的研发进展和发展趋势,以期为工业丝状真菌研究提供参考. ...
... 相关的研究发现[19],在基因组上特定的DNA位点发生双链断裂(double-strand breaks,DSBs)以后,其发生同源重组效率将会极大提升,这一发现给基因组编辑研究带来了新的突破.因此,近年来研究学者陆续研发了人工特异性核酸内切酶技术[8-15],包括ZFNs技术、TALENs技术和CRISPR/Cas9技术,从而能够在基因组特异性的DNA位点上进行双链断裂(图1).这些核酸内切酶介导的基因组编辑均是利用DSBs后细胞的修复机制进行操作的,双链断裂后会诱导细胞体内两种修复途径(NHEJ或HR)对断裂的双链DNA进行编辑修复[图1(a)],来实现对基因组上特异性位点的精确编辑. ...
... 2013年,第三代基因组编辑系统CRISPR/Cas9系统开始出现并迅速崛起[8-9].与之前的ZFNs及TALENs技术相比,CRISPR/Cas9系统具有显著优点,设计简单和改造方便,向导RNA(single guideRNA, sgRNA)非常容易获得而且成本很低,其识别序列仅需要一个紧随PAM的20bp的序列[图1(d)],可以利用体外转录和体内表达等方式进行操作,能同时作用于多个靶点实现多基因同时打靶.因此,CRISPR/Cas9系统自出现以后就受到广泛的关注和应用[10-16],是本文接下来重点综述内容. ...
... 目前Class Ⅱ 的Type Ⅱ型中CRISPR-Cas9系统是被改造得最为成功的核酸酶,研究表明仅Cas9单一蛋白就能发挥切割的功能[34].Cas9蛋白共有2个核酸酶结构域RuvC和HNH,这一发现为CRISPR-Cas9核酸酶技术的研发奠定了基础.引导Cas9的RNA由两部分组成,一部分是靶向目标系列的crRNA,另一部分是tracrRNA.crRNA与tracrRNA结合后再与Cas9蛋白形成复合体,对原型间隔序列邻近基序(protospacer adjacent motif,PAM)(5'-NGG-3')区域附近的位点进行双链断裂,进而激活细胞的修复机制.2012年,Jinek等[35]将crRNA与tracrRNA结构进行重新设计,形成了一个简便的引导RNA(single guide RNA),从此sgRNA的操作使用都更为简易.2013年,Church实验室、张锋实验室报道了在动物细胞中CRISPR-Cas9新一代核酸酶技术的研发和建立,其编辑效率不低于ZFNs和TALENs,有的甚至高于TALENs [36-37].随后,CRISPR/Cas9系统进入快速的黄金发展时期,目前已经在相当多的物种(包括人类及其他动物、植物和微生物)中成功得到了高效的研发和应用[8-16]. ...
Genome editing. The new frontier of genome engineering with CRISPR-Cas9
1
2014
... 2013年,第三代基因组编辑系统CRISPR/Cas9系统开始出现并迅速崛起[8-9].与之前的ZFNs及TALENs技术相比,CRISPR/Cas9系统具有显著优点,设计简单和改造方便,向导RNA(single guideRNA, sgRNA)非常容易获得而且成本很低,其识别序列仅需要一个紧随PAM的20bp的序列[图1(d)],可以利用体外转录和体内表达等方式进行操作,能同时作用于多个靶点实现多基因同时打靶.因此,CRISPR/Cas9系统自出现以后就受到广泛的关注和应用[10-16],是本文接下来重点综述内容. ...
High-throughput functional genomics using CRISPR-Cas9
1
2015
... 2013年,第三代基因组编辑系统CRISPR/Cas9系统开始出现并迅速崛起[8-9].与之前的ZFNs及TALENs技术相比,CRISPR/Cas9系统具有显著优点,设计简单和改造方便,向导RNA(single guideRNA, sgRNA)非常容易获得而且成本很低,其识别序列仅需要一个紧随PAM的20bp的序列[图1(d)],可以利用体外转录和体内表达等方式进行操作,能同时作用于多个靶点实现多基因同时打靶.因此,CRISPR/Cas9系统自出现以后就受到广泛的关注和应用[10-16],是本文接下来重点综述内容. ...
Applications of CRISPR genome engineering in cell biology
0
2016
Biology and applications of CRISPR Systems: harnessing nature's toolbox for genome engineering
1
2016
... 基因组编辑(genome editing)是基于位点特异性的核酸酶对基因组双链DNA进行切割,从而激活特定位点的插入、删除或者替换的技术.目前应用最广泛的核酸酶主要有三种,包括锌指核酸酶技术(zinc-finger nucleases,ZFNs)、转录激活因子类似的核酸酶(transcription activator-like effector nuclease proteins,TALENs)以及成簇的规律间隔的短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR),如图1所示.其中CRISPR/Cas9系统是目前使用最为广泛的技术[8-12].基因组编辑技术作为关键的反向遗传学研究方法,是功能基因组研究与遗传改造重要的手段,显著促进了合成生物学研究的迅猛发展,在真菌遗传育种方面具有重要应用前景,是当前真菌合成生物学的研究热点[13-14].本文将对基因组编辑技术的类型和作用机理进行简要综述,重点介绍工业丝状真菌(industrial filamentous fungi)中CRISPR系统介导的基因组编辑技术的研发进展和发展趋势,以期为工业丝状真菌研究提供参考. ...
Applications of CRISPR technologies in research and beyond
1
2016
... 基因组编辑(genome editing)是基于位点特异性的核酸酶对基因组双链DNA进行切割,从而激活特定位点的插入、删除或者替换的技术.目前应用最广泛的核酸酶主要有三种,包括锌指核酸酶技术(zinc-finger nucleases,ZFNs)、转录激活因子类似的核酸酶(transcription activator-like effector nuclease proteins,TALENs)以及成簇的规律间隔的短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR),如图1所示.其中CRISPR/Cas9系统是目前使用最为广泛的技术[8-12].基因组编辑技术作为关键的反向遗传学研究方法,是功能基因组研究与遗传改造重要的手段,显著促进了合成生物学研究的迅猛发展,在真菌遗传育种方面具有重要应用前景,是当前真菌合成生物学的研究热点[13-14].本文将对基因组编辑技术的类型和作用机理进行简要综述,重点介绍工业丝状真菌(industrial filamentous fungi)中CRISPR系统介导的基因组编辑技术的研发进展和发展趋势,以期为工业丝状真菌研究提供参考. ...
The promise and challenge of therapeutic genome editing
1
2020
... 基因组编辑(genome editing)是基于位点特异性的核酸酶对基因组双链DNA进行切割,从而激活特定位点的插入、删除或者替换的技术.目前应用最广泛的核酸酶主要有三种,包括锌指核酸酶技术(zinc-finger nucleases,ZFNs)、转录激活因子类似的核酸酶(transcription activator-like effector nuclease proteins,TALENs)以及成簇的规律间隔的短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR),如图1所示.其中CRISPR/Cas9系统是目前使用最为广泛的技术[8-12].基因组编辑技术作为关键的反向遗传学研究方法,是功能基因组研究与遗传改造重要的手段,显著促进了合成生物学研究的迅猛发展,在真菌遗传育种方面具有重要应用前景,是当前真菌合成生物学的研究热点[13-14].本文将对基因组编辑技术的类型和作用机理进行简要综述,重点介绍工业丝状真菌(industrial filamentous fungi)中CRISPR系统介导的基因组编辑技术的研发进展和发展趋势,以期为工业丝状真菌研究提供参考. ...
Chemical biology approaches to genome editing: understanding, controlling, and delivering programmable nucleases
2
2016
... 基因组编辑是指基因组中靶标DNA序列上进行的核苷酸的删除、片段替换和插入、定点突变和碱基易位等,从而实现基因的定点编辑.早期的基因组编辑技术主要是利用同源重组(homologous recombination,HR)原理进行基因打靶,即在外源DNA序列两侧添加同源臂,从而实现外源序列的精确整合[15-16].但在真核生物中,同源重组发生频率非常低,外源DNA序列更容易随机整合到基因组上其他位点,造成脱靶效应,这主要是因为它们存在着占有主导地位的Ku70/Ku80蛋白介导的非同源末端连接的修复机制,即NHEJ(non-homologous ending-joining),并且两端的同源臂序列和插入片段的长度也对整合有很大的影响,从而限制了该技术的应用[17-18].虽然NHEJ介导随机整合的策略不需要经过同源重组且效率较高,但却远不能满足精确编辑基因组的要求. ...
... 相关的研究发现[19],在基因组上特定的DNA位点发生双链断裂(double-strand breaks,DSBs)以后,其发生同源重组效率将会极大提升,这一发现给基因组编辑研究带来了新的突破.因此,近年来研究学者陆续研发了人工特异性核酸内切酶技术[8-15],包括ZFNs技术、TALENs技术和CRISPR/Cas9技术,从而能够在基因组特异性的DNA位点上进行双链断裂(图1).这些核酸内切酶介导的基因组编辑均是利用DSBs后细胞的修复机制进行操作的,双链断裂后会诱导细胞体内两种修复途径(NHEJ或HR)对断裂的双链DNA进行编辑修复[图1(a)],来实现对基因组上特异性位点的精确编辑. ...
CRISPR-based technologies for the manipulation of eukaryotic genomes
3
2017
... 基因组编辑是指基因组中靶标DNA序列上进行的核苷酸的删除、片段替换和插入、定点突变和碱基易位等,从而实现基因的定点编辑.早期的基因组编辑技术主要是利用同源重组(homologous recombination,HR)原理进行基因打靶,即在外源DNA序列两侧添加同源臂,从而实现外源序列的精确整合[15-16].但在真核生物中,同源重组发生频率非常低,外源DNA序列更容易随机整合到基因组上其他位点,造成脱靶效应,这主要是因为它们存在着占有主导地位的Ku70/Ku80蛋白介导的非同源末端连接的修复机制,即NHEJ(non-homologous ending-joining),并且两端的同源臂序列和插入片段的长度也对整合有很大的影响,从而限制了该技术的应用[17-18].虽然NHEJ介导随机整合的策略不需要经过同源重组且效率较高,但却远不能满足精确编辑基因组的要求. ...
... 2013年,第三代基因组编辑系统CRISPR/Cas9系统开始出现并迅速崛起[8-9].与之前的ZFNs及TALENs技术相比,CRISPR/Cas9系统具有显著优点,设计简单和改造方便,向导RNA(single guideRNA, sgRNA)非常容易获得而且成本很低,其识别序列仅需要一个紧随PAM的20bp的序列[图1(d)],可以利用体外转录和体内表达等方式进行操作,能同时作用于多个靶点实现多基因同时打靶.因此,CRISPR/Cas9系统自出现以后就受到广泛的关注和应用[10-16],是本文接下来重点综述内容. ...
... 目前Class Ⅱ 的Type Ⅱ型中CRISPR-Cas9系统是被改造得最为成功的核酸酶,研究表明仅Cas9单一蛋白就能发挥切割的功能[34].Cas9蛋白共有2个核酸酶结构域RuvC和HNH,这一发现为CRISPR-Cas9核酸酶技术的研发奠定了基础.引导Cas9的RNA由两部分组成,一部分是靶向目标系列的crRNA,另一部分是tracrRNA.crRNA与tracrRNA结合后再与Cas9蛋白形成复合体,对原型间隔序列邻近基序(protospacer adjacent motif,PAM)(5'-NGG-3')区域附近的位点进行双链断裂,进而激活细胞的修复机制.2012年,Jinek等[35]将crRNA与tracrRNA结构进行重新设计,形成了一个简便的引导RNA(single guide RNA),从此sgRNA的操作使用都更为简易.2013年,Church实验室、张锋实验室报道了在动物细胞中CRISPR-Cas9新一代核酸酶技术的研发和建立,其编辑效率不低于ZFNs和TALENs,有的甚至高于TALENs [36-37].随后,CRISPR/Cas9系统进入快速的黄金发展时期,目前已经在相当多的物种(包括人类及其他动物、植物和微生物)中成功得到了高效的研发和应用[8-16]. ...
Regulation of DNA double-strand break repair pathway choice
1
2008
... 基因组编辑是指基因组中靶标DNA序列上进行的核苷酸的删除、片段替换和插入、定点突变和碱基易位等,从而实现基因的定点编辑.早期的基因组编辑技术主要是利用同源重组(homologous recombination,HR)原理进行基因打靶,即在外源DNA序列两侧添加同源臂,从而实现外源序列的精确整合[15-16].但在真核生物中,同源重组发生频率非常低,外源DNA序列更容易随机整合到基因组上其他位点,造成脱靶效应,这主要是因为它们存在着占有主导地位的Ku70/Ku80蛋白介导的非同源末端连接的修复机制,即NHEJ(non-homologous ending-joining),并且两端的同源臂序列和插入片段的长度也对整合有很大的影响,从而限制了该技术的应用[17-18].虽然NHEJ介导随机整合的策略不需要经过同源重组且效率较高,但却远不能满足精确编辑基因组的要求. ...
Genetic engineering of filamentous fungi—progress, obstacles and future trends
1
2008
... 基因组编辑是指基因组中靶标DNA序列上进行的核苷酸的删除、片段替换和插入、定点突变和碱基易位等,从而实现基因的定点编辑.早期的基因组编辑技术主要是利用同源重组(homologous recombination,HR)原理进行基因打靶,即在外源DNA序列两侧添加同源臂,从而实现外源序列的精确整合[15-16].但在真核生物中,同源重组发生频率非常低,外源DNA序列更容易随机整合到基因组上其他位点,造成脱靶效应,这主要是因为它们存在着占有主导地位的Ku70/Ku80蛋白介导的非同源末端连接的修复机制,即NHEJ(non-homologous ending-joining),并且两端的同源臂序列和插入片段的长度也对整合有很大的影响,从而限制了该技术的应用[17-18].虽然NHEJ介导随机整合的策略不需要经过同源重组且效率较高,但却远不能满足精确编辑基因组的要求. ...
Chimeric nucleases stimulate gene targeting in human cells
1
2003
... 相关的研究发现[19],在基因组上特定的DNA位点发生双链断裂(double-strand breaks,DSBs)以后,其发生同源重组效率将会极大提升,这一发现给基因组编辑研究带来了新的突破.因此,近年来研究学者陆续研发了人工特异性核酸内切酶技术[8-15],包括ZFNs技术、TALENs技术和CRISPR/Cas9技术,从而能够在基因组特异性的DNA位点上进行双链断裂(图1).这些核酸内切酶介导的基因组编辑均是利用DSBs后细胞的修复机制进行操作的,双链断裂后会诱导细胞体内两种修复途径(NHEJ或HR)对断裂的双链DNA进行编辑修复[图1(a)],来实现对基因组上特异性位点的精确编辑. ...
The discovery of zinc fingers and their applications in gene regulation and genome manipulation
1
2010
... ZFNs作为第一代基因编辑技术,是两部分结构融合而成,一部分是由锌指蛋白的核苷酸结合域(ZFA),另一部分是核酸内切酶(Fok I)的切割结构域 [20].ZFA的核苷酸结合域是由3~6个Cys2-His2锌指结构串联而成的,每个锌指结构能够特异识别DNA单链上3个连续的碱基,并结合到识别的DNA上,由多个锌指单元串联形成的ZFA则可识别更长的靶序列,能增加DNA的识别特异性.FokⅠ是来源于海床黄杆菌(Flavobacterium okeanokoites)的一种限制性核酸内切酶,连接到ZFA的C端,Fok Ⅰ只在二聚体的形式下才能发挥剪切活性,因此需要在靶标位点的两端设计正反向排列且间隔5~7 bp的成对ZFNs,从而实现目的基因位点产生DSB[图1(b)].但ZFNs设计复杂成本较高,构建周期长,工作量较大,且序列的上下文依赖效应会降低编辑效率,限制了该技术的发展. ...
A simple cipher governs DNA recognition by TAL effectors
2
2009
... 随后在2011年,研究人员开发了第二代基因组编辑技术TALENs,它是将改造后能够靶向特异DNA序列的TALE蛋白和非特异性的Fok Ⅰ的切割结构域融合而得到的人工核酸酶[21].与ZFNs不同的是,TALENs用于识别特异性目标序列是来自黄单胞菌的TALE蛋白,经过人工改造后能够靶向任意DNA序列.TALE蛋白是由N端转运信号、转录激活结构域、DNA特异性识别结合结构域和C端核定位信号肽组成.TALENs技术的特异性位点核心识别结构域由一系列重复串联序列组成,这些串联序列高度保守,每个重复结构由33~35个氨基酸组成,该重复结构的核苷酸特异性识别位点是通过第12、13位氨基酸的重复可变双氨基酸残基(repeat variable diresidues,RVD)确定的,也就是“两个氨基酸与一个核苷酸相对应”.与ZFNs相似,TALENs的TALE单体特异性识别DNA序列并结合在双链上,两个单体融合的Fok Ⅰ二聚体对目的DNA序列进行切割产生DSBs[图1(c)],两个相邻TALEN靶序列间隔一般为15 bp左右.由于TALENs的设计性更强和构建简便的优势,TALENs比ZFNs在基因编辑和转录调控中有着更为广阔的应用潜力,在多个物种中被成功应用[21-22].2011年,人工核酸酶介导的基因组编辑被Nature选入最受关注的技术成果,并在2012年,以TALEN为代表的核酸酶被Science列入年度十大科学进展. ...
... [21-22].2011年,人工核酸酶介导的基因组编辑被Nature选入最受关注的技术成果,并在2012年,以TALEN为代表的核酸酶被Science列入年度十大科学进展. ...
TALENs: customizable molecular DNA scissors for genome engineering of plants
1
2013
... 随后在2011年,研究人员开发了第二代基因组编辑技术TALENs,它是将改造后能够靶向特异DNA序列的TALE蛋白和非特异性的Fok Ⅰ的切割结构域融合而得到的人工核酸酶[21].与ZFNs不同的是,TALENs用于识别特异性目标序列是来自黄单胞菌的TALE蛋白,经过人工改造后能够靶向任意DNA序列.TALE蛋白是由N端转运信号、转录激活结构域、DNA特异性识别结合结构域和C端核定位信号肽组成.TALENs技术的特异性位点核心识别结构域由一系列重复串联序列组成,这些串联序列高度保守,每个重复结构由33~35个氨基酸组成,该重复结构的核苷酸特异性识别位点是通过第12、13位氨基酸的重复可变双氨基酸残基(repeat variable diresidues,RVD)确定的,也就是“两个氨基酸与一个核苷酸相对应”.与ZFNs相似,TALENs的TALE单体特异性识别DNA序列并结合在双链上,两个单体融合的Fok Ⅰ二聚体对目的DNA序列进行切割产生DSBs[图1(c)],两个相邻TALEN靶序列间隔一般为15 bp左右.由于TALENs的设计性更强和构建简便的优势,TALENs比ZFNs在基因编辑和转录调控中有着更为广阔的应用潜力,在多个物种中被成功应用[21-22].2011年,人工核酸酶介导的基因组编辑被Nature选入最受关注的技术成果,并在2012年,以TALEN为代表的核酸酶被Science列入年度十大科学进展. ...
Tailor-made TALEN system for highly efficient targeted gene replacement in the rice blast fungus
1
2015
... 2015年3月,日本明治大学Shigeru Kuwata教授团队率先在植物病源真菌-稻瘟病菌(M. oryzae)中研发并构建了Platinum-Fungal TALENs(PtFg TALENs)技术体系,该研究是最早在丝状真菌中进行核酸酶介导的基因组编辑技术的报道[23].该团队将PtFg TALEN表达元件和donor DNA通过转生质体法转化进入稻瘟病菌细胞中,成功对靶标基因SDH和SRS2开展了基因敲除,其敲除效率高达100%.该研究表明,TALENs系统能够在丝状真菌中进行高效的基因组编辑. ...
Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product
1
1987
... CRISPR-Cas系统是细菌或者古细菌等原核生物在长期进化中形成的用来抵抗入侵病毒或外源DNA的适应性免疫机制[24],2002年将其称为CRISPR,它是由高度保守的短重复序列(repeats)与长度相似的间隔序列(spacers)构成,位点附近是其相关的效应蛋白(CRISPR-associated Cas effector proteins).CRISPR序列位点和Cas蛋白共同组成CRISPR-Cas系统,其作为一种获得性免疫系统,通过编码Cas蛋白及RNA共同对抗噬菌体和质粒等外来DNA的入侵[25]. ...
CRISPR provides acquired resistance against viruses in prokaryotes
1
2007
... CRISPR-Cas系统是细菌或者古细菌等原核生物在长期进化中形成的用来抵抗入侵病毒或外源DNA的适应性免疫机制[24],2002年将其称为CRISPR,它是由高度保守的短重复序列(repeats)与长度相似的间隔序列(spacers)构成,位点附近是其相关的效应蛋白(CRISPR-associated Cas effector proteins).CRISPR序列位点和Cas蛋白共同组成CRISPR-Cas系统,其作为一种获得性免疫系统,通过编码Cas蛋白及RNA共同对抗噬菌体和质粒等外来DNA的入侵[25]. ...
Evolutionary conservation of sequence and secondary structures in CRISPR repeats
1
2007
... 典型的CRISPR/Cas由3部分组成[26-29]:①前导序列(leader sequence),是转录CRISPR序列表达的启动子,它通常在CRISPR位点的最前面;②重复序列,其序列长度仅为几十个核苷酸,具有调控和加工的作用,其部分碱基能够形成互补对称,因此加工成熟的crRNA(CRISPR targeting RNA)能够成茎环;③间隔序列,其在两个repeats之间,repeats和spacers串联组成不同长度的重复序列,间隔序列与重复序列一同转录,并加工形成crRNA,部分CRISPR/Cas系统还需要反式激活CRISPR RNA(tracrRNA)的参与,这些crRNA和Cas蛋白共同组成免疫防御CRISPR系统.该免疫防御过程的作用机制[28-31]为3个阶段,在外来DNA入侵起始阶段,细菌将外源DNA中的原型间隔序列(protospacer)整合到两个重复序列中间,在CRISPR系统的作用下转录形成pre-crRNA,进而被剪切加工形成成熟的crRNA,然后crRNA与多个Cas蛋白复合体结合,可以识别入侵的外源DNA的间隔前体附近的序列,进行碱基配对,进而切割外源DNA分子,如图2所示. ...
CRISPR families of the crenarchaeal genus sulfolobus: bidirectional transcription and dynamic properties
0
2009
The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli
1
2011
... 典型的CRISPR/Cas由3部分组成[26-29]:①前导序列(leader sequence),是转录CRISPR序列表达的启动子,它通常在CRISPR位点的最前面;②重复序列,其序列长度仅为几十个核苷酸,具有调控和加工的作用,其部分碱基能够形成互补对称,因此加工成熟的crRNA(CRISPR targeting RNA)能够成茎环;③间隔序列,其在两个repeats之间,repeats和spacers串联组成不同长度的重复序列,间隔序列与重复序列一同转录,并加工形成crRNA,部分CRISPR/Cas系统还需要反式激活CRISPR RNA(tracrRNA)的参与,这些crRNA和Cas蛋白共同组成免疫防御CRISPR系统.该免疫防御过程的作用机制[28-31]为3个阶段,在外来DNA入侵起始阶段,细菌将外源DNA中的原型间隔序列(protospacer)整合到两个重复序列中间,在CRISPR系统的作用下转录形成pre-crRNA,进而被剪切加工形成成熟的crRNA,然后crRNA与多个Cas蛋白复合体结合,可以识别入侵的外源DNA的间隔前体附近的序列,进行碱基配对,进而切割外源DNA分子,如图2所示. ...
CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III
1
2011
... 典型的CRISPR/Cas由3部分组成[26-29]:①前导序列(leader sequence),是转录CRISPR序列表达的启动子,它通常在CRISPR位点的最前面;②重复序列,其序列长度仅为几十个核苷酸,具有调控和加工的作用,其部分碱基能够形成互补对称,因此加工成熟的crRNA(CRISPR targeting RNA)能够成茎环;③间隔序列,其在两个repeats之间,repeats和spacers串联组成不同长度的重复序列,间隔序列与重复序列一同转录,并加工形成crRNA,部分CRISPR/Cas系统还需要反式激活CRISPR RNA(tracrRNA)的参与,这些crRNA和Cas蛋白共同组成免疫防御CRISPR系统.该免疫防御过程的作用机制[28-31]为3个阶段,在外来DNA入侵起始阶段,细菌将外源DNA中的原型间隔序列(protospacer)整合到两个重复序列中间,在CRISPR系统的作用下转录形成pre-crRNA,进而被剪切加工形成成熟的crRNA,然后crRNA与多个Cas蛋白复合体结合,可以识别入侵的外源DNA的间隔前体附近的序列,进行碱基配对,进而切割外源DNA分子,如图2所示. ...
CRISPR/Cas system and its role in phage-bacteria interactions
0
2010
The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA
1
2010
... 典型的CRISPR/Cas由3部分组成[26-29]:①前导序列(leader sequence),是转录CRISPR序列表达的启动子,它通常在CRISPR位点的最前面;②重复序列,其序列长度仅为几十个核苷酸,具有调控和加工的作用,其部分碱基能够形成互补对称,因此加工成熟的crRNA(CRISPR targeting RNA)能够成茎环;③间隔序列,其在两个repeats之间,repeats和spacers串联组成不同长度的重复序列,间隔序列与重复序列一同转录,并加工形成crRNA,部分CRISPR/Cas系统还需要反式激活CRISPR RNA(tracrRNA)的参与,这些crRNA和Cas蛋白共同组成免疫防御CRISPR系统.该免疫防御过程的作用机制[28-31]为3个阶段,在外来DNA入侵起始阶段,细菌将外源DNA中的原型间隔序列(protospacer)整合到两个重复序列中间,在CRISPR系统的作用下转录形成pre-crRNA,进而被剪切加工形成成熟的crRNA,然后crRNA与多个Cas蛋白复合体结合,可以识别入侵的外源DNA的间隔前体附近的序列,进行碱基配对,进而切割外源DNA分子,如图2所示. ...
Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems
2
2016
... CRISPR-Cas系统有两大类(Class Ⅰ和Class Ⅱ)和6种类型[Type Ⅰ~Ⅵ],进而又可分为多个亚型(Subtype)[32-34].Class I系统在干扰靶基因时需要多个Cas蛋白形成复合体协同工作,包括Type Ⅰ、Type Ⅲ和Type Ⅳ型;而Class Ⅱ仅需一个Cas蛋白就能够干扰靶基因,包括Type Ⅱ、Type V和Type Ⅵ类型,这类系统较为简单,研究也更加透彻,经过人工改造构建成一种高效的基因组编辑工具.目前应用最为广泛的CRISPR/Cas9系统为Class Ⅱ的Type Ⅱ型系统,而新兴的CRISPR-Cas12a(Cpf1)/Cas12b和CRISPR-Cas14系统属于Class Ⅱ 的Type V型,能对RNA进行编辑的CRISPR/Cas13系统属于新发现的Class Ⅱ的Type Ⅵ型. ...
... 尽管CRISPR-Cas9系统已经发展成为目前广泛应用的第三代基因组编辑工具,但仍存在一些弊端和技术上的挑战,例如该系统存在编辑位点受限、脱靶情况较多、在某些细胞中有毒性、PAM序列的限制等缺陷,因此优化CRISPR-Cas9系统与建立新的CRISPR基因组编辑系统一直是研究热点.Cas12a(之前被称为Cpf1)与Cas9都是第2类CRISPR系统,但是Cas12a蛋白是Ⅴ型,仅一个Cas12a核酸酶就能造成双链断裂,其引导RNA元件和切割方式与Cas9完全不同[32-34].与Cas9相比,首先CRISPR-Cas12a系统在pre-crRNA的加工上,没有tracrRNA,且crRNA更短,Cas12a系统不需要RNase而是由其本身的RNase结构域就能完成整个crRNA的加工过程;其次,Cas12a仅有RuvC核酸内切酶结构域,不含有HNH结构域;第三,Cas12a在靶标DNA的PAM序列下游18~23 bp处对DNA双链进行切割,形成的是黏性末端,且Cas12a特异识别富含T的PAM序列(5'-TTTV-3'),扩充了CRISPR/Cas系统及其衍生技术体系的基因组编辑范围[38].CRISPR-Cas12a目前主要应用为多位点基因编辑和大片段的删除,利用Cas12a系统则可以在一个质粒上仅用一个启动子串联多个crRNA,并在Cas12a的作用下加工出单独的多个成熟crRNA,极大简化了引导RNA的构建,并缩减了多个crRNA片段合成的长度[39].近5年来,Cas12a核酸酶已被研究学者广泛关注,并在许多物种中构建了高效的CRISPR-Cas12a基因组编辑体系,Cas12a与Cas9系统交替使用,相互补充,丰富了基因组编辑技术中特异性位点选择的灵活性[38-43]. ...
Diversity, classification and evolution of CRISPR-Cas systems
0
2017
Diversity and evolution of class 2 CRISPR-Cas systems
3
2017
... CRISPR-Cas系统有两大类(Class Ⅰ和Class Ⅱ)和6种类型[Type Ⅰ~Ⅵ],进而又可分为多个亚型(Subtype)[32-34].Class I系统在干扰靶基因时需要多个Cas蛋白形成复合体协同工作,包括Type Ⅰ、Type Ⅲ和Type Ⅳ型;而Class Ⅱ仅需一个Cas蛋白就能够干扰靶基因,包括Type Ⅱ、Type V和Type Ⅵ类型,这类系统较为简单,研究也更加透彻,经过人工改造构建成一种高效的基因组编辑工具.目前应用最为广泛的CRISPR/Cas9系统为Class Ⅱ的Type Ⅱ型系统,而新兴的CRISPR-Cas12a(Cpf1)/Cas12b和CRISPR-Cas14系统属于Class Ⅱ 的Type V型,能对RNA进行编辑的CRISPR/Cas13系统属于新发现的Class Ⅱ的Type Ⅵ型. ...
... 目前Class Ⅱ 的Type Ⅱ型中CRISPR-Cas9系统是被改造得最为成功的核酸酶,研究表明仅Cas9单一蛋白就能发挥切割的功能[34].Cas9蛋白共有2个核酸酶结构域RuvC和HNH,这一发现为CRISPR-Cas9核酸酶技术的研发奠定了基础.引导Cas9的RNA由两部分组成,一部分是靶向目标系列的crRNA,另一部分是tracrRNA.crRNA与tracrRNA结合后再与Cas9蛋白形成复合体,对原型间隔序列邻近基序(protospacer adjacent motif,PAM)(5'-NGG-3')区域附近的位点进行双链断裂,进而激活细胞的修复机制.2012年,Jinek等[35]将crRNA与tracrRNA结构进行重新设计,形成了一个简便的引导RNA(single guide RNA),从此sgRNA的操作使用都更为简易.2013年,Church实验室、张锋实验室报道了在动物细胞中CRISPR-Cas9新一代核酸酶技术的研发和建立,其编辑效率不低于ZFNs和TALENs,有的甚至高于TALENs [36-37].随后,CRISPR/Cas9系统进入快速的黄金发展时期,目前已经在相当多的物种(包括人类及其他动物、植物和微生物)中成功得到了高效的研发和应用[8-16]. ...
... 尽管CRISPR-Cas9系统已经发展成为目前广泛应用的第三代基因组编辑工具,但仍存在一些弊端和技术上的挑战,例如该系统存在编辑位点受限、脱靶情况较多、在某些细胞中有毒性、PAM序列的限制等缺陷,因此优化CRISPR-Cas9系统与建立新的CRISPR基因组编辑系统一直是研究热点.Cas12a(之前被称为Cpf1)与Cas9都是第2类CRISPR系统,但是Cas12a蛋白是Ⅴ型,仅一个Cas12a核酸酶就能造成双链断裂,其引导RNA元件和切割方式与Cas9完全不同[32-34].与Cas9相比,首先CRISPR-Cas12a系统在pre-crRNA的加工上,没有tracrRNA,且crRNA更短,Cas12a系统不需要RNase而是由其本身的RNase结构域就能完成整个crRNA的加工过程;其次,Cas12a仅有RuvC核酸内切酶结构域,不含有HNH结构域;第三,Cas12a在靶标DNA的PAM序列下游18~23 bp处对DNA双链进行切割,形成的是黏性末端,且Cas12a特异识别富含T的PAM序列(5'-TTTV-3'),扩充了CRISPR/Cas系统及其衍生技术体系的基因组编辑范围[38].CRISPR-Cas12a目前主要应用为多位点基因编辑和大片段的删除,利用Cas12a系统则可以在一个质粒上仅用一个启动子串联多个crRNA,并在Cas12a的作用下加工出单独的多个成熟crRNA,极大简化了引导RNA的构建,并缩减了多个crRNA片段合成的长度[39].近5年来,Cas12a核酸酶已被研究学者广泛关注,并在许多物种中构建了高效的CRISPR-Cas12a基因组编辑体系,Cas12a与Cas9系统交替使用,相互补充,丰富了基因组编辑技术中特异性位点选择的灵活性[38-43]. ...
A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity
1
2012
... 目前Class Ⅱ 的Type Ⅱ型中CRISPR-Cas9系统是被改造得最为成功的核酸酶,研究表明仅Cas9单一蛋白就能发挥切割的功能[34].Cas9蛋白共有2个核酸酶结构域RuvC和HNH,这一发现为CRISPR-Cas9核酸酶技术的研发奠定了基础.引导Cas9的RNA由两部分组成,一部分是靶向目标系列的crRNA,另一部分是tracrRNA.crRNA与tracrRNA结合后再与Cas9蛋白形成复合体,对原型间隔序列邻近基序(protospacer adjacent motif,PAM)(5'-NGG-3')区域附近的位点进行双链断裂,进而激活细胞的修复机制.2012年,Jinek等[35]将crRNA与tracrRNA结构进行重新设计,形成了一个简便的引导RNA(single guide RNA),从此sgRNA的操作使用都更为简易.2013年,Church实验室、张锋实验室报道了在动物细胞中CRISPR-Cas9新一代核酸酶技术的研发和建立,其编辑效率不低于ZFNs和TALENs,有的甚至高于TALENs [36-37].随后,CRISPR/Cas9系统进入快速的黄金发展时期,目前已经在相当多的物种(包括人类及其他动物、植物和微生物)中成功得到了高效的研发和应用[8-16]. ...
Multiplex genome engineering using CRISPR/Cas systems
1
2013
... 目前Class Ⅱ 的Type Ⅱ型中CRISPR-Cas9系统是被改造得最为成功的核酸酶,研究表明仅Cas9单一蛋白就能发挥切割的功能[34].Cas9蛋白共有2个核酸酶结构域RuvC和HNH,这一发现为CRISPR-Cas9核酸酶技术的研发奠定了基础.引导Cas9的RNA由两部分组成,一部分是靶向目标系列的crRNA,另一部分是tracrRNA.crRNA与tracrRNA结合后再与Cas9蛋白形成复合体,对原型间隔序列邻近基序(protospacer adjacent motif,PAM)(5'-NGG-3')区域附近的位点进行双链断裂,进而激活细胞的修复机制.2012年,Jinek等[35]将crRNA与tracrRNA结构进行重新设计,形成了一个简便的引导RNA(single guide RNA),从此sgRNA的操作使用都更为简易.2013年,Church实验室、张锋实验室报道了在动物细胞中CRISPR-Cas9新一代核酸酶技术的研发和建立,其编辑效率不低于ZFNs和TALENs,有的甚至高于TALENs [36-37].随后,CRISPR/Cas9系统进入快速的黄金发展时期,目前已经在相当多的物种(包括人类及其他动物、植物和微生物)中成功得到了高效的研发和应用[8-16]. ...
RNA-guided human genome engineering via Cas9
1
2013
... 目前Class Ⅱ 的Type Ⅱ型中CRISPR-Cas9系统是被改造得最为成功的核酸酶,研究表明仅Cas9单一蛋白就能发挥切割的功能[34].Cas9蛋白共有2个核酸酶结构域RuvC和HNH,这一发现为CRISPR-Cas9核酸酶技术的研发奠定了基础.引导Cas9的RNA由两部分组成,一部分是靶向目标系列的crRNA,另一部分是tracrRNA.crRNA与tracrRNA结合后再与Cas9蛋白形成复合体,对原型间隔序列邻近基序(protospacer adjacent motif,PAM)(5'-NGG-3')区域附近的位点进行双链断裂,进而激活细胞的修复机制.2012年,Jinek等[35]将crRNA与tracrRNA结构进行重新设计,形成了一个简便的引导RNA(single guide RNA),从此sgRNA的操作使用都更为简易.2013年,Church实验室、张锋实验室报道了在动物细胞中CRISPR-Cas9新一代核酸酶技术的研发和建立,其编辑效率不低于ZFNs和TALENs,有的甚至高于TALENs [36-37].随后,CRISPR/Cas9系统进入快速的黄金发展时期,目前已经在相当多的物种(包括人类及其他动物、植物和微生物)中成功得到了高效的研发和应用[8-16]. ...
Cpf1 is a single RNA-guided endonuclease of a Class 2 CRISPR-Cas system
2
2015
... 尽管CRISPR-Cas9系统已经发展成为目前广泛应用的第三代基因组编辑工具,但仍存在一些弊端和技术上的挑战,例如该系统存在编辑位点受限、脱靶情况较多、在某些细胞中有毒性、PAM序列的限制等缺陷,因此优化CRISPR-Cas9系统与建立新的CRISPR基因组编辑系统一直是研究热点.Cas12a(之前被称为Cpf1)与Cas9都是第2类CRISPR系统,但是Cas12a蛋白是Ⅴ型,仅一个Cas12a核酸酶就能造成双链断裂,其引导RNA元件和切割方式与Cas9完全不同[32-34].与Cas9相比,首先CRISPR-Cas12a系统在pre-crRNA的加工上,没有tracrRNA,且crRNA更短,Cas12a系统不需要RNase而是由其本身的RNase结构域就能完成整个crRNA的加工过程;其次,Cas12a仅有RuvC核酸内切酶结构域,不含有HNH结构域;第三,Cas12a在靶标DNA的PAM序列下游18~23 bp处对DNA双链进行切割,形成的是黏性末端,且Cas12a特异识别富含T的PAM序列(5'-TTTV-3'),扩充了CRISPR/Cas系统及其衍生技术体系的基因组编辑范围[38].CRISPR-Cas12a目前主要应用为多位点基因编辑和大片段的删除,利用Cas12a系统则可以在一个质粒上仅用一个启动子串联多个crRNA,并在Cas12a的作用下加工出单独的多个成熟crRNA,极大简化了引导RNA的构建,并缩减了多个crRNA片段合成的长度[39].近5年来,Cas12a核酸酶已被研究学者广泛关注,并在许多物种中构建了高效的CRISPR-Cas12a基因组编辑体系,Cas12a与Cas9系统交替使用,相互补充,丰富了基因组编辑技术中特异性位点选择的灵活性[38-43]. ...
... [38-43]. ...
Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array
1
2017
... 尽管CRISPR-Cas9系统已经发展成为目前广泛应用的第三代基因组编辑工具,但仍存在一些弊端和技术上的挑战,例如该系统存在编辑位点受限、脱靶情况较多、在某些细胞中有毒性、PAM序列的限制等缺陷,因此优化CRISPR-Cas9系统与建立新的CRISPR基因组编辑系统一直是研究热点.Cas12a(之前被称为Cpf1)与Cas9都是第2类CRISPR系统,但是Cas12a蛋白是Ⅴ型,仅一个Cas12a核酸酶就能造成双链断裂,其引导RNA元件和切割方式与Cas9完全不同[32-34].与Cas9相比,首先CRISPR-Cas12a系统在pre-crRNA的加工上,没有tracrRNA,且crRNA更短,Cas12a系统不需要RNase而是由其本身的RNase结构域就能完成整个crRNA的加工过程;其次,Cas12a仅有RuvC核酸内切酶结构域,不含有HNH结构域;第三,Cas12a在靶标DNA的PAM序列下游18~23 bp处对DNA双链进行切割,形成的是黏性末端,且Cas12a特异识别富含T的PAM序列(5'-TTTV-3'),扩充了CRISPR/Cas系统及其衍生技术体系的基因组编辑范围[38].CRISPR-Cas12a目前主要应用为多位点基因编辑和大片段的删除,利用Cas12a系统则可以在一个质粒上仅用一个启动子串联多个crRNA,并在Cas12a的作用下加工出单独的多个成熟crRNA,极大简化了引导RNA的构建,并缩减了多个crRNA片段合成的长度[39].近5年来,Cas12a核酸酶已被研究学者广泛关注,并在许多物种中构建了高效的CRISPR-Cas12a基因组编辑体系,Cas12a与Cas9系统交替使用,相互补充,丰富了基因组编辑技术中特异性位点选择的灵活性[38-43]. ...
A CRISPR-Cpf1 system for efficient genome editing and transcriptional repression in plants
0
2017
CRISPR-Cpf1 assisted genome editing of Corynebacterium glutamicum
0
2017
The CCTL (Cpf1-assisted Cutting and Taq DNA ligase-assisted Ligation) method for efficient editing of large DNA constructs in vitro
0
2017
FnCpf1: a novel and efficient genome editing tool for Saccharomyces cerevisiae
1
2017
... 尽管CRISPR-Cas9系统已经发展成为目前广泛应用的第三代基因组编辑工具,但仍存在一些弊端和技术上的挑战,例如该系统存在编辑位点受限、脱靶情况较多、在某些细胞中有毒性、PAM序列的限制等缺陷,因此优化CRISPR-Cas9系统与建立新的CRISPR基因组编辑系统一直是研究热点.Cas12a(之前被称为Cpf1)与Cas9都是第2类CRISPR系统,但是Cas12a蛋白是Ⅴ型,仅一个Cas12a核酸酶就能造成双链断裂,其引导RNA元件和切割方式与Cas9完全不同[32-34].与Cas9相比,首先CRISPR-Cas12a系统在pre-crRNA的加工上,没有tracrRNA,且crRNA更短,Cas12a系统不需要RNase而是由其本身的RNase结构域就能完成整个crRNA的加工过程;其次,Cas12a仅有RuvC核酸内切酶结构域,不含有HNH结构域;第三,Cas12a在靶标DNA的PAM序列下游18~23 bp处对DNA双链进行切割,形成的是黏性末端,且Cas12a特异识别富含T的PAM序列(5'-TTTV-3'),扩充了CRISPR/Cas系统及其衍生技术体系的基因组编辑范围[38].CRISPR-Cas12a目前主要应用为多位点基因编辑和大片段的删除,利用Cas12a系统则可以在一个质粒上仅用一个启动子串联多个crRNA,并在Cas12a的作用下加工出单独的多个成熟crRNA,极大简化了引导RNA的构建,并缩减了多个crRNA片段合成的长度[39].近5年来,Cas12a核酸酶已被研究学者广泛关注,并在许多物种中构建了高效的CRISPR-Cas12a基因组编辑体系,Cas12a与Cas9系统交替使用,相互补充,丰富了基因组编辑技术中特异性位点选择的灵活性[38-43]. ...
CRISPR-cas system: history and prospects as a genome editing tool in microorganisms
1
2018
... CRISPR-Cas9技术在人类细胞和动植物细胞改造研究火热的同时,研究者对细菌、酵母和丝状真菌等进行基因编辑也取得了相应的成功[44].2015年,CRISPR-Cas9系统介导的真菌基因组编辑技术开始出现,该技术在丝状真菌分子生物学和遗传改造方面潜力巨大,一些重要的模式丝状真菌CRISPR/Cas9系统陆续被成功开发和报道[45-50],包括构巢曲霉和粗糙脉孢菌等模式菌株,里氏木霉、黑曲霉、米曲霉、产黄青霉和嗜热毁丝霉等工业真菌,以及以稻瘟病菌和烟曲霉等病原真菌,自此丝状真菌进入基因组编辑新时代. ...
CRISPR system in filamentous fungi: current achievements and future directions
2
2017
... CRISPR-Cas9技术在人类细胞和动植物细胞改造研究火热的同时,研究者对细菌、酵母和丝状真菌等进行基因编辑也取得了相应的成功[44].2015年,CRISPR-Cas9系统介导的真菌基因组编辑技术开始出现,该技术在丝状真菌分子生物学和遗传改造方面潜力巨大,一些重要的模式丝状真菌CRISPR/Cas9系统陆续被成功开发和报道[45-50],包括构巢曲霉和粗糙脉孢菌等模式菌株,里氏木霉、黑曲霉、米曲霉、产黄青霉和嗜热毁丝霉等工业真菌,以及以稻瘟病菌和烟曲霉等病原真菌,自此丝状真菌进入基因组编辑新时代. ...
... 2015年之前,CRISPR-Cas9系统尚未应用于真菌,研究者一直使用经典的基因工程手段来研究丝状真菌体系,尽管传统手段编辑丝状真菌基因可以达到多种研究目的,但仅限于一些模式真菌,其效率较低,编辑位点较少,且花费大量的时间和劳动力.CRISPR-Cas系统作为新一代基因组编辑技术,自出现以来,经过多种巧妙的改造,在多个物种中的基因编辑能力被相继证实,得到迅速而广泛的应用,为生命科学和医学研究带来前所未有的革新.自2015年起,CRISPR-Cas9系统被国内外研究学者陆续在丝状真菌体系中进行研发和应用,在短短几年内飞速发展,在不同的丝状真菌中各种高效的、CRISPR-Cas体系已经被陆续建立起来[45-50].近三年来,一些大型真菌的CRISPR-Cas9编辑系统也开始发展并建立,例如灵芝[88]、灰盖鬼伞[89]、蛹虫草[58, 90]和竹黄菌[91]等.CRISPR基因组编辑技术能对真菌基因组特定基因位点进行精准、高效的改造,在真菌细胞工厂构建和优化改造中展示了强大的功能,对真菌功能基因组研究和合成生物学的发展起着非常重要的作用. ...
Molecular tools for gene manipulation in filamentous fungi
0
2017
CRISPR/Cas9-based genome editing of the filamentous fungi: the state of the art
0
2017
CRISPR-Cas9 genome editing approaches in filamentous fungi and oomycetes
0
2019
Recent Advances of CRISPR/Cas9-based genetic engineering and transcriptional regulation in industrial Biology
0
2019
CRISPR/Cas9 genome editing technology in filamentous fungi: progress and perspective
2
2019
... CRISPR-Cas9技术在人类细胞和动植物细胞改造研究火热的同时,研究者对细菌、酵母和丝状真菌等进行基因编辑也取得了相应的成功[44].2015年,CRISPR-Cas9系统介导的真菌基因组编辑技术开始出现,该技术在丝状真菌分子生物学和遗传改造方面潜力巨大,一些重要的模式丝状真菌CRISPR/Cas9系统陆续被成功开发和报道[45-50],包括构巢曲霉和粗糙脉孢菌等模式菌株,里氏木霉、黑曲霉、米曲霉、产黄青霉和嗜热毁丝霉等工业真菌,以及以稻瘟病菌和烟曲霉等病原真菌,自此丝状真菌进入基因组编辑新时代. ...
... 2015年之前,CRISPR-Cas9系统尚未应用于真菌,研究者一直使用经典的基因工程手段来研究丝状真菌体系,尽管传统手段编辑丝状真菌基因可以达到多种研究目的,但仅限于一些模式真菌,其效率较低,编辑位点较少,且花费大量的时间和劳动力.CRISPR-Cas系统作为新一代基因组编辑技术,自出现以来,经过多种巧妙的改造,在多个物种中的基因编辑能力被相继证实,得到迅速而广泛的应用,为生命科学和医学研究带来前所未有的革新.自2015年起,CRISPR-Cas9系统被国内外研究学者陆续在丝状真菌体系中进行研发和应用,在短短几年内飞速发展,在不同的丝状真菌中各种高效的、CRISPR-Cas体系已经被陆续建立起来[45-50].近三年来,一些大型真菌的CRISPR-Cas9编辑系统也开始发展并建立,例如灵芝[88]、灰盖鬼伞[89]、蛹虫草[58, 90]和竹黄菌[91]等.CRISPR基因组编辑技术能对真菌基因组特定基因位点进行精准、高效的改造,在真菌细胞工厂构建和优化改造中展示了强大的功能,对真菌功能基因组研究和合成生物学的发展起着非常重要的作用. ...
Efficient gene editing in Neurospora crassa with CRISPR technology
1
2015
... 粗糙脉孢菌(N. crassa)作为遗传学研究模式生物已有近百年的历史,在细胞融合、生物钟周期以及表观遗传学研究等领域有着深入的研究.粗糙脉孢菌是第一个被测序的丝状真菌,其全基因组序列于2003年被公开报道,随后开展了全基因组范围基因敲除突变体库的构建.早期的粗糙脉孢菌基因敲除是利用潮霉素抗性基因(hph)将Ku70/Ku80的同源基因(mus-51/mus-52)敲除后,再通过同源重组实现的,对于单基因的敲除是有效的,但是对多基因的敲除仍然很难操作.随着第三代基因组编辑技术的发展,研究人员开始尝试在粗糙脉孢菌中进行新型基因编辑系统的开发,2015年,Matsu-ura等[51]利用CRISPR/Cas9系统在粗糙脉孢菌中成功编辑基因组,这是较早报道的丝状真菌CRISPR/Cas9基因组编辑技术的研究之一.Matsu-ura等用来源于构巢曲霉的trpC基因(AN0648)启动子和终止子来表达Cas9-NLS基因,同时利用酿酒酵母SNR52启动子转录sgRNA,将Cas9、sgRNA表达载体和供体DNA同时电击共转化进入宿主细胞,对基因组上的csr-1和clr-2位点进行基因的定点插入.研究结果表明, Cas9和gRNA载体的浓度增加,转化效率提升.荧光强度和PCR结果显示成功将csr-1位点进行luciferase基因的替换,同时clr-2的启动子替换为β-tubulin持续性表达的启动子,从而构建了clr-2基因过表达菌株,极大提高了clr-2自身表达水平和关键的纤维素酶基因cbh-1、gh5-1和gh6-2的转录水平.这一研究表明,CRISPR/Cas9介导基因编辑技术能够在模式丝状真菌——粗糙脉孢菌中成功实现,这为后续开展其他丝状真菌CRISPR-Cas9系统的研究提供了一定的参考. ...
A CRISPR-Cas9 system for genetic engineering of filamentous fungi
3
2015
... 构巢曲霉(A. nidulans)是丝状真菌研究中常用的曲霉属模式物种,广泛用于真核微生物细胞生物学等基础研究.2015年,丹麦技术大学Uffe Hasbro Mortensen教授团队[52]首次在曲霉属的6个物种中建立了通用的CRISPR/Cas9基因组编辑体系,这些物种包括构巢曲霉(A. nidulans)、黑曲霉(A. niger)、棘孢曲霉(A. aculeatus)、巴西曲霉(A. brasiliensis)、炭黑曲霉菌(A. carbonarius)和泡盛曲霉(A. luchuensis).该团队首先基于黑曲霉密码子偏好性优化了SpCas9的核苷酸序列,并使用构巢曲霉tef1基因的启动子和终止子来表达SpCas9-SV40NLS;他们在sgRNA两侧添加了能被核酸酶切割的5′-HH和3′-HDV序列,构成的融合表达元件被构巢曲霉gpdA启动子和trpC终止子来转录表达,并通过核酸酶切割获得sgRNA,该通用的曲霉CRISPR系统包含Cas9蛋白、引导sgRNA和标记基因,且这些元件被装载在一个自我复制的AMA1质粒上,筛选标记可以选择4种不同的抗性标记,从而通用于亲缘关系相近的丝状真菌.该系统被报道在这6个曲霉物种都可以实现基因编辑的作用,其中在构巢曲霉中yA基因的编辑效率可以高达到90%.2018年,Mortensen团队[53]改造并设计出更加强大的曲霉CRISPR-Cas9基因组编辑体系(TAPE).他们使用NHEJ修复缺陷的菌株作为CRISPR-Cas9转化的受体菌株,并将寡核苷酸作为修复模板实现了3种曲霉(构巢曲霉、黑曲霉和米曲霉)的高效编辑,通过加入含有终止子点突变的单链DNA修复模板,可以在靶标位点进行原位点突变,且效率接近100%.同时,他们还使用来源于烟曲霉的U6启动子构建了tRNA介导的3个sgRNA串联表达体系,简化了多个sgRNA构建的步骤,缩短了时间.利用这一体系,该团队对三个靶标基因argB、pyrG和nkuA开展编辑,成功在一个转化子中引入两个原位点突变和一个插入突变. ...
... 黑曲霉(A. niger)作为一类重要的工业丝状真菌,广泛用于糖化酶和柠檬酸等大宗发酵产品的生产.除了上述的Mortensen团队陆续开发的曲霉CRISPR-Cas9基因组编辑技术[52-53]以外,国内外其他研究团队也陆续构建和优化了黑曲霉CRISPR-Cas9编辑系统.2018年,中国科学院天津工业生物技术研究所郑平研究员团队[54]利用黑曲霉自身的PanU6启动子、外源的PhU6和PyU6启动子转录sgRNA,结果表明这3个U6启动子均能够发挥作用,并通过40 bp同源臂的模板DNA实现了albA基因的编辑,敲除效率达到79%,缩短了供体DNA的构建时间.随后,该团队[55]又构建了一种基于5S rRNA高效的黑曲霉CRISPR/Cas9编辑技术体系.在真核生物中,5S rRNA是通过RNA聚合酶Ⅲ转录出来的一类小的rRNA,5S rRNA只需要进行简单的加工或者根本不需要进行加工.5S rRNA在生物体中高度保守,并且在细胞中含量很高.5S rRNA具有易于鉴定和高转录水平等优点,这启发了该团队尝试使用内源性5S rRNA来转录sgRNA的表达.结果表明,基于5S rRNA转录sgRNA表达的策略能够显著提高黑曲霉CRISPR/Cas9系统的编辑效率.该系统对靶标基因编辑效率高达100%,同时建立了黑曲霉高效基因组编辑工具包,以40 bp的短同源臂模板就可以实现单位点、多位点的基因敲入以及长至48 kb的大片段DNA敲除等基因组精准编辑.基于5S rRNA的sgRNA表达策略有效解决了黑曲霉sgRNA转录问题,为其他丝状真菌CRISPR/Cas9编辑系统的构建提供了一种新思路.2019年华南理工大学的潘力教授团队[56]基于标记基因可回收利用的技术再次对黑曲霉CRISPR-Cas9编辑系统进行了改造,构建了CRISPR-HDR(CRISPR/Cas9 homologous direct repair)技术系统.该系统利用5-FOA反筛的技术将携带标记基因pyrG和Cas9表达元件的自主复制质粒进行回收去除,并通过共转化donor-DNA,成功实现对基因组中0kb、2kb、10kb和50kb大片段的删除;并将葡萄糖氧化酶基因(goxC)整合到amyR和glaA位点中,实现了goxC多拷贝的过表达敲入.随后,潘力团队利用构建的CRISPR-HDR技术体系将来源于嗜热毁丝霉的酸性海藻糖水解酶基因(MthT)成功整合到两个amyR位点进行双拷贝的过表达[57],并将纯化所得的重组海藻糖酶MthT作为添加剂在酿酒酵母乙醇发酵过程中使用,结果表明在高糖的环境下,乙醇的转化率显著提高.与此同时,该团队还建立了黑曲霉胞嘧啶碱基编辑系统(CRISPR/Cas9-rAPOBEC1)[58],该系统成功将尿嘧啶营养缺陷型基因pyrG和色素基因fwnA进行定点突变,其突变的效率为47.36%~100%;并对非表型的基因prtT也成功实现了C到T的单碱基编辑,结果表明其编辑效率仍然高达60%,该系统编辑的窗口范围是C2~C9这8个碱基,该系统的建立为黑曲霉的基因编辑系统提供了另一种高效、新型的技术工具.本文作者所在团队也对黑曲霉基因组编辑技术进行了研究,利用从黑曲霉基因组挖掘的全新内源的RNA聚合酶Ⅲ型U6启动子(Anp)来构建sgRNA表达载体,将密码子优化的NLS-Cas9-NLS基因分别放置于持续性强启动子P tef1、P gpdA 和诱导型启动子P glaA 下进行高效表达.将Cas9表达框和Anp-target-sgRNA表达框共转化进入黑曲霉CBS113.46菌株原生质体细胞后,通过NHEJ机制对pyrG位点进行了不精确修复,进而得到pyrG突变株;在添加了donor-pyrG共转化后,通过同源重组获得了pyrG突变株,其同源重组效率高达97.2%,随后对moc基因和laeA基因开展了编辑,并获得moc和laeA单、双突变的黑曲霉突变株,从而成功构建了黑曲霉工业菌种基因组编辑系统[59]. ...
... Applycations of CRISPR-Cas systems in industrial filamentous fungi
Tab. 1| 丝状真菌 | Cas 类型 | Cas启动子 | sgRNA启动子 | 靶标基因 | 编辑效率 | 参考文献 |
|---|
| 黑曲霉 | Cas9 | P tef1 | P gpdA | albA | 高效 | [52] |
| P tef1 | Af_U6-1p | albA | 高效 | [53] |
| P tef1 | PanU6 | albA | 79% | [54] |
| P gpdA | 5S rRNA | alba, fum5, fum1 | 高达100% | [55] |
| P tef1 | AoU6, AfU6 | fwnA, amyA, glaA | 单基因高达80% | [56-58] |
| P tef1, P gpdA, P glaA | Anp | pyrG, moc, laeA | 高达97.2% | [59] |
| LbCpf1 | P tef1 | Af_U3p | albA | 80% | [60] |
| 米曲霉 | Cas9 | P amyB | 米曲霉U6p | wA, pyrG, yA | 10%~100% | [61] |
| P Aotef1, AMA1-自主复制质粒 | PU6p,AMA1-自主复制质粒 | wA, pyrG, yA | 55.6%~100% | [62] |
| 里氏木霉 | Cas9 | P pdc, P cbh1 | 体外转录合成 | ura5, lae1, vib1, clr2 | 单基因高达93%~100%;多基因同时编辑效率4.2%~45% | [63] |
| 体外转录合成 | 体外转录合成 | cbh-1 | 40% | [64] |
| P pdc | 里氏木霉PU61, PU62 | ura5 | 高效 | [65] |
| 体外转录合成 | 体外转录合成 | Trura5,Trlae1,Trcbh1,Trcbh2, Treg1 | 56.52%~100% | [66] |
| 产黄青霉 | Cas9 | P xyl 或体外 转录合成 | U6, 145 tRNA, Utp25或体外转录合成 | pks17, roqA, lovF,pcbAB, penDE, hcpA | 高达60%~100% | [67, 68] |
| 嗜热毁丝霉 | Cas9 | P tef1 | 嗜热毁丝霉MtU6p | amd S, cre1, res1, alp1, gh1-1, rca1, hcr1, ap3, prk6 | 单基因高达95%;双、三、四基因同时编辑效率22%~70% | [75] |
| SpCas9, FnCpf1, AsCpf1 | P tef1 或体外 转录合成 | U6p或体外 转录合成 | pks4.2, alp1, snc1, ptf1 | 单基因高达100%;多基因同时编辑时单基因效率5%~100% | [76] |
| AsCas12a | P tef1 | MtU6p | cre1, res1, alp1, gh1-1, rca1,neo, bar, hcr1, ap3, prk6 | 单基因高达90%;三、四基因同时编辑效率22%~41% | [77] |
| 棉阿舒囊霉 | SpCas9 | 酿酒酵母P tef1 | P snr52 | ade2, A754, fmp27 | 36%~80% | [78] |
| LbCpf1 | P tsa1 | P snr52 | his3, ade2, trp1, leu2, ura3 | 多基因编辑效率10.4%~77.2% | [79] |
3.3 CRISPR-Cas技术在其他丝状真菌中的应用3.3.1 稻瘟病菌2015年,日本明治大学Shigeru Kuwata教授团队[80]采用共转化载体Cas9表达框和sgRNA表达元件的策略成功在稻瘟病菌(M. oryzae)进行CRISPR-Cas9系统的构建.该团队同时使用稻瘟病菌自身的启动子U6p和构巢曲霉的启动子P trpC 分别转录表达sgRNA,对其编辑效率进行比较评价,成功对基因组中的SDH和SRS2基因进行敲除.2018年,英国埃克塞特大学Nicholas J. Talbot教授团队构建了CRISPR-Cas9 RNPs介导的稻瘟病菌基因组编辑技术体系[81],在不添加模板DNA时,以NHEJ的修复方式对靶标基因alb1进行编辑,并且不引入标记基因,建立了CRISPR-Cas9-RNP介导的无标记基因编辑系统;以HR修复方式时,仅用80 bp单链核苷酸为模板与RNPs共转化就实现了碱基的替换,或者80 bp双链DNA与RNPs共转化就能高效进行双基因同时编辑,并基于带有的苯菌灵抗性的突变菌株(E198A)对乙霉威(diethofencarb)的敏感性,设计了反筛体系,成功去除了标记基因,从而构建了CRISPR-Cas9-RNP介导的无标记基因的编辑系统.2019年,Shigeru Kuwata团队又基于单交叉介导的同源重组修复,改造升级了稻瘟病菌CRISPR-Cas9基因编辑系统,仅使用100 bp同源臂就实现了碱基的替换和GFP基因的敲入[82]. ...
Efficient oligo nucleotide mediated CRISPR-Cas9 gene editing in Aspergilli
3
2018
... 构巢曲霉(A. nidulans)是丝状真菌研究中常用的曲霉属模式物种,广泛用于真核微生物细胞生物学等基础研究.2015年,丹麦技术大学Uffe Hasbro Mortensen教授团队[52]首次在曲霉属的6个物种中建立了通用的CRISPR/Cas9基因组编辑体系,这些物种包括构巢曲霉(A. nidulans)、黑曲霉(A. niger)、棘孢曲霉(A. aculeatus)、巴西曲霉(A. brasiliensis)、炭黑曲霉菌(A. carbonarius)和泡盛曲霉(A. luchuensis).该团队首先基于黑曲霉密码子偏好性优化了SpCas9的核苷酸序列,并使用构巢曲霉tef1基因的启动子和终止子来表达SpCas9-SV40NLS;他们在sgRNA两侧添加了能被核酸酶切割的5′-HH和3′-HDV序列,构成的融合表达元件被构巢曲霉gpdA启动子和trpC终止子来转录表达,并通过核酸酶切割获得sgRNA,该通用的曲霉CRISPR系统包含Cas9蛋白、引导sgRNA和标记基因,且这些元件被装载在一个自我复制的AMA1质粒上,筛选标记可以选择4种不同的抗性标记,从而通用于亲缘关系相近的丝状真菌.该系统被报道在这6个曲霉物种都可以实现基因编辑的作用,其中在构巢曲霉中yA基因的编辑效率可以高达到90%.2018年,Mortensen团队[53]改造并设计出更加强大的曲霉CRISPR-Cas9基因组编辑体系(TAPE).他们使用NHEJ修复缺陷的菌株作为CRISPR-Cas9转化的受体菌株,并将寡核苷酸作为修复模板实现了3种曲霉(构巢曲霉、黑曲霉和米曲霉)的高效编辑,通过加入含有终止子点突变的单链DNA修复模板,可以在靶标位点进行原位点突变,且效率接近100%.同时,他们还使用来源于烟曲霉的U6启动子构建了tRNA介导的3个sgRNA串联表达体系,简化了多个sgRNA构建的步骤,缩短了时间.利用这一体系,该团队对三个靶标基因argB、pyrG和nkuA开展编辑,成功在一个转化子中引入两个原位点突变和一个插入突变. ...
... 黑曲霉(A. niger)作为一类重要的工业丝状真菌,广泛用于糖化酶和柠檬酸等大宗发酵产品的生产.除了上述的Mortensen团队陆续开发的曲霉CRISPR-Cas9基因组编辑技术[52-53]以外,国内外其他研究团队也陆续构建和优化了黑曲霉CRISPR-Cas9编辑系统.2018年,中国科学院天津工业生物技术研究所郑平研究员团队[54]利用黑曲霉自身的PanU6启动子、外源的PhU6和PyU6启动子转录sgRNA,结果表明这3个U6启动子均能够发挥作用,并通过40 bp同源臂的模板DNA实现了albA基因的编辑,敲除效率达到79%,缩短了供体DNA的构建时间.随后,该团队[55]又构建了一种基于5S rRNA高效的黑曲霉CRISPR/Cas9编辑技术体系.在真核生物中,5S rRNA是通过RNA聚合酶Ⅲ转录出来的一类小的rRNA,5S rRNA只需要进行简单的加工或者根本不需要进行加工.5S rRNA在生物体中高度保守,并且在细胞中含量很高.5S rRNA具有易于鉴定和高转录水平等优点,这启发了该团队尝试使用内源性5S rRNA来转录sgRNA的表达.结果表明,基于5S rRNA转录sgRNA表达的策略能够显著提高黑曲霉CRISPR/Cas9系统的编辑效率.该系统对靶标基因编辑效率高达100%,同时建立了黑曲霉高效基因组编辑工具包,以40 bp的短同源臂模板就可以实现单位点、多位点的基因敲入以及长至48 kb的大片段DNA敲除等基因组精准编辑.基于5S rRNA的sgRNA表达策略有效解决了黑曲霉sgRNA转录问题,为其他丝状真菌CRISPR/Cas9编辑系统的构建提供了一种新思路.2019年华南理工大学的潘力教授团队[56]基于标记基因可回收利用的技术再次对黑曲霉CRISPR-Cas9编辑系统进行了改造,构建了CRISPR-HDR(CRISPR/Cas9 homologous direct repair)技术系统.该系统利用5-FOA反筛的技术将携带标记基因pyrG和Cas9表达元件的自主复制质粒进行回收去除,并通过共转化donor-DNA,成功实现对基因组中0kb、2kb、10kb和50kb大片段的删除;并将葡萄糖氧化酶基因(goxC)整合到amyR和glaA位点中,实现了goxC多拷贝的过表达敲入.随后,潘力团队利用构建的CRISPR-HDR技术体系将来源于嗜热毁丝霉的酸性海藻糖水解酶基因(MthT)成功整合到两个amyR位点进行双拷贝的过表达[57],并将纯化所得的重组海藻糖酶MthT作为添加剂在酿酒酵母乙醇发酵过程中使用,结果表明在高糖的环境下,乙醇的转化率显著提高.与此同时,该团队还建立了黑曲霉胞嘧啶碱基编辑系统(CRISPR/Cas9-rAPOBEC1)[58],该系统成功将尿嘧啶营养缺陷型基因pyrG和色素基因fwnA进行定点突变,其突变的效率为47.36%~100%;并对非表型的基因prtT也成功实现了C到T的单碱基编辑,结果表明其编辑效率仍然高达60%,该系统编辑的窗口范围是C2~C9这8个碱基,该系统的建立为黑曲霉的基因编辑系统提供了另一种高效、新型的技术工具.本文作者所在团队也对黑曲霉基因组编辑技术进行了研究,利用从黑曲霉基因组挖掘的全新内源的RNA聚合酶Ⅲ型U6启动子(Anp)来构建sgRNA表达载体,将密码子优化的NLS-Cas9-NLS基因分别放置于持续性强启动子P tef1、P gpdA 和诱导型启动子P glaA 下进行高效表达.将Cas9表达框和Anp-target-sgRNA表达框共转化进入黑曲霉CBS113.46菌株原生质体细胞后,通过NHEJ机制对pyrG位点进行了不精确修复,进而得到pyrG突变株;在添加了donor-pyrG共转化后,通过同源重组获得了pyrG突变株,其同源重组效率高达97.2%,随后对moc基因和laeA基因开展了编辑,并获得moc和laeA单、双突变的黑曲霉突变株,从而成功构建了黑曲霉工业菌种基因组编辑系统[59]. ...
... Applycations of CRISPR-Cas systems in industrial filamentous fungi
Tab. 1| 丝状真菌 | Cas 类型 | Cas启动子 | sgRNA启动子 | 靶标基因 | 编辑效率 | 参考文献 |
|---|
| 黑曲霉 | Cas9 | P tef1 | P gpdA | albA | 高效 | [52] |
| P tef1 | Af_U6-1p | albA | 高效 | [53] |
| P tef1 | PanU6 | albA | 79% | [54] |
| P gpdA | 5S rRNA | alba, fum5, fum1 | 高达100% | [55] |
| P tef1 | AoU6, AfU6 | fwnA, amyA, glaA | 单基因高达80% | [56-58] |
| P tef1, P gpdA, P glaA | Anp | pyrG, moc, laeA | 高达97.2% | [59] |
| LbCpf1 | P tef1 | Af_U3p | albA | 80% | [60] |
| 米曲霉 | Cas9 | P amyB | 米曲霉U6p | wA, pyrG, yA | 10%~100% | [61] |
| P Aotef1, AMA1-自主复制质粒 | PU6p,AMA1-自主复制质粒 | wA, pyrG, yA | 55.6%~100% | [62] |
| 里氏木霉 | Cas9 | P pdc, P cbh1 | 体外转录合成 | ura5, lae1, vib1, clr2 | 单基因高达93%~100%;多基因同时编辑效率4.2%~45% | [63] |
| 体外转录合成 | 体外转录合成 | cbh-1 | 40% | [64] |
| P pdc | 里氏木霉PU61, PU62 | ura5 | 高效 | [65] |
| 体外转录合成 | 体外转录合成 | Trura5,Trlae1,Trcbh1,Trcbh2, Treg1 | 56.52%~100% | [66] |
| 产黄青霉 | Cas9 | P xyl 或体外 转录合成 | U6, 145 tRNA, Utp25或体外转录合成 | pks17, roqA, lovF,pcbAB, penDE, hcpA | 高达60%~100% | [67, 68] |
| 嗜热毁丝霉 | Cas9 | P tef1 | 嗜热毁丝霉MtU6p | amd S, cre1, res1, alp1, gh1-1, rca1, hcr1, ap3, prk6 | 单基因高达95%;双、三、四基因同时编辑效率22%~70% | [75] |
| SpCas9, FnCpf1, AsCpf1 | P tef1 或体外 转录合成 | U6p或体外 转录合成 | pks4.2, alp1, snc1, ptf1 | 单基因高达100%;多基因同时编辑时单基因效率5%~100% | [76] |
| AsCas12a | P tef1 | MtU6p | cre1, res1, alp1, gh1-1, rca1,neo, bar, hcr1, ap3, prk6 | 单基因高达90%;三、四基因同时编辑效率22%~41% | [77] |
| 棉阿舒囊霉 | SpCas9 | 酿酒酵母P tef1 | P snr52 | ade2, A754, fmp27 | 36%~80% | [78] |
| LbCpf1 | P tsa1 | P snr52 | his3, ade2, trp1, leu2, ura3 | 多基因编辑效率10.4%~77.2% | [79] |
3.3 CRISPR-Cas技术在其他丝状真菌中的应用3.3.1 稻瘟病菌2015年,日本明治大学Shigeru Kuwata教授团队[80]采用共转化载体Cas9表达框和sgRNA表达元件的策略成功在稻瘟病菌(M. oryzae)进行CRISPR-Cas9系统的构建.该团队同时使用稻瘟病菌自身的启动子U6p和构巢曲霉的启动子P trpC 分别转录表达sgRNA,对其编辑效率进行比较评价,成功对基因组中的SDH和SRS2基因进行敲除.2018年,英国埃克塞特大学Nicholas J. Talbot教授团队构建了CRISPR-Cas9 RNPs介导的稻瘟病菌基因组编辑技术体系[81],在不添加模板DNA时,以NHEJ的修复方式对靶标基因alb1进行编辑,并且不引入标记基因,建立了CRISPR-Cas9-RNP介导的无标记基因编辑系统;以HR修复方式时,仅用80 bp单链核苷酸为模板与RNPs共转化就实现了碱基的替换,或者80 bp双链DNA与RNPs共转化就能高效进行双基因同时编辑,并基于带有的苯菌灵抗性的突变菌株(E198A)对乙霉威(diethofencarb)的敏感性,设计了反筛体系,成功去除了标记基因,从而构建了CRISPR-Cas9-RNP介导的无标记基因的编辑系统.2019年,Shigeru Kuwata团队又基于单交叉介导的同源重组修复,改造升级了稻瘟病菌CRISPR-Cas9基因编辑系统,仅使用100 bp同源臂就实现了碱基的替换和GFP基因的敲入[82]. ...
Heterologous and endogenous U6 snRNA promoters enable CRISPR/Cas9 mediated genome editing in Aspergillus niger
2
2018
... 黑曲霉(A. niger)作为一类重要的工业丝状真菌,广泛用于糖化酶和柠檬酸等大宗发酵产品的生产.除了上述的Mortensen团队陆续开发的曲霉CRISPR-Cas9基因组编辑技术[52-53]以外,国内外其他研究团队也陆续构建和优化了黑曲霉CRISPR-Cas9编辑系统.2018年,中国科学院天津工业生物技术研究所郑平研究员团队[54]利用黑曲霉自身的PanU6启动子、外源的PhU6和PyU6启动子转录sgRNA,结果表明这3个U6启动子均能够发挥作用,并通过40 bp同源臂的模板DNA实现了albA基因的编辑,敲除效率达到79%,缩短了供体DNA的构建时间.随后,该团队[55]又构建了一种基于5S rRNA高效的黑曲霉CRISPR/Cas9编辑技术体系.在真核生物中,5S rRNA是通过RNA聚合酶Ⅲ转录出来的一类小的rRNA,5S rRNA只需要进行简单的加工或者根本不需要进行加工.5S rRNA在生物体中高度保守,并且在细胞中含量很高.5S rRNA具有易于鉴定和高转录水平等优点,这启发了该团队尝试使用内源性5S rRNA来转录sgRNA的表达.结果表明,基于5S rRNA转录sgRNA表达的策略能够显著提高黑曲霉CRISPR/Cas9系统的编辑效率.该系统对靶标基因编辑效率高达100%,同时建立了黑曲霉高效基因组编辑工具包,以40 bp的短同源臂模板就可以实现单位点、多位点的基因敲入以及长至48 kb的大片段DNA敲除等基因组精准编辑.基于5S rRNA的sgRNA表达策略有效解决了黑曲霉sgRNA转录问题,为其他丝状真菌CRISPR/Cas9编辑系统的构建提供了一种新思路.2019年华南理工大学的潘力教授团队[56]基于标记基因可回收利用的技术再次对黑曲霉CRISPR-Cas9编辑系统进行了改造,构建了CRISPR-HDR(CRISPR/Cas9 homologous direct repair)技术系统.该系统利用5-FOA反筛的技术将携带标记基因pyrG和Cas9表达元件的自主复制质粒进行回收去除,并通过共转化donor-DNA,成功实现对基因组中0kb、2kb、10kb和50kb大片段的删除;并将葡萄糖氧化酶基因(goxC)整合到amyR和glaA位点中,实现了goxC多拷贝的过表达敲入.随后,潘力团队利用构建的CRISPR-HDR技术体系将来源于嗜热毁丝霉的酸性海藻糖水解酶基因(MthT)成功整合到两个amyR位点进行双拷贝的过表达[57],并将纯化所得的重组海藻糖酶MthT作为添加剂在酿酒酵母乙醇发酵过程中使用,结果表明在高糖的环境下,乙醇的转化率显著提高.与此同时,该团队还建立了黑曲霉胞嘧啶碱基编辑系统(CRISPR/Cas9-rAPOBEC1)[58],该系统成功将尿嘧啶营养缺陷型基因pyrG和色素基因fwnA进行定点突变,其突变的效率为47.36%~100%;并对非表型的基因prtT也成功实现了C到T的单碱基编辑,结果表明其编辑效率仍然高达60%,该系统编辑的窗口范围是C2~C9这8个碱基,该系统的建立为黑曲霉的基因编辑系统提供了另一种高效、新型的技术工具.本文作者所在团队也对黑曲霉基因组编辑技术进行了研究,利用从黑曲霉基因组挖掘的全新内源的RNA聚合酶Ⅲ型U6启动子(Anp)来构建sgRNA表达载体,将密码子优化的NLS-Cas9-NLS基因分别放置于持续性强启动子P tef1、P gpdA 和诱导型启动子P glaA 下进行高效表达.将Cas9表达框和Anp-target-sgRNA表达框共转化进入黑曲霉CBS113.46菌株原生质体细胞后,通过NHEJ机制对pyrG位点进行了不精确修复,进而得到pyrG突变株;在添加了donor-pyrG共转化后,通过同源重组获得了pyrG突变株,其同源重组效率高达97.2%,随后对moc基因和laeA基因开展了编辑,并获得moc和laeA单、双突变的黑曲霉突变株,从而成功构建了黑曲霉工业菌种基因组编辑系统[59]. ...
... Applycations of CRISPR-Cas systems in industrial filamentous fungi
Tab. 1| 丝状真菌 | Cas 类型 | Cas启动子 | sgRNA启动子 | 靶标基因 | 编辑效率 | 参考文献 |
|---|
| 黑曲霉 | Cas9 | P tef1 | P gpdA | albA | 高效 | [52] |
| P tef1 | Af_U6-1p | albA | 高效 | [53] |
| P tef1 | PanU6 | albA | 79% | [54] |
| P gpdA | 5S rRNA | alba, fum5, fum1 | 高达100% | [55] |
| P tef1 | AoU6, AfU6 | fwnA, amyA, glaA | 单基因高达80% | [56-58] |
| P tef1, P gpdA, P glaA | Anp | pyrG, moc, laeA | 高达97.2% | [59] |
| LbCpf1 | P tef1 | Af_U3p | albA | 80% | [60] |
| 米曲霉 | Cas9 | P amyB | 米曲霉U6p | wA, pyrG, yA | 10%~100% | [61] |
| P Aotef1, AMA1-自主复制质粒 | PU6p,AMA1-自主复制质粒 | wA, pyrG, yA | 55.6%~100% | [62] |
| 里氏木霉 | Cas9 | P pdc, P cbh1 | 体外转录合成 | ura5, lae1, vib1, clr2 | 单基因高达93%~100%;多基因同时编辑效率4.2%~45% | [63] |
| 体外转录合成 | 体外转录合成 | cbh-1 | 40% | [64] |
| P pdc | 里氏木霉PU61, PU62 | ura5 | 高效 | [65] |
| 体外转录合成 | 体外转录合成 | Trura5,Trlae1,Trcbh1,Trcbh2, Treg1 | 56.52%~100% | [66] |
| 产黄青霉 | Cas9 | P xyl 或体外 转录合成 | U6, 145 tRNA, Utp25或体外转录合成 | pks17, roqA, lovF,pcbAB, penDE, hcpA | 高达60%~100% | [67, 68] |
| 嗜热毁丝霉 | Cas9 | P tef1 | 嗜热毁丝霉MtU6p | amd S, cre1, res1, alp1, gh1-1, rca1, hcr1, ap3, prk6 | 单基因高达95%;双、三、四基因同时编辑效率22%~70% | [75] |
| SpCas9, FnCpf1, AsCpf1 | P tef1 或体外 转录合成 | U6p或体外 转录合成 | pks4.2, alp1, snc1, ptf1 | 单基因高达100%;多基因同时编辑时单基因效率5%~100% | [76] |
| AsCas12a | P tef1 | MtU6p | cre1, res1, alp1, gh1-1, rca1,neo, bar, hcr1, ap3, prk6 | 单基因高达90%;三、四基因同时编辑效率22%~41% | [77] |
| 棉阿舒囊霉 | SpCas9 | 酿酒酵母P tef1 | P snr52 | ade2, A754, fmp27 | 36%~80% | [78] |
| LbCpf1 | P tsa1 | P snr52 | his3, ade2, trp1, leu2, ura3 | 多基因编辑效率10.4%~77.2% | [79] |
3.3 CRISPR-Cas技术在其他丝状真菌中的应用3.3.1 稻瘟病菌2015年,日本明治大学Shigeru Kuwata教授团队[80]采用共转化载体Cas9表达框和sgRNA表达元件的策略成功在稻瘟病菌(M. oryzae)进行CRISPR-Cas9系统的构建.该团队同时使用稻瘟病菌自身的启动子U6p和构巢曲霉的启动子P trpC 分别转录表达sgRNA,对其编辑效率进行比较评价,成功对基因组中的SDH和SRS2基因进行敲除.2018年,英国埃克塞特大学Nicholas J. Talbot教授团队构建了CRISPR-Cas9 RNPs介导的稻瘟病菌基因组编辑技术体系[81],在不添加模板DNA时,以NHEJ的修复方式对靶标基因alb1进行编辑,并且不引入标记基因,建立了CRISPR-Cas9-RNP介导的无标记基因编辑系统;以HR修复方式时,仅用80 bp单链核苷酸为模板与RNPs共转化就实现了碱基的替换,或者80 bp双链DNA与RNPs共转化就能高效进行双基因同时编辑,并基于带有的苯菌灵抗性的突变菌株(E198A)对乙霉威(diethofencarb)的敏感性,设计了反筛体系,成功去除了标记基因,从而构建了CRISPR-Cas9-RNP介导的无标记基因的编辑系统.2019年,Shigeru Kuwata团队又基于单交叉介导的同源重组修复,改造升级了稻瘟病菌CRISPR-Cas9基因编辑系统,仅使用100 bp同源臂就实现了碱基的替换和GFP基因的敲入[82]. ...
5S rRNA Promoter for guide RNA expression enabled highly efficient CRISPR/Cas9 genome editing in Aspergillus niger
2
2019
... 黑曲霉(A. niger)作为一类重要的工业丝状真菌,广泛用于糖化酶和柠檬酸等大宗发酵产品的生产.除了上述的Mortensen团队陆续开发的曲霉CRISPR-Cas9基因组编辑技术[52-53]以外,国内外其他研究团队也陆续构建和优化了黑曲霉CRISPR-Cas9编辑系统.2018年,中国科学院天津工业生物技术研究所郑平研究员团队[54]利用黑曲霉自身的PanU6启动子、外源的PhU6和PyU6启动子转录sgRNA,结果表明这3个U6启动子均能够发挥作用,并通过40 bp同源臂的模板DNA实现了albA基因的编辑,敲除效率达到79%,缩短了供体DNA的构建时间.随后,该团队[55]又构建了一种基于5S rRNA高效的黑曲霉CRISPR/Cas9编辑技术体系.在真核生物中,5S rRNA是通过RNA聚合酶Ⅲ转录出来的一类小的rRNA,5S rRNA只需要进行简单的加工或者根本不需要进行加工.5S rRNA在生物体中高度保守,并且在细胞中含量很高.5S rRNA具有易于鉴定和高转录水平等优点,这启发了该团队尝试使用内源性5S rRNA来转录sgRNA的表达.结果表明,基于5S rRNA转录sgRNA表达的策略能够显著提高黑曲霉CRISPR/Cas9系统的编辑效率.该系统对靶标基因编辑效率高达100%,同时建立了黑曲霉高效基因组编辑工具包,以40 bp的短同源臂模板就可以实现单位点、多位点的基因敲入以及长至48 kb的大片段DNA敲除等基因组精准编辑.基于5S rRNA的sgRNA表达策略有效解决了黑曲霉sgRNA转录问题,为其他丝状真菌CRISPR/Cas9编辑系统的构建提供了一种新思路.2019年华南理工大学的潘力教授团队[56]基于标记基因可回收利用的技术再次对黑曲霉CRISPR-Cas9编辑系统进行了改造,构建了CRISPR-HDR(CRISPR/Cas9 homologous direct repair)技术系统.该系统利用5-FOA反筛的技术将携带标记基因pyrG和Cas9表达元件的自主复制质粒进行回收去除,并通过共转化donor-DNA,成功实现对基因组中0kb、2kb、10kb和50kb大片段的删除;并将葡萄糖氧化酶基因(goxC)整合到amyR和glaA位点中,实现了goxC多拷贝的过表达敲入.随后,潘力团队利用构建的CRISPR-HDR技术体系将来源于嗜热毁丝霉的酸性海藻糖水解酶基因(MthT)成功整合到两个amyR位点进行双拷贝的过表达[57],并将纯化所得的重组海藻糖酶MthT作为添加剂在酿酒酵母乙醇发酵过程中使用,结果表明在高糖的环境下,乙醇的转化率显著提高.与此同时,该团队还建立了黑曲霉胞嘧啶碱基编辑系统(CRISPR/Cas9-rAPOBEC1)[58],该系统成功将尿嘧啶营养缺陷型基因pyrG和色素基因fwnA进行定点突变,其突变的效率为47.36%~100%;并对非表型的基因prtT也成功实现了C到T的单碱基编辑,结果表明其编辑效率仍然高达60%,该系统编辑的窗口范围是C2~C9这8个碱基,该系统的建立为黑曲霉的基因编辑系统提供了另一种高效、新型的技术工具.本文作者所在团队也对黑曲霉基因组编辑技术进行了研究,利用从黑曲霉基因组挖掘的全新内源的RNA聚合酶Ⅲ型U6启动子(Anp)来构建sgRNA表达载体,将密码子优化的NLS-Cas9-NLS基因分别放置于持续性强启动子P tef1、P gpdA 和诱导型启动子P glaA 下进行高效表达.将Cas9表达框和Anp-target-sgRNA表达框共转化进入黑曲霉CBS113.46菌株原生质体细胞后,通过NHEJ机制对pyrG位点进行了不精确修复,进而得到pyrG突变株;在添加了donor-pyrG共转化后,通过同源重组获得了pyrG突变株,其同源重组效率高达97.2%,随后对moc基因和laeA基因开展了编辑,并获得moc和laeA单、双突变的黑曲霉突变株,从而成功构建了黑曲霉工业菌种基因组编辑系统[59]. ...
... Applycations of CRISPR-Cas systems in industrial filamentous fungi
Tab. 1| 丝状真菌 | Cas 类型 | Cas启动子 | sgRNA启动子 | 靶标基因 | 编辑效率 | 参考文献 |
|---|
| 黑曲霉 | Cas9 | P tef1 | P gpdA | albA | 高效 | [52] |
| P tef1 | Af_U6-1p | albA | 高效 | [53] |
| P tef1 | PanU6 | albA | 79% | [54] |
| P gpdA | 5S rRNA | alba, fum5, fum1 | 高达100% | [55] |
| P tef1 | AoU6, AfU6 | fwnA, amyA, glaA | 单基因高达80% | [56-58] |
| P tef1, P gpdA, P glaA | Anp | pyrG, moc, laeA | 高达97.2% | [59] |
| LbCpf1 | P tef1 | Af_U3p | albA | 80% | [60] |
| 米曲霉 | Cas9 | P amyB | 米曲霉U6p | wA, pyrG, yA | 10%~100% | [61] |
| P Aotef1, AMA1-自主复制质粒 | PU6p,AMA1-自主复制质粒 | wA, pyrG, yA | 55.6%~100% | [62] |
| 里氏木霉 | Cas9 | P pdc, P cbh1 | 体外转录合成 | ura5, lae1, vib1, clr2 | 单基因高达93%~100%;多基因同时编辑效率4.2%~45% | [63] |
| 体外转录合成 | 体外转录合成 | cbh-1 | 40% | [64] |
| P pdc | 里氏木霉PU61, PU62 | ura5 | 高效 | [65] |
| 体外转录合成 | 体外转录合成 | Trura5,Trlae1,Trcbh1,Trcbh2, Treg1 | 56.52%~100% | [66] |
| 产黄青霉 | Cas9 | P xyl 或体外 转录合成 | U6, 145 tRNA, Utp25或体外转录合成 | pks17, roqA, lovF,pcbAB, penDE, hcpA | 高达60%~100% | [67, 68] |
| 嗜热毁丝霉 | Cas9 | P tef1 | 嗜热毁丝霉MtU6p | amd S, cre1, res1, alp1, gh1-1, rca1, hcr1, ap3, prk6 | 单基因高达95%;双、三、四基因同时编辑效率22%~70% | [75] |
| SpCas9, FnCpf1, AsCpf1 | P tef1 或体外 转录合成 | U6p或体外 转录合成 | pks4.2, alp1, snc1, ptf1 | 单基因高达100%;多基因同时编辑时单基因效率5%~100% | [76] |
| AsCas12a | P tef1 | MtU6p | cre1, res1, alp1, gh1-1, rca1,neo, bar, hcr1, ap3, prk6 | 单基因高达90%;三、四基因同时编辑效率22%~41% | [77] |
| 棉阿舒囊霉 | SpCas9 | 酿酒酵母P tef1 | P snr52 | ade2, A754, fmp27 | 36%~80% | [78] |
| LbCpf1 | P tsa1 | P snr52 | his3, ade2, trp1, leu2, ura3 | 多基因编辑效率10.4%~77.2% | [79] |
3.3 CRISPR-Cas技术在其他丝状真菌中的应用3.3.1 稻瘟病菌2015年,日本明治大学Shigeru Kuwata教授团队[80]采用共转化载体Cas9表达框和sgRNA表达元件的策略成功在稻瘟病菌(M. oryzae)进行CRISPR-Cas9系统的构建.该团队同时使用稻瘟病菌自身的启动子U6p和构巢曲霉的启动子P trpC 分别转录表达sgRNA,对其编辑效率进行比较评价,成功对基因组中的SDH和SRS2基因进行敲除.2018年,英国埃克塞特大学Nicholas J. Talbot教授团队构建了CRISPR-Cas9 RNPs介导的稻瘟病菌基因组编辑技术体系[81],在不添加模板DNA时,以NHEJ的修复方式对靶标基因alb1进行编辑,并且不引入标记基因,建立了CRISPR-Cas9-RNP介导的无标记基因编辑系统;以HR修复方式时,仅用80 bp单链核苷酸为模板与RNPs共转化就实现了碱基的替换,或者80 bp双链DNA与RNPs共转化就能高效进行双基因同时编辑,并基于带有的苯菌灵抗性的突变菌株(E198A)对乙霉威(diethofencarb)的敏感性,设计了反筛体系,成功去除了标记基因,从而构建了CRISPR-Cas9-RNP介导的无标记基因的编辑系统.2019年,Shigeru Kuwata团队又基于单交叉介导的同源重组修复,改造升级了稻瘟病菌CRISPR-Cas9基因编辑系统,仅使用100 bp同源臂就实现了碱基的替换和GFP基因的敲入[82]. ...
Efficient genome editing in Aspergillus niger with an improved recyclable CRISPR-HDR toolbox and its application in introducing multiple copies of heterologous genes
2
2019
... 黑曲霉(A. niger)作为一类重要的工业丝状真菌,广泛用于糖化酶和柠檬酸等大宗发酵产品的生产.除了上述的Mortensen团队陆续开发的曲霉CRISPR-Cas9基因组编辑技术[52-53]以外,国内外其他研究团队也陆续构建和优化了黑曲霉CRISPR-Cas9编辑系统.2018年,中国科学院天津工业生物技术研究所郑平研究员团队[54]利用黑曲霉自身的PanU6启动子、外源的PhU6和PyU6启动子转录sgRNA,结果表明这3个U6启动子均能够发挥作用,并通过40 bp同源臂的模板DNA实现了albA基因的编辑,敲除效率达到79%,缩短了供体DNA的构建时间.随后,该团队[55]又构建了一种基于5S rRNA高效的黑曲霉CRISPR/Cas9编辑技术体系.在真核生物中,5S rRNA是通过RNA聚合酶Ⅲ转录出来的一类小的rRNA,5S rRNA只需要进行简单的加工或者根本不需要进行加工.5S rRNA在生物体中高度保守,并且在细胞中含量很高.5S rRNA具有易于鉴定和高转录水平等优点,这启发了该团队尝试使用内源性5S rRNA来转录sgRNA的表达.结果表明,基于5S rRNA转录sgRNA表达的策略能够显著提高黑曲霉CRISPR/Cas9系统的编辑效率.该系统对靶标基因编辑效率高达100%,同时建立了黑曲霉高效基因组编辑工具包,以40 bp的短同源臂模板就可以实现单位点、多位点的基因敲入以及长至48 kb的大片段DNA敲除等基因组精准编辑.基于5S rRNA的sgRNA表达策略有效解决了黑曲霉sgRNA转录问题,为其他丝状真菌CRISPR/Cas9编辑系统的构建提供了一种新思路.2019年华南理工大学的潘力教授团队[56]基于标记基因可回收利用的技术再次对黑曲霉CRISPR-Cas9编辑系统进行了改造,构建了CRISPR-HDR(CRISPR/Cas9 homologous direct repair)技术系统.该系统利用5-FOA反筛的技术将携带标记基因pyrG和Cas9表达元件的自主复制质粒进行回收去除,并通过共转化donor-DNA,成功实现对基因组中0kb、2kb、10kb和50kb大片段的删除;并将葡萄糖氧化酶基因(goxC)整合到amyR和glaA位点中,实现了goxC多拷贝的过表达敲入.随后,潘力团队利用构建的CRISPR-HDR技术体系将来源于嗜热毁丝霉的酸性海藻糖水解酶基因(MthT)成功整合到两个amyR位点进行双拷贝的过表达[57],并将纯化所得的重组海藻糖酶MthT作为添加剂在酿酒酵母乙醇发酵过程中使用,结果表明在高糖的环境下,乙醇的转化率显著提高.与此同时,该团队还建立了黑曲霉胞嘧啶碱基编辑系统(CRISPR/Cas9-rAPOBEC1)[58],该系统成功将尿嘧啶营养缺陷型基因pyrG和色素基因fwnA进行定点突变,其突变的效率为47.36%~100%;并对非表型的基因prtT也成功实现了C到T的单碱基编辑,结果表明其编辑效率仍然高达60%,该系统编辑的窗口范围是C2~C9这8个碱基,该系统的建立为黑曲霉的基因编辑系统提供了另一种高效、新型的技术工具.本文作者所在团队也对黑曲霉基因组编辑技术进行了研究,利用从黑曲霉基因组挖掘的全新内源的RNA聚合酶Ⅲ型U6启动子(Anp)来构建sgRNA表达载体,将密码子优化的NLS-Cas9-NLS基因分别放置于持续性强启动子P tef1、P gpdA 和诱导型启动子P glaA 下进行高效表达.将Cas9表达框和Anp-target-sgRNA表达框共转化进入黑曲霉CBS113.46菌株原生质体细胞后,通过NHEJ机制对pyrG位点进行了不精确修复,进而得到pyrG突变株;在添加了donor-pyrG共转化后,通过同源重组获得了pyrG突变株,其同源重组效率高达97.2%,随后对moc基因和laeA基因开展了编辑,并获得moc和laeA单、双突变的黑曲霉突变株,从而成功构建了黑曲霉工业菌种基因组编辑系统[59]. ...
... Applycations of CRISPR-Cas systems in industrial filamentous fungi
Tab. 1| 丝状真菌 | Cas 类型 | Cas启动子 | sgRNA启动子 | 靶标基因 | 编辑效率 | 参考文献 |
|---|
| 黑曲霉 | Cas9 | P tef1 | P gpdA | albA | 高效 | [52] |
| P tef1 | Af_U6-1p | albA | 高效 | [53] |
| P tef1 | PanU6 | albA | 79% | [54] |
| P gpdA | 5S rRNA | alba, fum5, fum1 | 高达100% | [55] |
| P tef1 | AoU6, AfU6 | fwnA, amyA, glaA | 单基因高达80% | [56-58] |
| P tef1, P gpdA, P glaA | Anp | pyrG, moc, laeA | 高达97.2% | [59] |
| LbCpf1 | P tef1 | Af_U3p | albA | 80% | [60] |
| 米曲霉 | Cas9 | P amyB | 米曲霉U6p | wA, pyrG, yA | 10%~100% | [61] |
| P Aotef1, AMA1-自主复制质粒 | PU6p,AMA1-自主复制质粒 | wA, pyrG, yA | 55.6%~100% | [62] |
| 里氏木霉 | Cas9 | P pdc, P cbh1 | 体外转录合成 | ura5, lae1, vib1, clr2 | 单基因高达93%~100%;多基因同时编辑效率4.2%~45% | [63] |
| 体外转录合成 | 体外转录合成 | cbh-1 | 40% | [64] |
| P pdc | 里氏木霉PU61, PU62 | ura5 | 高效 | [65] |
| 体外转录合成 | 体外转录合成 | Trura5,Trlae1,Trcbh1,Trcbh2, Treg1 | 56.52%~100% | [66] |
| 产黄青霉 | Cas9 | P xyl 或体外 转录合成 | U6, 145 tRNA, Utp25或体外转录合成 | pks17, roqA, lovF,pcbAB, penDE, hcpA | 高达60%~100% | [67, 68] |
| 嗜热毁丝霉 | Cas9 | P tef1 | 嗜热毁丝霉MtU6p | amd S, cre1, res1, alp1, gh1-1, rca1, hcr1, ap3, prk6 | 单基因高达95%;双、三、四基因同时编辑效率22%~70% | [75] |
| SpCas9, FnCpf1, AsCpf1 | P tef1 或体外 转录合成 | U6p或体外 转录合成 | pks4.2, alp1, snc1, ptf1 | 单基因高达100%;多基因同时编辑时单基因效率5%~100% | [76] |
| AsCas12a | P tef1 | MtU6p | cre1, res1, alp1, gh1-1, rca1,neo, bar, hcr1, ap3, prk6 | 单基因高达90%;三、四基因同时编辑效率22%~41% | [77] |
| 棉阿舒囊霉 | SpCas9 | 酿酒酵母P tef1 | P snr52 | ade2, A754, fmp27 | 36%~80% | [78] |
| LbCpf1 | P tsa1 | P snr52 | his3, ade2, trp1, leu2, ura3 | 多基因编辑效率10.4%~77.2% | [79] |
3.3 CRISPR-Cas技术在其他丝状真菌中的应用3.3.1 稻瘟病菌2015年,日本明治大学Shigeru Kuwata教授团队[80]采用共转化载体Cas9表达框和sgRNA表达元件的策略成功在稻瘟病菌(M. oryzae)进行CRISPR-Cas9系统的构建.该团队同时使用稻瘟病菌自身的启动子U6p和构巢曲霉的启动子P trpC 分别转录表达sgRNA,对其编辑效率进行比较评价,成功对基因组中的SDH和SRS2基因进行敲除.2018年,英国埃克塞特大学Nicholas J. Talbot教授团队构建了CRISPR-Cas9 RNPs介导的稻瘟病菌基因组编辑技术体系[81],在不添加模板DNA时,以NHEJ的修复方式对靶标基因alb1进行编辑,并且不引入标记基因,建立了CRISPR-Cas9-RNP介导的无标记基因编辑系统;以HR修复方式时,仅用80 bp单链核苷酸为模板与RNPs共转化就实现了碱基的替换,或者80 bp双链DNA与RNPs共转化就能高效进行双基因同时编辑,并基于带有的苯菌灵抗性的突变菌株(E198A)对乙霉威(diethofencarb)的敏感性,设计了反筛体系,成功去除了标记基因,从而构建了CRISPR-Cas9-RNP介导的无标记基因的编辑系统.2019年,Shigeru Kuwata团队又基于单交叉介导的同源重组修复,改造升级了稻瘟病菌CRISPR-Cas9基因编辑系统,仅使用100 bp同源臂就实现了碱基的替换和GFP基因的敲入[82]. ...
High-level expression of highly active and thermostable trehalase from Myceliophthora thermophila in Aspergillus niger by using the CRISPR/Cas9 tool and its application in ethanol fermentation
1
2020
... 黑曲霉(A. niger)作为一类重要的工业丝状真菌,广泛用于糖化酶和柠檬酸等大宗发酵产品的生产.除了上述的Mortensen团队陆续开发的曲霉CRISPR-Cas9基因组编辑技术[52-53]以外,国内外其他研究团队也陆续构建和优化了黑曲霉CRISPR-Cas9编辑系统.2018年,中国科学院天津工业生物技术研究所郑平研究员团队[54]利用黑曲霉自身的PanU6启动子、外源的PhU6和PyU6启动子转录sgRNA,结果表明这3个U6启动子均能够发挥作用,并通过40 bp同源臂的模板DNA实现了albA基因的编辑,敲除效率达到79%,缩短了供体DNA的构建时间.随后,该团队[55]又构建了一种基于5S rRNA高效的黑曲霉CRISPR/Cas9编辑技术体系.在真核生物中,5S rRNA是通过RNA聚合酶Ⅲ转录出来的一类小的rRNA,5S rRNA只需要进行简单的加工或者根本不需要进行加工.5S rRNA在生物体中高度保守,并且在细胞中含量很高.5S rRNA具有易于鉴定和高转录水平等优点,这启发了该团队尝试使用内源性5S rRNA来转录sgRNA的表达.结果表明,基于5S rRNA转录sgRNA表达的策略能够显著提高黑曲霉CRISPR/Cas9系统的编辑效率.该系统对靶标基因编辑效率高达100%,同时建立了黑曲霉高效基因组编辑工具包,以40 bp的短同源臂模板就可以实现单位点、多位点的基因敲入以及长至48 kb的大片段DNA敲除等基因组精准编辑.基于5S rRNA的sgRNA表达策略有效解决了黑曲霉sgRNA转录问题,为其他丝状真菌CRISPR/Cas9编辑系统的构建提供了一种新思路.2019年华南理工大学的潘力教授团队[56]基于标记基因可回收利用的技术再次对黑曲霉CRISPR-Cas9编辑系统进行了改造,构建了CRISPR-HDR(CRISPR/Cas9 homologous direct repair)技术系统.该系统利用5-FOA反筛的技术将携带标记基因pyrG和Cas9表达元件的自主复制质粒进行回收去除,并通过共转化donor-DNA,成功实现对基因组中0kb、2kb、10kb和50kb大片段的删除;并将葡萄糖氧化酶基因(goxC)整合到amyR和glaA位点中,实现了goxC多拷贝的过表达敲入.随后,潘力团队利用构建的CRISPR-HDR技术体系将来源于嗜热毁丝霉的酸性海藻糖水解酶基因(MthT)成功整合到两个amyR位点进行双拷贝的过表达[57],并将纯化所得的重组海藻糖酶MthT作为添加剂在酿酒酵母乙醇发酵过程中使用,结果表明在高糖的环境下,乙醇的转化率显著提高.与此同时,该团队还建立了黑曲霉胞嘧啶碱基编辑系统(CRISPR/Cas9-rAPOBEC1)[58],该系统成功将尿嘧啶营养缺陷型基因pyrG和色素基因fwnA进行定点突变,其突变的效率为47.36%~100%;并对非表型的基因prtT也成功实现了C到T的单碱基编辑,结果表明其编辑效率仍然高达60%,该系统编辑的窗口范围是C2~C9这8个碱基,该系统的建立为黑曲霉的基因编辑系统提供了另一种高效、新型的技术工具.本文作者所在团队也对黑曲霉基因组编辑技术进行了研究,利用从黑曲霉基因组挖掘的全新内源的RNA聚合酶Ⅲ型U6启动子(Anp)来构建sgRNA表达载体,将密码子优化的NLS-Cas9-NLS基因分别放置于持续性强启动子P tef1、P gpdA 和诱导型启动子P glaA 下进行高效表达.将Cas9表达框和Anp-target-sgRNA表达框共转化进入黑曲霉CBS113.46菌株原生质体细胞后,通过NHEJ机制对pyrG位点进行了不精确修复,进而得到pyrG突变株;在添加了donor-pyrG共转化后,通过同源重组获得了pyrG突变株,其同源重组效率高达97.2%,随后对moc基因和laeA基因开展了编辑,并获得moc和laeA单、双突变的黑曲霉突变株,从而成功构建了黑曲霉工业菌种基因组编辑系统[59]. ...
Highly efficient single base editing in Aspergillus niger with CRISPR/Cas9 cytidine deaminase fusion
3
2019
... 黑曲霉(A. niger)作为一类重要的工业丝状真菌,广泛用于糖化酶和柠檬酸等大宗发酵产品的生产.除了上述的Mortensen团队陆续开发的曲霉CRISPR-Cas9基因组编辑技术[52-53]以外,国内外其他研究团队也陆续构建和优化了黑曲霉CRISPR-Cas9编辑系统.2018年,中国科学院天津工业生物技术研究所郑平研究员团队[54]利用黑曲霉自身的PanU6启动子、外源的PhU6和PyU6启动子转录sgRNA,结果表明这3个U6启动子均能够发挥作用,并通过40 bp同源臂的模板DNA实现了albA基因的编辑,敲除效率达到79%,缩短了供体DNA的构建时间.随后,该团队[55]又构建了一种基于5S rRNA高效的黑曲霉CRISPR/Cas9编辑技术体系.在真核生物中,5S rRNA是通过RNA聚合酶Ⅲ转录出来的一类小的rRNA,5S rRNA只需要进行简单的加工或者根本不需要进行加工.5S rRNA在生物体中高度保守,并且在细胞中含量很高.5S rRNA具有易于鉴定和高转录水平等优点,这启发了该团队尝试使用内源性5S rRNA来转录sgRNA的表达.结果表明,基于5S rRNA转录sgRNA表达的策略能够显著提高黑曲霉CRISPR/Cas9系统的编辑效率.该系统对靶标基因编辑效率高达100%,同时建立了黑曲霉高效基因组编辑工具包,以40 bp的短同源臂模板就可以实现单位点、多位点的基因敲入以及长至48 kb的大片段DNA敲除等基因组精准编辑.基于5S rRNA的sgRNA表达策略有效解决了黑曲霉sgRNA转录问题,为其他丝状真菌CRISPR/Cas9编辑系统的构建提供了一种新思路.2019年华南理工大学的潘力教授团队[56]基于标记基因可回收利用的技术再次对黑曲霉CRISPR-Cas9编辑系统进行了改造,构建了CRISPR-HDR(CRISPR/Cas9 homologous direct repair)技术系统.该系统利用5-FOA反筛的技术将携带标记基因pyrG和Cas9表达元件的自主复制质粒进行回收去除,并通过共转化donor-DNA,成功实现对基因组中0kb、2kb、10kb和50kb大片段的删除;并将葡萄糖氧化酶基因(goxC)整合到amyR和glaA位点中,实现了goxC多拷贝的过表达敲入.随后,潘力团队利用构建的CRISPR-HDR技术体系将来源于嗜热毁丝霉的酸性海藻糖水解酶基因(MthT)成功整合到两个amyR位点进行双拷贝的过表达[57],并将纯化所得的重组海藻糖酶MthT作为添加剂在酿酒酵母乙醇发酵过程中使用,结果表明在高糖的环境下,乙醇的转化率显著提高.与此同时,该团队还建立了黑曲霉胞嘧啶碱基编辑系统(CRISPR/Cas9-rAPOBEC1)[58],该系统成功将尿嘧啶营养缺陷型基因pyrG和色素基因fwnA进行定点突变,其突变的效率为47.36%~100%;并对非表型的基因prtT也成功实现了C到T的单碱基编辑,结果表明其编辑效率仍然高达60%,该系统编辑的窗口范围是C2~C9这8个碱基,该系统的建立为黑曲霉的基因编辑系统提供了另一种高效、新型的技术工具.本文作者所在团队也对黑曲霉基因组编辑技术进行了研究,利用从黑曲霉基因组挖掘的全新内源的RNA聚合酶Ⅲ型U6启动子(Anp)来构建sgRNA表达载体,将密码子优化的NLS-Cas9-NLS基因分别放置于持续性强启动子P tef1、P gpdA 和诱导型启动子P glaA 下进行高效表达.将Cas9表达框和Anp-target-sgRNA表达框共转化进入黑曲霉CBS113.46菌株原生质体细胞后,通过NHEJ机制对pyrG位点进行了不精确修复,进而得到pyrG突变株;在添加了donor-pyrG共转化后,通过同源重组获得了pyrG突变株,其同源重组效率高达97.2%,随后对moc基因和laeA基因开展了编辑,并获得moc和laeA单、双突变的黑曲霉突变株,从而成功构建了黑曲霉工业菌种基因组编辑系统[59]. ...
... Applycations of CRISPR-Cas systems in industrial filamentous fungi
Tab. 1| 丝状真菌 | Cas 类型 | Cas启动子 | sgRNA启动子 | 靶标基因 | 编辑效率 | 参考文献 |
|---|
| 黑曲霉 | Cas9 | P tef1 | P gpdA | albA | 高效 | [52] |
| P tef1 | Af_U6-1p | albA | 高效 | [53] |
| P tef1 | PanU6 | albA | 79% | [54] |
| P gpdA | 5S rRNA | alba, fum5, fum1 | 高达100% | [55] |
| P tef1 | AoU6, AfU6 | fwnA, amyA, glaA | 单基因高达80% | [56-58] |
| P tef1, P gpdA, P glaA | Anp | pyrG, moc, laeA | 高达97.2% | [59] |
| LbCpf1 | P tef1 | Af_U3p | albA | 80% | [60] |
| 米曲霉 | Cas9 | P amyB | 米曲霉U6p | wA, pyrG, yA | 10%~100% | [61] |
| P Aotef1, AMA1-自主复制质粒 | PU6p,AMA1-自主复制质粒 | wA, pyrG, yA | 55.6%~100% | [62] |
| 里氏木霉 | Cas9 | P pdc, P cbh1 | 体外转录合成 | ura5, lae1, vib1, clr2 | 单基因高达93%~100%;多基因同时编辑效率4.2%~45% | [63] |
| 体外转录合成 | 体外转录合成 | cbh-1 | 40% | [64] |
| P pdc | 里氏木霉PU61, PU62 | ura5 | 高效 | [65] |
| 体外转录合成 | 体外转录合成 | Trura5,Trlae1,Trcbh1,Trcbh2, Treg1 | 56.52%~100% | [66] |
| 产黄青霉 | Cas9 | P xyl 或体外 转录合成 | U6, 145 tRNA, Utp25或体外转录合成 | pks17, roqA, lovF,pcbAB, penDE, hcpA | 高达60%~100% | [67, 68] |
| 嗜热毁丝霉 | Cas9 | P tef1 | 嗜热毁丝霉MtU6p | amd S, cre1, res1, alp1, gh1-1, rca1, hcr1, ap3, prk6 | 单基因高达95%;双、三、四基因同时编辑效率22%~70% | [75] |
| SpCas9, FnCpf1, AsCpf1 | P tef1 或体外 转录合成 | U6p或体外 转录合成 | pks4.2, alp1, snc1, ptf1 | 单基因高达100%;多基因同时编辑时单基因效率5%~100% | [76] |
| AsCas12a | P tef1 | MtU6p | cre1, res1, alp1, gh1-1, rca1,neo, bar, hcr1, ap3, prk6 | 单基因高达90%;三、四基因同时编辑效率22%~41% | [77] |
| 棉阿舒囊霉 | SpCas9 | 酿酒酵母P tef1 | P snr52 | ade2, A754, fmp27 | 36%~80% | [78] |
| LbCpf1 | P tsa1 | P snr52 | his3, ade2, trp1, leu2, ura3 | 多基因编辑效率10.4%~77.2% | [79] |
3.3 CRISPR-Cas技术在其他丝状真菌中的应用3.3.1 稻瘟病菌2015年,日本明治大学Shigeru Kuwata教授团队[80]采用共转化载体Cas9表达框和sgRNA表达元件的策略成功在稻瘟病菌(M. oryzae)进行CRISPR-Cas9系统的构建.该团队同时使用稻瘟病菌自身的启动子U6p和构巢曲霉的启动子P trpC 分别转录表达sgRNA,对其编辑效率进行比较评价,成功对基因组中的SDH和SRS2基因进行敲除.2018年,英国埃克塞特大学Nicholas J. Talbot教授团队构建了CRISPR-Cas9 RNPs介导的稻瘟病菌基因组编辑技术体系[81],在不添加模板DNA时,以NHEJ的修复方式对靶标基因alb1进行编辑,并且不引入标记基因,建立了CRISPR-Cas9-RNP介导的无标记基因编辑系统;以HR修复方式时,仅用80 bp单链核苷酸为模板与RNPs共转化就实现了碱基的替换,或者80 bp双链DNA与RNPs共转化就能高效进行双基因同时编辑,并基于带有的苯菌灵抗性的突变菌株(E198A)对乙霉威(diethofencarb)的敏感性,设计了反筛体系,成功去除了标记基因,从而构建了CRISPR-Cas9-RNP介导的无标记基因的编辑系统.2019年,Shigeru Kuwata团队又基于单交叉介导的同源重组修复,改造升级了稻瘟病菌CRISPR-Cas9基因编辑系统,仅使用100 bp同源臂就实现了碱基的替换和GFP基因的敲入[82]. ...
... 2015年之前,CRISPR-Cas9系统尚未应用于真菌,研究者一直使用经典的基因工程手段来研究丝状真菌体系,尽管传统手段编辑丝状真菌基因可以达到多种研究目的,但仅限于一些模式真菌,其效率较低,编辑位点较少,且花费大量的时间和劳动力.CRISPR-Cas系统作为新一代基因组编辑技术,自出现以来,经过多种巧妙的改造,在多个物种中的基因编辑能力被相继证实,得到迅速而广泛的应用,为生命科学和医学研究带来前所未有的革新.自2015年起,CRISPR-Cas9系统被国内外研究学者陆续在丝状真菌体系中进行研发和应用,在短短几年内飞速发展,在不同的丝状真菌中各种高效的、CRISPR-Cas体系已经被陆续建立起来[45-50].近三年来,一些大型真菌的CRISPR-Cas9编辑系统也开始发展并建立,例如灵芝[88]、灰盖鬼伞[89]、蛹虫草[58, 90]和竹黄菌[91]等.CRISPR基因组编辑技术能对真菌基因组特定基因位点进行精准、高效的改造,在真菌细胞工厂构建和优化改造中展示了强大的功能,对真菌功能基因组研究和合成生物学的发展起着非常重要的作用. ...
一种调控sgRNA转录的启动子、表达载体,及其基因组编辑系统和应用
2
... 黑曲霉(A. niger)作为一类重要的工业丝状真菌,广泛用于糖化酶和柠檬酸等大宗发酵产品的生产.除了上述的Mortensen团队陆续开发的曲霉CRISPR-Cas9基因组编辑技术[52-53]以外,国内外其他研究团队也陆续构建和优化了黑曲霉CRISPR-Cas9编辑系统.2018年,中国科学院天津工业生物技术研究所郑平研究员团队[54]利用黑曲霉自身的PanU6启动子、外源的PhU6和PyU6启动子转录sgRNA,结果表明这3个U6启动子均能够发挥作用,并通过40 bp同源臂的模板DNA实现了albA基因的编辑,敲除效率达到79%,缩短了供体DNA的构建时间.随后,该团队[55]又构建了一种基于5S rRNA高效的黑曲霉CRISPR/Cas9编辑技术体系.在真核生物中,5S rRNA是通过RNA聚合酶Ⅲ转录出来的一类小的rRNA,5S rRNA只需要进行简单的加工或者根本不需要进行加工.5S rRNA在生物体中高度保守,并且在细胞中含量很高.5S rRNA具有易于鉴定和高转录水平等优点,这启发了该团队尝试使用内源性5S rRNA来转录sgRNA的表达.结果表明,基于5S rRNA转录sgRNA表达的策略能够显著提高黑曲霉CRISPR/Cas9系统的编辑效率.该系统对靶标基因编辑效率高达100%,同时建立了黑曲霉高效基因组编辑工具包,以40 bp的短同源臂模板就可以实现单位点、多位点的基因敲入以及长至48 kb的大片段DNA敲除等基因组精准编辑.基于5S rRNA的sgRNA表达策略有效解决了黑曲霉sgRNA转录问题,为其他丝状真菌CRISPR/Cas9编辑系统的构建提供了一种新思路.2019年华南理工大学的潘力教授团队[56]基于标记基因可回收利用的技术再次对黑曲霉CRISPR-Cas9编辑系统进行了改造,构建了CRISPR-HDR(CRISPR/Cas9 homologous direct repair)技术系统.该系统利用5-FOA反筛的技术将携带标记基因pyrG和Cas9表达元件的自主复制质粒进行回收去除,并通过共转化donor-DNA,成功实现对基因组中0kb、2kb、10kb和50kb大片段的删除;并将葡萄糖氧化酶基因(goxC)整合到amyR和glaA位点中,实现了goxC多拷贝的过表达敲入.随后,潘力团队利用构建的CRISPR-HDR技术体系将来源于嗜热毁丝霉的酸性海藻糖水解酶基因(MthT)成功整合到两个amyR位点进行双拷贝的过表达[57],并将纯化所得的重组海藻糖酶MthT作为添加剂在酿酒酵母乙醇发酵过程中使用,结果表明在高糖的环境下,乙醇的转化率显著提高.与此同时,该团队还建立了黑曲霉胞嘧啶碱基编辑系统(CRISPR/Cas9-rAPOBEC1)[58],该系统成功将尿嘧啶营养缺陷型基因pyrG和色素基因fwnA进行定点突变,其突变的效率为47.36%~100%;并对非表型的基因prtT也成功实现了C到T的单碱基编辑,结果表明其编辑效率仍然高达60%,该系统编辑的窗口范围是C2~C9这8个碱基,该系统的建立为黑曲霉的基因编辑系统提供了另一种高效、新型的技术工具.本文作者所在团队也对黑曲霉基因组编辑技术进行了研究,利用从黑曲霉基因组挖掘的全新内源的RNA聚合酶Ⅲ型U6启动子(Anp)来构建sgRNA表达载体,将密码子优化的NLS-Cas9-NLS基因分别放置于持续性强启动子P tef1、P gpdA 和诱导型启动子P glaA 下进行高效表达.将Cas9表达框和Anp-target-sgRNA表达框共转化进入黑曲霉CBS113.46菌株原生质体细胞后,通过NHEJ机制对pyrG位点进行了不精确修复,进而得到pyrG突变株;在添加了donor-pyrG共转化后,通过同源重组获得了pyrG突变株,其同源重组效率高达97.2%,随后对moc基因和laeA基因开展了编辑,并获得moc和laeA单、双突变的黑曲霉突变株,从而成功构建了黑曲霉工业菌种基因组编辑系统[59]. ...
... Applycations of CRISPR-Cas systems in industrial filamentous fungi
Tab. 1| 丝状真菌 | Cas 类型 | Cas启动子 | sgRNA启动子 | 靶标基因 | 编辑效率 | 参考文献 |
|---|
| 黑曲霉 | Cas9 | P tef1 | P gpdA | albA | 高效 | [52] |
| P tef1 | Af_U6-1p | albA | 高效 | [53] |
| P tef1 | PanU6 | albA | 79% | [54] |
| P gpdA | 5S rRNA | alba, fum5, fum1 | 高达100% | [55] |
| P tef1 | AoU6, AfU6 | fwnA, amyA, glaA | 单基因高达80% | [56-58] |
| P tef1, P gpdA, P glaA | Anp | pyrG, moc, laeA | 高达97.2% | [59] |
| LbCpf1 | P tef1 | Af_U3p | albA | 80% | [60] |
| 米曲霉 | Cas9 | P amyB | 米曲霉U6p | wA, pyrG, yA | 10%~100% | [61] |
| P Aotef1, AMA1-自主复制质粒 | PU6p,AMA1-自主复制质粒 | wA, pyrG, yA | 55.6%~100% | [62] |
| 里氏木霉 | Cas9 | P pdc, P cbh1 | 体外转录合成 | ura5, lae1, vib1, clr2 | 单基因高达93%~100%;多基因同时编辑效率4.2%~45% | [63] |
| 体外转录合成 | 体外转录合成 | cbh-1 | 40% | [64] |
| P pdc | 里氏木霉PU61, PU62 | ura5 | 高效 | [65] |
| 体外转录合成 | 体外转录合成 | Trura5,Trlae1,Trcbh1,Trcbh2, Treg1 | 56.52%~100% | [66] |
| 产黄青霉 | Cas9 | P xyl 或体外 转录合成 | U6, 145 tRNA, Utp25或体外转录合成 | pks17, roqA, lovF,pcbAB, penDE, hcpA | 高达60%~100% | [67, 68] |
| 嗜热毁丝霉 | Cas9 | P tef1 | 嗜热毁丝霉MtU6p | amd S, cre1, res1, alp1, gh1-1, rca1, hcr1, ap3, prk6 | 单基因高达95%;双、三、四基因同时编辑效率22%~70% | [75] |
| SpCas9, FnCpf1, AsCpf1 | P tef1 或体外 转录合成 | U6p或体外 转录合成 | pks4.2, alp1, snc1, ptf1 | 单基因高达100%;多基因同时编辑时单基因效率5%~100% | [76] |
| AsCas12a | P tef1 | MtU6p | cre1, res1, alp1, gh1-1, rca1,neo, bar, hcr1, ap3, prk6 | 单基因高达90%;三、四基因同时编辑效率22%~41% | [77] |
| 棉阿舒囊霉 | SpCas9 | 酿酒酵母P tef1 | P snr52 | ade2, A754, fmp27 | 36%~80% | [78] |
| LbCpf1 | P tsa1 | P snr52 | his3, ade2, trp1, leu2, ura3 | 多基因编辑效率10.4%~77.2% | [79] |
3.3 CRISPR-Cas技术在其他丝状真菌中的应用3.3.1 稻瘟病菌2015年,日本明治大学Shigeru Kuwata教授团队[80]采用共转化载体Cas9表达框和sgRNA表达元件的策略成功在稻瘟病菌(M. oryzae)进行CRISPR-Cas9系统的构建.该团队同时使用稻瘟病菌自身的启动子U6p和构巢曲霉的启动子P trpC 分别转录表达sgRNA,对其编辑效率进行比较评价,成功对基因组中的SDH和SRS2基因进行敲除.2018年,英国埃克塞特大学Nicholas J. Talbot教授团队构建了CRISPR-Cas9 RNPs介导的稻瘟病菌基因组编辑技术体系[81],在不添加模板DNA时,以NHEJ的修复方式对靶标基因alb1进行编辑,并且不引入标记基因,建立了CRISPR-Cas9-RNP介导的无标记基因编辑系统;以HR修复方式时,仅用80 bp单链核苷酸为模板与RNPs共转化就实现了碱基的替换,或者80 bp双链DNA与RNPs共转化就能高效进行双基因同时编辑,并基于带有的苯菌灵抗性的突变菌株(E198A)对乙霉威(diethofencarb)的敏感性,设计了反筛体系,成功去除了标记基因,从而构建了CRISPR-Cas9-RNP介导的无标记基因的编辑系统.2019年,Shigeru Kuwata团队又基于单交叉介导的同源重组修复,改造升级了稻瘟病菌CRISPR-Cas9基因编辑系统,仅使用100 bp同源臂就实现了碱基的替换和GFP基因的敲入[82]. ...
A promoter and expression vector that regulate the transcription of sgRNA and its genome editing system and application
2
... 黑曲霉(A. niger)作为一类重要的工业丝状真菌,广泛用于糖化酶和柠檬酸等大宗发酵产品的生产.除了上述的Mortensen团队陆续开发的曲霉CRISPR-Cas9基因组编辑技术[52-53]以外,国内外其他研究团队也陆续构建和优化了黑曲霉CRISPR-Cas9编辑系统.2018年,中国科学院天津工业生物技术研究所郑平研究员团队[54]利用黑曲霉自身的PanU6启动子、外源的PhU6和PyU6启动子转录sgRNA,结果表明这3个U6启动子均能够发挥作用,并通过40 bp同源臂的模板DNA实现了albA基因的编辑,敲除效率达到79%,缩短了供体DNA的构建时间.随后,该团队[55]又构建了一种基于5S rRNA高效的黑曲霉CRISPR/Cas9编辑技术体系.在真核生物中,5S rRNA是通过RNA聚合酶Ⅲ转录出来的一类小的rRNA,5S rRNA只需要进行简单的加工或者根本不需要进行加工.5S rRNA在生物体中高度保守,并且在细胞中含量很高.5S rRNA具有易于鉴定和高转录水平等优点,这启发了该团队尝试使用内源性5S rRNA来转录sgRNA的表达.结果表明,基于5S rRNA转录sgRNA表达的策略能够显著提高黑曲霉CRISPR/Cas9系统的编辑效率.该系统对靶标基因编辑效率高达100%,同时建立了黑曲霉高效基因组编辑工具包,以40 bp的短同源臂模板就可以实现单位点、多位点的基因敲入以及长至48 kb的大片段DNA敲除等基因组精准编辑.基于5S rRNA的sgRNA表达策略有效解决了黑曲霉sgRNA转录问题,为其他丝状真菌CRISPR/Cas9编辑系统的构建提供了一种新思路.2019年华南理工大学的潘力教授团队[56]基于标记基因可回收利用的技术再次对黑曲霉CRISPR-Cas9编辑系统进行了改造,构建了CRISPR-HDR(CRISPR/Cas9 homologous direct repair)技术系统.该系统利用5-FOA反筛的技术将携带标记基因pyrG和Cas9表达元件的自主复制质粒进行回收去除,并通过共转化donor-DNA,成功实现对基因组中0kb、2kb、10kb和50kb大片段的删除;并将葡萄糖氧化酶基因(goxC)整合到amyR和glaA位点中,实现了goxC多拷贝的过表达敲入.随后,潘力团队利用构建的CRISPR-HDR技术体系将来源于嗜热毁丝霉的酸性海藻糖水解酶基因(MthT)成功整合到两个amyR位点进行双拷贝的过表达[57],并将纯化所得的重组海藻糖酶MthT作为添加剂在酿酒酵母乙醇发酵过程中使用,结果表明在高糖的环境下,乙醇的转化率显著提高.与此同时,该团队还建立了黑曲霉胞嘧啶碱基编辑系统(CRISPR/Cas9-rAPOBEC1)[58],该系统成功将尿嘧啶营养缺陷型基因pyrG和色素基因fwnA进行定点突变,其突变的效率为47.36%~100%;并对非表型的基因prtT也成功实现了C到T的单碱基编辑,结果表明其编辑效率仍然高达60%,该系统编辑的窗口范围是C2~C9这8个碱基,该系统的建立为黑曲霉的基因编辑系统提供了另一种高效、新型的技术工具.本文作者所在团队也对黑曲霉基因组编辑技术进行了研究,利用从黑曲霉基因组挖掘的全新内源的RNA聚合酶Ⅲ型U6启动子(Anp)来构建sgRNA表达载体,将密码子优化的NLS-Cas9-NLS基因分别放置于持续性强启动子P tef1、P gpdA 和诱导型启动子P glaA 下进行高效表达.将Cas9表达框和Anp-target-sgRNA表达框共转化进入黑曲霉CBS113.46菌株原生质体细胞后,通过NHEJ机制对pyrG位点进行了不精确修复,进而得到pyrG突变株;在添加了donor-pyrG共转化后,通过同源重组获得了pyrG突变株,其同源重组效率高达97.2%,随后对moc基因和laeA基因开展了编辑,并获得moc和laeA单、双突变的黑曲霉突变株,从而成功构建了黑曲霉工业菌种基因组编辑系统[59]. ...
... Applycations of CRISPR-Cas systems in industrial filamentous fungi
Tab. 1| 丝状真菌 | Cas 类型 | Cas启动子 | sgRNA启动子 | 靶标基因 | 编辑效率 | 参考文献 |
|---|
| 黑曲霉 | Cas9 | P tef1 | P gpdA | albA | 高效 | [52] |
| P tef1 | Af_U6-1p | albA | 高效 | [53] |
| P tef1 | PanU6 | albA | 79% | [54] |
| P gpdA | 5S rRNA | alba, fum5, fum1 | 高达100% | [55] |
| P tef1 | AoU6, AfU6 | fwnA, amyA, glaA | 单基因高达80% | [56-58] |
| P tef1, P gpdA, P glaA | Anp | pyrG, moc, laeA | 高达97.2% | [59] |
| LbCpf1 | P tef1 | Af_U3p | albA | 80% | [60] |
| 米曲霉 | Cas9 | P amyB | 米曲霉U6p | wA, pyrG, yA | 10%~100% | [61] |
| P Aotef1, AMA1-自主复制质粒 | PU6p,AMA1-自主复制质粒 | wA, pyrG, yA | 55.6%~100% | [62] |
| 里氏木霉 | Cas9 | P pdc, P cbh1 | 体外转录合成 | ura5, lae1, vib1, clr2 | 单基因高达93%~100%;多基因同时编辑效率4.2%~45% | [63] |
| 体外转录合成 | 体外转录合成 | cbh-1 | 40% | [64] |
| P pdc | 里氏木霉PU61, PU62 | ura5 | 高效 | [65] |
| 体外转录合成 | 体外转录合成 | Trura5,Trlae1,Trcbh1,Trcbh2, Treg1 | 56.52%~100% | [66] |
| 产黄青霉 | Cas9 | P xyl 或体外 转录合成 | U6, 145 tRNA, Utp25或体外转录合成 | pks17, roqA, lovF,pcbAB, penDE, hcpA | 高达60%~100% | [67, 68] |
| 嗜热毁丝霉 | Cas9 | P tef1 | 嗜热毁丝霉MtU6p | amd S, cre1, res1, alp1, gh1-1, rca1, hcr1, ap3, prk6 | 单基因高达95%;双、三、四基因同时编辑效率22%~70% | [75] |
| SpCas9, FnCpf1, AsCpf1 | P tef1 或体外 转录合成 | U6p或体外 转录合成 | pks4.2, alp1, snc1, ptf1 | 单基因高达100%;多基因同时编辑时单基因效率5%~100% | [76] |
| AsCas12a | P tef1 | MtU6p | cre1, res1, alp1, gh1-1, rca1,neo, bar, hcr1, ap3, prk6 | 单基因高达90%;三、四基因同时编辑效率22%~41% | [77] |
| 棉阿舒囊霉 | SpCas9 | 酿酒酵母P tef1 | P snr52 | ade2, A754, fmp27 | 36%~80% | [78] |
| LbCpf1 | P tsa1 | P snr52 | his3, ade2, trp1, leu2, ura3 | 多基因编辑效率10.4%~77.2% | [79] |
3.3 CRISPR-Cas技术在其他丝状真菌中的应用3.3.1 稻瘟病菌2015年,日本明治大学Shigeru Kuwata教授团队[80]采用共转化载体Cas9表达框和sgRNA表达元件的策略成功在稻瘟病菌(M. oryzae)进行CRISPR-Cas9系统的构建.该团队同时使用稻瘟病菌自身的启动子U6p和构巢曲霉的启动子P trpC 分别转录表达sgRNA,对其编辑效率进行比较评价,成功对基因组中的SDH和SRS2基因进行敲除.2018年,英国埃克塞特大学Nicholas J. Talbot教授团队构建了CRISPR-Cas9 RNPs介导的稻瘟病菌基因组编辑技术体系[81],在不添加模板DNA时,以NHEJ的修复方式对靶标基因alb1进行编辑,并且不引入标记基因,建立了CRISPR-Cas9-RNP介导的无标记基因编辑系统;以HR修复方式时,仅用80 bp单链核苷酸为模板与RNPs共转化就实现了碱基的替换,或者80 bp双链DNA与RNPs共转化就能高效进行双基因同时编辑,并基于带有的苯菌灵抗性的突变菌株(E198A)对乙霉威(diethofencarb)的敏感性,设计了反筛体系,成功去除了标记基因,从而构建了CRISPR-Cas9-RNP介导的无标记基因的编辑系统.2019年,Shigeru Kuwata团队又基于单交叉介导的同源重组修复,改造升级了稻瘟病菌CRISPR-Cas9基因编辑系统,仅使用100 bp同源臂就实现了碱基的替换和GFP基因的敲入[82]. ...
Cpf1 enables fast and efficient genome editing in Aspergilli
2
2019
... 2019年,Mortensen团队基于AMA1自主复制质粒,构建了含有P tef1 -LbCpf1-T trpC 、U3p-tRNA-sgRNA-tRNA-U3t和筛选标记pryG的Cpf1-CRISPR-tRNA表达系统,该系统成功对构巢曲霉中yA基因和黑曲霉中albA基因开展了编辑,分别以NHEJ和HR两种修复方式获得了突变菌株,并且通过仅含有60 bp同源臂的供体DNA,成功将红色荧光蛋白(mRFP)整合到yA或albA位点[60].该研究表明,新兴的CRISPR-Cas12a(Cpf1)系统能够在丝状真菌中高效进行基因组编辑. ...
... Applycations of CRISPR-Cas systems in industrial filamentous fungi
Tab. 1| 丝状真菌 | Cas 类型 | Cas启动子 | sgRNA启动子 | 靶标基因 | 编辑效率 | 参考文献 |
|---|
| 黑曲霉 | Cas9 | P tef1 | P gpdA | albA | 高效 | [52] |
| P tef1 | Af_U6-1p | albA | 高效 | [53] |
| P tef1 | PanU6 | albA | 79% | [54] |
| P gpdA | 5S rRNA | alba, fum5, fum1 | 高达100% | [55] |
| P tef1 | AoU6, AfU6 | fwnA, amyA, glaA | 单基因高达80% | [56-58] |
| P tef1, P gpdA, P glaA | Anp | pyrG, moc, laeA | 高达97.2% | [59] |
| LbCpf1 | P tef1 | Af_U3p | albA | 80% | [60] |
| 米曲霉 | Cas9 | P amyB | 米曲霉U6p | wA, pyrG, yA | 10%~100% | [61] |
| P Aotef1, AMA1-自主复制质粒 | PU6p,AMA1-自主复制质粒 | wA, pyrG, yA | 55.6%~100% | [62] |
| 里氏木霉 | Cas9 | P pdc, P cbh1 | 体外转录合成 | ura5, lae1, vib1, clr2 | 单基因高达93%~100%;多基因同时编辑效率4.2%~45% | [63] |
| 体外转录合成 | 体外转录合成 | cbh-1 | 40% | [64] |
| P pdc | 里氏木霉PU61, PU62 | ura5 | 高效 | [65] |
| 体外转录合成 | 体外转录合成 | Trura5,Trlae1,Trcbh1,Trcbh2, Treg1 | 56.52%~100% | [66] |
| 产黄青霉 | Cas9 | P xyl 或体外 转录合成 | U6, 145 tRNA, Utp25或体外转录合成 | pks17, roqA, lovF,pcbAB, penDE, hcpA | 高达60%~100% | [67, 68] |
| 嗜热毁丝霉 | Cas9 | P tef1 | 嗜热毁丝霉MtU6p | amd S, cre1, res1, alp1, gh1-1, rca1, hcr1, ap3, prk6 | 单基因高达95%;双、三、四基因同时编辑效率22%~70% | [75] |
| SpCas9, FnCpf1, AsCpf1 | P tef1 或体外 转录合成 | U6p或体外 转录合成 | pks4.2, alp1, snc1, ptf1 | 单基因高达100%;多基因同时编辑时单基因效率5%~100% | [76] |
| AsCas12a | P tef1 | MtU6p | cre1, res1, alp1, gh1-1, rca1,neo, bar, hcr1, ap3, prk6 | 单基因高达90%;三、四基因同时编辑效率22%~41% | [77] |
| 棉阿舒囊霉 | SpCas9 | 酿酒酵母P tef1 | P snr52 | ade2, A754, fmp27 | 36%~80% | [78] |
| LbCpf1 | P tsa1 | P snr52 | his3, ade2, trp1, leu2, ura3 | 多基因编辑效率10.4%~77.2% | [79] |
3.3 CRISPR-Cas技术在其他丝状真菌中的应用3.3.1 稻瘟病菌2015年,日本明治大学Shigeru Kuwata教授团队[80]采用共转化载体Cas9表达框和sgRNA表达元件的策略成功在稻瘟病菌(M. oryzae)进行CRISPR-Cas9系统的构建.该团队同时使用稻瘟病菌自身的启动子U6p和构巢曲霉的启动子P trpC 分别转录表达sgRNA,对其编辑效率进行比较评价,成功对基因组中的SDH和SRS2基因进行敲除.2018年,英国埃克塞特大学Nicholas J. Talbot教授团队构建了CRISPR-Cas9 RNPs介导的稻瘟病菌基因组编辑技术体系[81],在不添加模板DNA时,以NHEJ的修复方式对靶标基因alb1进行编辑,并且不引入标记基因,建立了CRISPR-Cas9-RNP介导的无标记基因编辑系统;以HR修复方式时,仅用80 bp单链核苷酸为模板与RNPs共转化就实现了碱基的替换,或者80 bp双链DNA与RNPs共转化就能高效进行双基因同时编辑,并基于带有的苯菌灵抗性的突变菌株(E198A)对乙霉威(diethofencarb)的敏感性,设计了反筛体系,成功去除了标记基因,从而构建了CRISPR-Cas9-RNP介导的无标记基因的编辑系统.2019年,Shigeru Kuwata团队又基于单交叉介导的同源重组修复,改造升级了稻瘟病菌CRISPR-Cas9基因编辑系统,仅使用100 bp同源臂就实现了碱基的替换和GFP基因的敲入[82]. ...
Development of a genome editing technique using the CRISPR/Cas9 system in the industrial filamentous fungus Aspergillus oryzae
2
2016
... 米曲霉(A. oryzae)作为丝状真菌中重要的工业菌种之一,具有高效的蛋白分泌表达能力,被认证为GRAS安全生产菌株,广泛应用于食品酿造行业和工业酶制剂行业.2016年,日本东京大学Maruyama团队在米曲霉中进行了CRISPR/Cas9编辑系统的构建[61].在该系统中,利用amyB强启动子和终止子来转录表达密码子优化的Cas9基因,Cas9的过表达并不影响米曲霉菌株的正常生长,sgRNA被置于来自于米曲霉自身的U6p启动子和U6终止子之间进行转录表达,并且将Cas9和sgRNA两个表达元件构建在一个质粒中,成功对wA、pyrG和yA这3个靶基因进行了编辑,利用NHEJ修复方式,在靶标位点产生1 bp的插入或者1~21 bp的删除突变,编辑效率为10%~100%.2019年,Maruyama团队基于携带抗性标记ptrA的AMA1自主复制质粒的可循环使用,对米曲霉的CRISPR/Cas9编辑系统进行了优化和升级[62],分别在野生型和工业菌株中显著提高了目标基因的编辑效率.该系统通过NHEJ修复方式将wA、pyrG和yA这3个靶基因的编辑效率提高到55.6%~100%,实现了wA和niaD双基因的同时敲除,编辑效率为68.1%;通过HR修复方式,添加不引入标记基因的环状donor-DNA质粒,对wA和niaD双基因进行了同时敲除,敲除效率为61.9%;该团队还进行了无筛选标记的外源基因敲入实验,结果表明,该体系成功将绿色荧光蛋白EGFP基因整合到wA基因位点,同时将mDsRed基因整合到niaD位点,敲入效率为84.6%,实现了基因的定点插入.改造升级后的CRISPR/Cas9 系统将能更加有效地促进米曲霉在工业上代谢工程改造. ...
... Applycations of CRISPR-Cas systems in industrial filamentous fungi
Tab. 1| 丝状真菌 | Cas 类型 | Cas启动子 | sgRNA启动子 | 靶标基因 | 编辑效率 | 参考文献 |
|---|
| 黑曲霉 | Cas9 | P tef1 | P gpdA | albA | 高效 | [52] |
| P tef1 | Af_U6-1p | albA | 高效 | [53] |
| P tef1 | PanU6 | albA | 79% | [54] |
| P gpdA | 5S rRNA | alba, fum5, fum1 | 高达100% | [55] |
| P tef1 | AoU6, AfU6 | fwnA, amyA, glaA | 单基因高达80% | [56-58] |
| P tef1, P gpdA, P glaA | Anp | pyrG, moc, laeA | 高达97.2% | [59] |
| LbCpf1 | P tef1 | Af_U3p | albA | 80% | [60] |
| 米曲霉 | Cas9 | P amyB | 米曲霉U6p | wA, pyrG, yA | 10%~100% | [61] |
| P Aotef1, AMA1-自主复制质粒 | PU6p,AMA1-自主复制质粒 | wA, pyrG, yA | 55.6%~100% | [62] |
| 里氏木霉 | Cas9 | P pdc, P cbh1 | 体外转录合成 | ura5, lae1, vib1, clr2 | 单基因高达93%~100%;多基因同时编辑效率4.2%~45% | [63] |
| 体外转录合成 | 体外转录合成 | cbh-1 | 40% | [64] |
| P pdc | 里氏木霉PU61, PU62 | ura5 | 高效 | [65] |
| 体外转录合成 | 体外转录合成 | Trura5,Trlae1,Trcbh1,Trcbh2, Treg1 | 56.52%~100% | [66] |
| 产黄青霉 | Cas9 | P xyl 或体外 转录合成 | U6, 145 tRNA, Utp25或体外转录合成 | pks17, roqA, lovF,pcbAB, penDE, hcpA | 高达60%~100% | [67, 68] |
| 嗜热毁丝霉 | Cas9 | P tef1 | 嗜热毁丝霉MtU6p | amd S, cre1, res1, alp1, gh1-1, rca1, hcr1, ap3, prk6 | 单基因高达95%;双、三、四基因同时编辑效率22%~70% | [75] |
| SpCas9, FnCpf1, AsCpf1 | P tef1 或体外 转录合成 | U6p或体外 转录合成 | pks4.2, alp1, snc1, ptf1 | 单基因高达100%;多基因同时编辑时单基因效率5%~100% | [76] |
| AsCas12a | P tef1 | MtU6p | cre1, res1, alp1, gh1-1, rca1,neo, bar, hcr1, ap3, prk6 | 单基因高达90%;三、四基因同时编辑效率22%~41% | [77] |
| 棉阿舒囊霉 | SpCas9 | 酿酒酵母P tef1 | P snr52 | ade2, A754, fmp27 | 36%~80% | [78] |
| LbCpf1 | P tsa1 | P snr52 | his3, ade2, trp1, leu2, ura3 | 多基因编辑效率10.4%~77.2% | [79] |
3.3 CRISPR-Cas技术在其他丝状真菌中的应用3.3.1 稻瘟病菌2015年,日本明治大学Shigeru Kuwata教授团队[80]采用共转化载体Cas9表达框和sgRNA表达元件的策略成功在稻瘟病菌(M. oryzae)进行CRISPR-Cas9系统的构建.该团队同时使用稻瘟病菌自身的启动子U6p和构巢曲霉的启动子P trpC 分别转录表达sgRNA,对其编辑效率进行比较评价,成功对基因组中的SDH和SRS2基因进行敲除.2018年,英国埃克塞特大学Nicholas J. Talbot教授团队构建了CRISPR-Cas9 RNPs介导的稻瘟病菌基因组编辑技术体系[81],在不添加模板DNA时,以NHEJ的修复方式对靶标基因alb1进行编辑,并且不引入标记基因,建立了CRISPR-Cas9-RNP介导的无标记基因编辑系统;以HR修复方式时,仅用80 bp单链核苷酸为模板与RNPs共转化就实现了碱基的替换,或者80 bp双链DNA与RNPs共转化就能高效进行双基因同时编辑,并基于带有的苯菌灵抗性的突变菌株(E198A)对乙霉威(diethofencarb)的敏感性,设计了反筛体系,成功去除了标记基因,从而构建了CRISPR-Cas9-RNP介导的无标记基因的编辑系统.2019年,Shigeru Kuwata团队又基于单交叉介导的同源重组修复,改造升级了稻瘟病菌CRISPR-Cas9基因编辑系统,仅使用100 bp同源臂就实现了碱基的替换和GFP基因的敲入[82]. ...
Forced recycling of an AMA1-based genome-editing plasmid allows for efficient multiple gene deletion/integration in the industrial filamentous fungus Aspergillus oryzae
2
2019
... 米曲霉(A. oryzae)作为丝状真菌中重要的工业菌种之一,具有高效的蛋白分泌表达能力,被认证为GRAS安全生产菌株,广泛应用于食品酿造行业和工业酶制剂行业.2016年,日本东京大学Maruyama团队在米曲霉中进行了CRISPR/Cas9编辑系统的构建[61].在该系统中,利用amyB强启动子和终止子来转录表达密码子优化的Cas9基因,Cas9的过表达并不影响米曲霉菌株的正常生长,sgRNA被置于来自于米曲霉自身的U6p启动子和U6终止子之间进行转录表达,并且将Cas9和sgRNA两个表达元件构建在一个质粒中,成功对wA、pyrG和yA这3个靶基因进行了编辑,利用NHEJ修复方式,在靶标位点产生1 bp的插入或者1~21 bp的删除突变,编辑效率为10%~100%.2019年,Maruyama团队基于携带抗性标记ptrA的AMA1自主复制质粒的可循环使用,对米曲霉的CRISPR/Cas9编辑系统进行了优化和升级[62],分别在野生型和工业菌株中显著提高了目标基因的编辑效率.该系统通过NHEJ修复方式将wA、pyrG和yA这3个靶基因的编辑效率提高到55.6%~100%,实现了wA和niaD双基因的同时敲除,编辑效率为68.1%;通过HR修复方式,添加不引入标记基因的环状donor-DNA质粒,对wA和niaD双基因进行了同时敲除,敲除效率为61.9%;该团队还进行了无筛选标记的外源基因敲入实验,结果表明,该体系成功将绿色荧光蛋白EGFP基因整合到wA基因位点,同时将mDsRed基因整合到niaD位点,敲入效率为84.6%,实现了基因的定点插入.改造升级后的CRISPR/Cas9 系统将能更加有效地促进米曲霉在工业上代谢工程改造. ...
... Applycations of CRISPR-Cas systems in industrial filamentous fungi
Tab. 1| 丝状真菌 | Cas 类型 | Cas启动子 | sgRNA启动子 | 靶标基因 | 编辑效率 | 参考文献 |
|---|
| 黑曲霉 | Cas9 | P tef1 | P gpdA | albA | 高效 | [52] |
| P tef1 | Af_U6-1p | albA | 高效 | [53] |
| P tef1 | PanU6 | albA | 79% | [54] |
| P gpdA | 5S rRNA | alba, fum5, fum1 | 高达100% | [55] |
| P tef1 | AoU6, AfU6 | fwnA, amyA, glaA | 单基因高达80% | [56-58] |
| P tef1, P gpdA, P glaA | Anp | pyrG, moc, laeA | 高达97.2% | [59] |
| LbCpf1 | P tef1 | Af_U3p | albA | 80% | [60] |
| 米曲霉 | Cas9 | P amyB | 米曲霉U6p | wA, pyrG, yA | 10%~100% | [61] |
| P Aotef1, AMA1-自主复制质粒 | PU6p,AMA1-自主复制质粒 | wA, pyrG, yA | 55.6%~100% | [62] |
| 里氏木霉 | Cas9 | P pdc, P cbh1 | 体外转录合成 | ura5, lae1, vib1, clr2 | 单基因高达93%~100%;多基因同时编辑效率4.2%~45% | [63] |
| 体外转录合成 | 体外转录合成 | cbh-1 | 40% | [64] |
| P pdc | 里氏木霉PU61, PU62 | ura5 | 高效 | [65] |
| 体外转录合成 | 体外转录合成 | Trura5,Trlae1,Trcbh1,Trcbh2, Treg1 | 56.52%~100% | [66] |
| 产黄青霉 | Cas9 | P xyl 或体外 转录合成 | U6, 145 tRNA, Utp25或体外转录合成 | pks17, roqA, lovF,pcbAB, penDE, hcpA | 高达60%~100% | [67, 68] |
| 嗜热毁丝霉 | Cas9 | P tef1 | 嗜热毁丝霉MtU6p | amd S, cre1, res1, alp1, gh1-1, rca1, hcr1, ap3, prk6 | 单基因高达95%;双、三、四基因同时编辑效率22%~70% | [75] |
| SpCas9, FnCpf1, AsCpf1 | P tef1 或体外 转录合成 | U6p或体外 转录合成 | pks4.2, alp1, snc1, ptf1 | 单基因高达100%;多基因同时编辑时单基因效率5%~100% | [76] |
| AsCas12a | P tef1 | MtU6p | cre1, res1, alp1, gh1-1, rca1,neo, bar, hcr1, ap3, prk6 | 单基因高达90%;三、四基因同时编辑效率22%~41% | [77] |
| 棉阿舒囊霉 | SpCas9 | 酿酒酵母P tef1 | P snr52 | ade2, A754, fmp27 | 36%~80% | [78] |
| LbCpf1 | P tsa1 | P snr52 | his3, ade2, trp1, leu2, ura3 | 多基因编辑效率10.4%~77.2% | [79] |
3.3 CRISPR-Cas技术在其他丝状真菌中的应用3.3.1 稻瘟病菌2015年,日本明治大学Shigeru Kuwata教授团队[80]采用共转化载体Cas9表达框和sgRNA表达元件的策略成功在稻瘟病菌(M. oryzae)进行CRISPR-Cas9系统的构建.该团队同时使用稻瘟病菌自身的启动子U6p和构巢曲霉的启动子P trpC 分别转录表达sgRNA,对其编辑效率进行比较评价,成功对基因组中的SDH和SRS2基因进行敲除.2018年,英国埃克塞特大学Nicholas J. Talbot教授团队构建了CRISPR-Cas9 RNPs介导的稻瘟病菌基因组编辑技术体系[81],在不添加模板DNA时,以NHEJ的修复方式对靶标基因alb1进行编辑,并且不引入标记基因,建立了CRISPR-Cas9-RNP介导的无标记基因编辑系统;以HR修复方式时,仅用80 bp单链核苷酸为模板与RNPs共转化就实现了碱基的替换,或者80 bp双链DNA与RNPs共转化就能高效进行双基因同时编辑,并基于带有的苯菌灵抗性的突变菌株(E198A)对乙霉威(diethofencarb)的敏感性,设计了反筛体系,成功去除了标记基因,从而构建了CRISPR-Cas9-RNP介导的无标记基因的编辑系统.2019年,Shigeru Kuwata团队又基于单交叉介导的同源重组修复,改造升级了稻瘟病菌CRISPR-Cas9基因编辑系统,仅使用100 bp同源臂就实现了碱基的替换和GFP基因的敲入[82]. ...
Efficient genome editing in filamentous fungus Trichoderma reesei using the CRISPR/Cas9 system
2
2015
... 2015年,中国科学院上海生命科学研究院周志华研究员团队率先在工业纤维素酶生产真菌里氏木霉(T. reesei)中开展了CRISPR/Cas9系统的研发[63],这是工业丝状真菌中CRISPR-Cas9技术最早的报道.他们成功将密码子优化的Cas9基因分别放置于持续性强启动子P pdc 和诱导型cbh1启动子下进行组成型和诱导型表达,利用体外转录sgRNA,成功对野生型菌株Qm6a和工业菌株Rut-C30中的靶标基因ura5进行编辑,获得NHEJ修复的突变菌株.同时,基于同源重组,他们研究了donor DNA的同源臂长度对靶标基因敲除效率的影响,结果表明donor DNA的同源臂仅为0.2 kb就能成功进行基因敲除,同源重组效率高达93%.随后,他们通过优化gRNA和donor DNA分子摩尔配比,成功对目标基因lae1、vib1和clr2进行单基因敲除、双基因和三基因的同时敲除,获得三基因突变菌株.这是首次在丝状真菌中利用CRISPR/Cas9技术对多个基因同时进行编辑的报道,表明CRISPR/Cas9系统能显著提高工业真菌的遗传编辑效率,促进工业菌株的功能基因和代谢工程改造的研究.2019年,中国农业科学院苏小运团队对里氏木霉CRISPR/Cas9系统进行进一步研究[64],他们在体外组装Cas9/gRNA核糖核蛋白复合体(ribonucleoproteins,RNPs),并将核糖核蛋白复合体和携带pyr4抗性标记的载体直接转化受体菌株,成功对靶基因cbh-1进行敲除,表明体外转录的Cas9/gRNA复合体能够对里氏木霉基因组序列进行切割编辑.2020年,华东理工大学魏东芝团队 基于嗜热毁丝霉U6 snRNA的基因序列在里氏木霉基因组中进行生物信息学比对,查找出2个U6基因(TrU6-1和TrU6-2),并用这2个U6基因的启动子转录sgRNA的表达,均能成功对靶标基因ura5进行NHEJ修复的编辑,表明里氏木霉体内转录的sgRNA也能够成功介导Cas9蛋白对双链DNA进行切割[65].近期,周志华研究员团队再次升级了里氏木霉的CRISPR/Cas9技术系统[66].他们通过添加Triton X-100、延长原生质体与RNPs的孵育时间来优化RNPs技术,提高了RNPs的转化效率,显著增加了里氏木霉和蛹虫草(Cordyceps militaris)的基因编辑效率,其中单基因Trura5和Cmura5的编辑效率高达100%.通过使用优化后的RNP技术,他们仅用20 bp的同源臂就实现了Trlae1的基因敲除,同源重组效率增加到56.52%.该团队通过加入添加肌醇或苯菌灵来抑制有丝分裂和细胞分裂,进一步优化了RNPs技术,显著提高了里氏木霉和米曲霉进行单核原生质体转化的效率,从而提高了获得纯合转化子的效率,进而消除了转化后期的多轮单孢分离过程.优化的RNPs技术将有助于在丝状真菌中建立更有效的不依赖转基因的基因组编辑技术,从而广泛应用于食品和医药行业. ...
... Applycations of CRISPR-Cas systems in industrial filamentous fungi
Tab. 1| 丝状真菌 | Cas 类型 | Cas启动子 | sgRNA启动子 | 靶标基因 | 编辑效率 | 参考文献 |
|---|
| 黑曲霉 | Cas9 | P tef1 | P gpdA | albA | 高效 | [52] |
| P tef1 | Af_U6-1p | albA | 高效 | [53] |
| P tef1 | PanU6 | albA | 79% | [54] |
| P gpdA | 5S rRNA | alba, fum5, fum1 | 高达100% | [55] |
| P tef1 | AoU6, AfU6 | fwnA, amyA, glaA | 单基因高达80% | [56-58] |
| P tef1, P gpdA, P glaA | Anp | pyrG, moc, laeA | 高达97.2% | [59] |
| LbCpf1 | P tef1 | Af_U3p | albA | 80% | [60] |
| 米曲霉 | Cas9 | P amyB | 米曲霉U6p | wA, pyrG, yA | 10%~100% | [61] |
| P Aotef1, AMA1-自主复制质粒 | PU6p,AMA1-自主复制质粒 | wA, pyrG, yA | 55.6%~100% | [62] |
| 里氏木霉 | Cas9 | P pdc, P cbh1 | 体外转录合成 | ura5, lae1, vib1, clr2 | 单基因高达93%~100%;多基因同时编辑效率4.2%~45% | [63] |
| 体外转录合成 | 体外转录合成 | cbh-1 | 40% | [64] |
| P pdc | 里氏木霉PU61, PU62 | ura5 | 高效 | [65] |
| 体外转录合成 | 体外转录合成 | Trura5,Trlae1,Trcbh1,Trcbh2, Treg1 | 56.52%~100% | [66] |
| 产黄青霉 | Cas9 | P xyl 或体外 转录合成 | U6, 145 tRNA, Utp25或体外转录合成 | pks17, roqA, lovF,pcbAB, penDE, hcpA | 高达60%~100% | [67, 68] |
| 嗜热毁丝霉 | Cas9 | P tef1 | 嗜热毁丝霉MtU6p | amd S, cre1, res1, alp1, gh1-1, rca1, hcr1, ap3, prk6 | 单基因高达95%;双、三、四基因同时编辑效率22%~70% | [75] |
| SpCas9, FnCpf1, AsCpf1 | P tef1 或体外 转录合成 | U6p或体外 转录合成 | pks4.2, alp1, snc1, ptf1 | 单基因高达100%;多基因同时编辑时单基因效率5%~100% | [76] |
| AsCas12a | P tef1 | MtU6p | cre1, res1, alp1, gh1-1, rca1,neo, bar, hcr1, ap3, prk6 | 单基因高达90%;三、四基因同时编辑效率22%~41% | [77] |
| 棉阿舒囊霉 | SpCas9 | 酿酒酵母P tef1 | P snr52 | ade2, A754, fmp27 | 36%~80% | [78] |
| LbCpf1 | P tsa1 | P snr52 | his3, ade2, trp1, leu2, ura3 | 多基因编辑效率10.4%~77.2% | [79] |
3.3 CRISPR-Cas技术在其他丝状真菌中的应用3.3.1 稻瘟病菌2015年,日本明治大学Shigeru Kuwata教授团队[80]采用共转化载体Cas9表达框和sgRNA表达元件的策略成功在稻瘟病菌(M. oryzae)进行CRISPR-Cas9系统的构建.该团队同时使用稻瘟病菌自身的启动子U6p和构巢曲霉的启动子P trpC 分别转录表达sgRNA,对其编辑效率进行比较评价,成功对基因组中的SDH和SRS2基因进行敲除.2018年,英国埃克塞特大学Nicholas J. Talbot教授团队构建了CRISPR-Cas9 RNPs介导的稻瘟病菌基因组编辑技术体系[81],在不添加模板DNA时,以NHEJ的修复方式对靶标基因alb1进行编辑,并且不引入标记基因,建立了CRISPR-Cas9-RNP介导的无标记基因编辑系统;以HR修复方式时,仅用80 bp单链核苷酸为模板与RNPs共转化就实现了碱基的替换,或者80 bp双链DNA与RNPs共转化就能高效进行双基因同时编辑,并基于带有的苯菌灵抗性的突变菌株(E198A)对乙霉威(diethofencarb)的敏感性,设计了反筛体系,成功去除了标记基因,从而构建了CRISPR-Cas9-RNP介导的无标记基因的编辑系统.2019年,Shigeru Kuwata团队又基于单交叉介导的同源重组修复,改造升级了稻瘟病菌CRISPR-Cas9基因编辑系统,仅使用100 bp同源臂就实现了碱基的替换和GFP基因的敲入[82]. ...
Fast gene disruption in Trichoderma reesei using in vitro assembled Cas9/gRNA complex
2
2019
... 2015年,中国科学院上海生命科学研究院周志华研究员团队率先在工业纤维素酶生产真菌里氏木霉(T. reesei)中开展了CRISPR/Cas9系统的研发[63],这是工业丝状真菌中CRISPR-Cas9技术最早的报道.他们成功将密码子优化的Cas9基因分别放置于持续性强启动子P pdc 和诱导型cbh1启动子下进行组成型和诱导型表达,利用体外转录sgRNA,成功对野生型菌株Qm6a和工业菌株Rut-C30中的靶标基因ura5进行编辑,获得NHEJ修复的突变菌株.同时,基于同源重组,他们研究了donor DNA的同源臂长度对靶标基因敲除效率的影响,结果表明donor DNA的同源臂仅为0.2 kb就能成功进行基因敲除,同源重组效率高达93%.随后,他们通过优化gRNA和donor DNA分子摩尔配比,成功对目标基因lae1、vib1和clr2进行单基因敲除、双基因和三基因的同时敲除,获得三基因突变菌株.这是首次在丝状真菌中利用CRISPR/Cas9技术对多个基因同时进行编辑的报道,表明CRISPR/Cas9系统能显著提高工业真菌的遗传编辑效率,促进工业菌株的功能基因和代谢工程改造的研究.2019年,中国农业科学院苏小运团队对里氏木霉CRISPR/Cas9系统进行进一步研究[64],他们在体外组装Cas9/gRNA核糖核蛋白复合体(ribonucleoproteins,RNPs),并将核糖核蛋白复合体和携带pyr4抗性标记的载体直接转化受体菌株,成功对靶基因cbh-1进行敲除,表明体外转录的Cas9/gRNA复合体能够对里氏木霉基因组序列进行切割编辑.2020年,华东理工大学魏东芝团队 基于嗜热毁丝霉U6 snRNA的基因序列在里氏木霉基因组中进行生物信息学比对,查找出2个U6基因(TrU6-1和TrU6-2),并用这2个U6基因的启动子转录sgRNA的表达,均能成功对靶标基因ura5进行NHEJ修复的编辑,表明里氏木霉体内转录的sgRNA也能够成功介导Cas9蛋白对双链DNA进行切割[65].近期,周志华研究员团队再次升级了里氏木霉的CRISPR/Cas9技术系统[66].他们通过添加Triton X-100、延长原生质体与RNPs的孵育时间来优化RNPs技术,提高了RNPs的转化效率,显著增加了里氏木霉和蛹虫草(Cordyceps militaris)的基因编辑效率,其中单基因Trura5和Cmura5的编辑效率高达100%.通过使用优化后的RNP技术,他们仅用20 bp的同源臂就实现了Trlae1的基因敲除,同源重组效率增加到56.52%.该团队通过加入添加肌醇或苯菌灵来抑制有丝分裂和细胞分裂,进一步优化了RNPs技术,显著提高了里氏木霉和米曲霉进行单核原生质体转化的效率,从而提高了获得纯合转化子的效率,进而消除了转化后期的多轮单孢分离过程.优化的RNPs技术将有助于在丝状真菌中建立更有效的不依赖转基因的基因组编辑技术,从而广泛应用于食品和医药行业. ...
... Applycations of CRISPR-Cas systems in industrial filamentous fungi
Tab. 1| 丝状真菌 | Cas 类型 | Cas启动子 | sgRNA启动子 | 靶标基因 | 编辑效率 | 参考文献 |
|---|
| 黑曲霉 | Cas9 | P tef1 | P gpdA | albA | 高效 | [52] |
| P tef1 | Af_U6-1p | albA | 高效 | [53] |
| P tef1 | PanU6 | albA | 79% | [54] |
| P gpdA | 5S rRNA | alba, fum5, fum1 | 高达100% | [55] |
| P tef1 | AoU6, AfU6 | fwnA, amyA, glaA | 单基因高达80% | [56-58] |
| P tef1, P gpdA, P glaA | Anp | pyrG, moc, laeA | 高达97.2% | [59] |
| LbCpf1 | P tef1 | Af_U3p | albA | 80% | [60] |
| 米曲霉 | Cas9 | P amyB | 米曲霉U6p | wA, pyrG, yA | 10%~100% | [61] |
| P Aotef1, AMA1-自主复制质粒 | PU6p,AMA1-自主复制质粒 | wA, pyrG, yA | 55.6%~100% | [62] |
| 里氏木霉 | Cas9 | P pdc, P cbh1 | 体外转录合成 | ura5, lae1, vib1, clr2 | 单基因高达93%~100%;多基因同时编辑效率4.2%~45% | [63] |
| 体外转录合成 | 体外转录合成 | cbh-1 | 40% | [64] |
| P pdc | 里氏木霉PU61, PU62 | ura5 | 高效 | [65] |
| 体外转录合成 | 体外转录合成 | Trura5,Trlae1,Trcbh1,Trcbh2, Treg1 | 56.52%~100% | [66] |
| 产黄青霉 | Cas9 | P xyl 或体外 转录合成 | U6, 145 tRNA, Utp25或体外转录合成 | pks17, roqA, lovF,pcbAB, penDE, hcpA | 高达60%~100% | [67, 68] |
| 嗜热毁丝霉 | Cas9 | P tef1 | 嗜热毁丝霉MtU6p | amd S, cre1, res1, alp1, gh1-1, rca1, hcr1, ap3, prk6 | 单基因高达95%;双、三、四基因同时编辑效率22%~70% | [75] |
| SpCas9, FnCpf1, AsCpf1 | P tef1 或体外 转录合成 | U6p或体外 转录合成 | pks4.2, alp1, snc1, ptf1 | 单基因高达100%;多基因同时编辑时单基因效率5%~100% | [76] |
| AsCas12a | P tef1 | MtU6p | cre1, res1, alp1, gh1-1, rca1,neo, bar, hcr1, ap3, prk6 | 单基因高达90%;三、四基因同时编辑效率22%~41% | [77] |
| 棉阿舒囊霉 | SpCas9 | 酿酒酵母P tef1 | P snr52 | ade2, A754, fmp27 | 36%~80% | [78] |
| LbCpf1 | P tsa1 | P snr52 | his3, ade2, trp1, leu2, ura3 | 多基因编辑效率10.4%~77.2% | [79] |
3.3 CRISPR-Cas技术在其他丝状真菌中的应用3.3.1 稻瘟病菌2015年,日本明治大学Shigeru Kuwata教授团队[80]采用共转化载体Cas9表达框和sgRNA表达元件的策略成功在稻瘟病菌(M. oryzae)进行CRISPR-Cas9系统的构建.该团队同时使用稻瘟病菌自身的启动子U6p和构巢曲霉的启动子P trpC 分别转录表达sgRNA,对其编辑效率进行比较评价,成功对基因组中的SDH和SRS2基因进行敲除.2018年,英国埃克塞特大学Nicholas J. Talbot教授团队构建了CRISPR-Cas9 RNPs介导的稻瘟病菌基因组编辑技术体系[81],在不添加模板DNA时,以NHEJ的修复方式对靶标基因alb1进行编辑,并且不引入标记基因,建立了CRISPR-Cas9-RNP介导的无标记基因编辑系统;以HR修复方式时,仅用80 bp单链核苷酸为模板与RNPs共转化就实现了碱基的替换,或者80 bp双链DNA与RNPs共转化就能高效进行双基因同时编辑,并基于带有的苯菌灵抗性的突变菌株(E198A)对乙霉威(diethofencarb)的敏感性,设计了反筛体系,成功去除了标记基因,从而构建了CRISPR-Cas9-RNP介导的无标记基因的编辑系统.2019年,Shigeru Kuwata团队又基于单交叉介导的同源重组修复,改造升级了稻瘟病菌CRISPR-Cas9基因编辑系统,仅使用100 bp同源臂就实现了碱基的替换和GFP基因的敲入[82]. ...
A simple approach to mediate genome editing in the filamentous fungus Trichoderma reesei by CRISPR/Cas9-coupled in vivo gRNA transcription
2
2020
... 2015年,中国科学院上海生命科学研究院周志华研究员团队率先在工业纤维素酶生产真菌里氏木霉(T. reesei)中开展了CRISPR/Cas9系统的研发[63],这是工业丝状真菌中CRISPR-Cas9技术最早的报道.他们成功将密码子优化的Cas9基因分别放置于持续性强启动子P pdc 和诱导型cbh1启动子下进行组成型和诱导型表达,利用体外转录sgRNA,成功对野生型菌株Qm6a和工业菌株Rut-C30中的靶标基因ura5进行编辑,获得NHEJ修复的突变菌株.同时,基于同源重组,他们研究了donor DNA的同源臂长度对靶标基因敲除效率的影响,结果表明donor DNA的同源臂仅为0.2 kb就能成功进行基因敲除,同源重组效率高达93%.随后,他们通过优化gRNA和donor DNA分子摩尔配比,成功对目标基因lae1、vib1和clr2进行单基因敲除、双基因和三基因的同时敲除,获得三基因突变菌株.这是首次在丝状真菌中利用CRISPR/Cas9技术对多个基因同时进行编辑的报道,表明CRISPR/Cas9系统能显著提高工业真菌的遗传编辑效率,促进工业菌株的功能基因和代谢工程改造的研究.2019年,中国农业科学院苏小运团队对里氏木霉CRISPR/Cas9系统进行进一步研究[64],他们在体外组装Cas9/gRNA核糖核蛋白复合体(ribonucleoproteins,RNPs),并将核糖核蛋白复合体和携带pyr4抗性标记的载体直接转化受体菌株,成功对靶基因cbh-1进行敲除,表明体外转录的Cas9/gRNA复合体能够对里氏木霉基因组序列进行切割编辑.2020年,华东理工大学魏东芝团队 基于嗜热毁丝霉U6 snRNA的基因序列在里氏木霉基因组中进行生物信息学比对,查找出2个U6基因(TrU6-1和TrU6-2),并用这2个U6基因的启动子转录sgRNA的表达,均能成功对靶标基因ura5进行NHEJ修复的编辑,表明里氏木霉体内转录的sgRNA也能够成功介导Cas9蛋白对双链DNA进行切割[65].近期,周志华研究员团队再次升级了里氏木霉的CRISPR/Cas9技术系统[66].他们通过添加Triton X-100、延长原生质体与RNPs的孵育时间来优化RNPs技术,提高了RNPs的转化效率,显著增加了里氏木霉和蛹虫草(Cordyceps militaris)的基因编辑效率,其中单基因Trura5和Cmura5的编辑效率高达100%.通过使用优化后的RNP技术,他们仅用20 bp的同源臂就实现了Trlae1的基因敲除,同源重组效率增加到56.52%.该团队通过加入添加肌醇或苯菌灵来抑制有丝分裂和细胞分裂,进一步优化了RNPs技术,显著提高了里氏木霉和米曲霉进行单核原生质体转化的效率,从而提高了获得纯合转化子的效率,进而消除了转化后期的多轮单孢分离过程.优化的RNPs技术将有助于在丝状真菌中建立更有效的不依赖转基因的基因组编辑技术,从而广泛应用于食品和医药行业. ...
... Applycations of CRISPR-Cas systems in industrial filamentous fungi
Tab. 1| 丝状真菌 | Cas 类型 | Cas启动子 | sgRNA启动子 | 靶标基因 | 编辑效率 | 参考文献 |
|---|
| 黑曲霉 | Cas9 | P tef1 | P gpdA | albA | 高效 | [52] |
| P tef1 | Af_U6-1p | albA | 高效 | [53] |
| P tef1 | PanU6 | albA | 79% | [54] |
| P gpdA | 5S rRNA | alba, fum5, fum1 | 高达100% | [55] |
| P tef1 | AoU6, AfU6 | fwnA, amyA, glaA | 单基因高达80% | [56-58] |
| P tef1, P gpdA, P glaA | Anp | pyrG, moc, laeA | 高达97.2% | [59] |
| LbCpf1 | P tef1 | Af_U3p | albA | 80% | [60] |
| 米曲霉 | Cas9 | P amyB | 米曲霉U6p | wA, pyrG, yA | 10%~100% | [61] |
| P Aotef1, AMA1-自主复制质粒 | PU6p,AMA1-自主复制质粒 | wA, pyrG, yA | 55.6%~100% | [62] |
| 里氏木霉 | Cas9 | P pdc, P cbh1 | 体外转录合成 | ura5, lae1, vib1, clr2 | 单基因高达93%~100%;多基因同时编辑效率4.2%~45% | [63] |
| 体外转录合成 | 体外转录合成 | cbh-1 | 40% | [64] |
| P pdc | 里氏木霉PU61, PU62 | ura5 | 高效 | [65] |
| 体外转录合成 | 体外转录合成 | Trura5,Trlae1,Trcbh1,Trcbh2, Treg1 | 56.52%~100% | [66] |
| 产黄青霉 | Cas9 | P xyl 或体外 转录合成 | U6, 145 tRNA, Utp25或体外转录合成 | pks17, roqA, lovF,pcbAB, penDE, hcpA | 高达60%~100% | [67, 68] |
| 嗜热毁丝霉 | Cas9 | P tef1 | 嗜热毁丝霉MtU6p | amd S, cre1, res1, alp1, gh1-1, rca1, hcr1, ap3, prk6 | 单基因高达95%;双、三、四基因同时编辑效率22%~70% | [75] |
| SpCas9, FnCpf1, AsCpf1 | P tef1 或体外 转录合成 | U6p或体外 转录合成 | pks4.2, alp1, snc1, ptf1 | 单基因高达100%;多基因同时编辑时单基因效率5%~100% | [76] |
| AsCas12a | P tef1 | MtU6p | cre1, res1, alp1, gh1-1, rca1,neo, bar, hcr1, ap3, prk6 | 单基因高达90%;三、四基因同时编辑效率22%~41% | [77] |
| 棉阿舒囊霉 | SpCas9 | 酿酒酵母P tef1 | P snr52 | ade2, A754, fmp27 | 36%~80% | [78] |
| LbCpf1 | P tsa1 | P snr52 | his3, ade2, trp1, leu2, ura3 | 多基因编辑效率10.4%~77.2% | [79] |
3.3 CRISPR-Cas技术在其他丝状真菌中的应用3.3.1 稻瘟病菌2015年,日本明治大学Shigeru Kuwata教授团队[80]采用共转化载体Cas9表达框和sgRNA表达元件的策略成功在稻瘟病菌(M. oryzae)进行CRISPR-Cas9系统的构建.该团队同时使用稻瘟病菌自身的启动子U6p和构巢曲霉的启动子P trpC 分别转录表达sgRNA,对其编辑效率进行比较评价,成功对基因组中的SDH和SRS2基因进行敲除.2018年,英国埃克塞特大学Nicholas J. Talbot教授团队构建了CRISPR-Cas9 RNPs介导的稻瘟病菌基因组编辑技术体系[81],在不添加模板DNA时,以NHEJ的修复方式对靶标基因alb1进行编辑,并且不引入标记基因,建立了CRISPR-Cas9-RNP介导的无标记基因编辑系统;以HR修复方式时,仅用80 bp单链核苷酸为模板与RNPs共转化就实现了碱基的替换,或者80 bp双链DNA与RNPs共转化就能高效进行双基因同时编辑,并基于带有的苯菌灵抗性的突变菌株(E198A)对乙霉威(diethofencarb)的敏感性,设计了反筛体系,成功去除了标记基因,从而构建了CRISPR-Cas9-RNP介导的无标记基因的编辑系统.2019年,Shigeru Kuwata团队又基于单交叉介导的同源重组修复,改造升级了稻瘟病菌CRISPR-Cas9基因编辑系统,仅使用100 bp同源臂就实现了碱基的替换和GFP基因的敲入[82]. ...
Efficient genome editing in filamentous fungi via an improved CRISPR-Cas9 ribonucleoprotein method facilitated by chemical reagents
2
2020
... 2015年,中国科学院上海生命科学研究院周志华研究员团队率先在工业纤维素酶生产真菌里氏木霉(T. reesei)中开展了CRISPR/Cas9系统的研发[63],这是工业丝状真菌中CRISPR-Cas9技术最早的报道.他们成功将密码子优化的Cas9基因分别放置于持续性强启动子P pdc 和诱导型cbh1启动子下进行组成型和诱导型表达,利用体外转录sgRNA,成功对野生型菌株Qm6a和工业菌株Rut-C30中的靶标基因ura5进行编辑,获得NHEJ修复的突变菌株.同时,基于同源重组,他们研究了donor DNA的同源臂长度对靶标基因敲除效率的影响,结果表明donor DNA的同源臂仅为0.2 kb就能成功进行基因敲除,同源重组效率高达93%.随后,他们通过优化gRNA和donor DNA分子摩尔配比,成功对目标基因lae1、vib1和clr2进行单基因敲除、双基因和三基因的同时敲除,获得三基因突变菌株.这是首次在丝状真菌中利用CRISPR/Cas9技术对多个基因同时进行编辑的报道,表明CRISPR/Cas9系统能显著提高工业真菌的遗传编辑效率,促进工业菌株的功能基因和代谢工程改造的研究.2019年,中国农业科学院苏小运团队对里氏木霉CRISPR/Cas9系统进行进一步研究[64],他们在体外组装Cas9/gRNA核糖核蛋白复合体(ribonucleoproteins,RNPs),并将核糖核蛋白复合体和携带pyr4抗性标记的载体直接转化受体菌株,成功对靶基因cbh-1进行敲除,表明体外转录的Cas9/gRNA复合体能够对里氏木霉基因组序列进行切割编辑.2020年,华东理工大学魏东芝团队 基于嗜热毁丝霉U6 snRNA的基因序列在里氏木霉基因组中进行生物信息学比对,查找出2个U6基因(TrU6-1和TrU6-2),并用这2个U6基因的启动子转录sgRNA的表达,均能成功对靶标基因ura5进行NHEJ修复的编辑,表明里氏木霉体内转录的sgRNA也能够成功介导Cas9蛋白对双链DNA进行切割[65].近期,周志华研究员团队再次升级了里氏木霉的CRISPR/Cas9技术系统[66].他们通过添加Triton X-100、延长原生质体与RNPs的孵育时间来优化RNPs技术,提高了RNPs的转化效率,显著增加了里氏木霉和蛹虫草(Cordyceps militaris)的基因编辑效率,其中单基因Trura5和Cmura5的编辑效率高达100%.通过使用优化后的RNP技术,他们仅用20 bp的同源臂就实现了Trlae1的基因敲除,同源重组效率增加到56.52%.该团队通过加入添加肌醇或苯菌灵来抑制有丝分裂和细胞分裂,进一步优化了RNPs技术,显著提高了里氏木霉和米曲霉进行单核原生质体转化的效率,从而提高了获得纯合转化子的效率,进而消除了转化后期的多轮单孢分离过程.优化的RNPs技术将有助于在丝状真菌中建立更有效的不依赖转基因的基因组编辑技术,从而广泛应用于食品和医药行业. ...
... Applycations of CRISPR-Cas systems in industrial filamentous fungi
Tab. 1| 丝状真菌 | Cas 类型 | Cas启动子 | sgRNA启动子 | 靶标基因 | 编辑效率 | 参考文献 |
|---|
| 黑曲霉 | Cas9 | P tef1 | P gpdA | albA | 高效 | [52] |
| P tef1 | Af_U6-1p | albA | 高效 | [53] |
| P tef1 | PanU6 | albA | 79% | [54] |
| P gpdA | 5S rRNA | alba, fum5, fum1 | 高达100% | [55] |
| P tef1 | AoU6, AfU6 | fwnA, amyA, glaA | 单基因高达80% | [56-58] |
| P tef1, P gpdA, P glaA | Anp | pyrG, moc, laeA | 高达97.2% | [59] |
| LbCpf1 | P tef1 | Af_U3p | albA | 80% | [60] |
| 米曲霉 | Cas9 | P amyB | 米曲霉U6p | wA, pyrG, yA | 10%~100% | [61] |
| P Aotef1, AMA1-自主复制质粒 | PU6p,AMA1-自主复制质粒 | wA, pyrG, yA | 55.6%~100% | [62] |
| 里氏木霉 | Cas9 | P pdc, P cbh1 | 体外转录合成 | ura5, lae1, vib1, clr2 | 单基因高达93%~100%;多基因同时编辑效率4.2%~45% | [63] |
| 体外转录合成 | 体外转录合成 | cbh-1 | 40% | [64] |
| P pdc | 里氏木霉PU61, PU62 | ura5 | 高效 | [65] |
| 体外转录合成 | 体外转录合成 | Trura5,Trlae1,Trcbh1,Trcbh2, Treg1 | 56.52%~100% | [66] |
| 产黄青霉 | Cas9 | P xyl 或体外 转录合成 | U6, 145 tRNA, Utp25或体外转录合成 | pks17, roqA, lovF,pcbAB, penDE, hcpA | 高达60%~100% | [67, 68] |
| 嗜热毁丝霉 | Cas9 | P tef1 | 嗜热毁丝霉MtU6p | amd S, cre1, res1, alp1, gh1-1, rca1, hcr1, ap3, prk6 | 单基因高达95%;双、三、四基因同时编辑效率22%~70% | [75] |
| SpCas9, FnCpf1, AsCpf1 | P tef1 或体外 转录合成 | U6p或体外 转录合成 | pks4.2, alp1, snc1, ptf1 | 单基因高达100%;多基因同时编辑时单基因效率5%~100% | [76] |
| AsCas12a | P tef1 | MtU6p | cre1, res1, alp1, gh1-1, rca1,neo, bar, hcr1, ap3, prk6 | 单基因高达90%;三、四基因同时编辑效率22%~41% | [77] |
| 棉阿舒囊霉 | SpCas9 | 酿酒酵母P tef1 | P snr52 | ade2, A754, fmp27 | 36%~80% | [78] |
| LbCpf1 | P tsa1 | P snr52 | his3, ade2, trp1, leu2, ura3 | 多基因编辑效率10.4%~77.2% | [79] |
3.3 CRISPR-Cas技术在其他丝状真菌中的应用3.3.1 稻瘟病菌2015年,日本明治大学Shigeru Kuwata教授团队[80]采用共转化载体Cas9表达框和sgRNA表达元件的策略成功在稻瘟病菌(M. oryzae)进行CRISPR-Cas9系统的构建.该团队同时使用稻瘟病菌自身的启动子U6p和构巢曲霉的启动子P trpC 分别转录表达sgRNA,对其编辑效率进行比较评价,成功对基因组中的SDH和SRS2基因进行敲除.2018年,英国埃克塞特大学Nicholas J. Talbot教授团队构建了CRISPR-Cas9 RNPs介导的稻瘟病菌基因组编辑技术体系[81],在不添加模板DNA时,以NHEJ的修复方式对靶标基因alb1进行编辑,并且不引入标记基因,建立了CRISPR-Cas9-RNP介导的无标记基因编辑系统;以HR修复方式时,仅用80 bp单链核苷酸为模板与RNPs共转化就实现了碱基的替换,或者80 bp双链DNA与RNPs共转化就能高效进行双基因同时编辑,并基于带有的苯菌灵抗性的突变菌株(E198A)对乙霉威(diethofencarb)的敏感性,设计了反筛体系,成功去除了标记基因,从而构建了CRISPR-Cas9-RNP介导的无标记基因的编辑系统.2019年,Shigeru Kuwata团队又基于单交叉介导的同源重组修复,改造升级了稻瘟病菌CRISPR-Cas9基因编辑系统,仅使用100 bp同源臂就实现了碱基的替换和GFP基因的敲入[82]. ...
CRISPR/Cas9 based genome editing of Penicillium chrysogenum
2
2016
... 产黄青霉(P. chrysogenum)是青霉素的主要工业生产菌种,作为一种重要的工业丝状真菌,可产生许多重要的次级代谢产物.因此,产黄青霉CRISPR/Cas9编辑系统的构建对提升青霉素以及其他次级代谢物的生产有重要意义.2016年,荷兰格罗宁根大学Nygård教授团队在产黄青霉中构建了3种不同方式的CRISPR/Cas9基因编辑系统[67],包括:含有Cas9和sgRNA表达元件的AMA1自主复制质粒;在细胞内表达Cas9蛋白后,通过转化体外合成的sgRNA;同时在体外转录合成sgRNA和Cas9蛋白,并将Cas9蛋白与sgRNA装配成核糖体核蛋白复合体(RNPs),以核糖体核蛋白复合体的形式通过原生质体转化进入细胞中.AMA1自主复制的质粒会在无抗性压力时丢失,因此该方法能在编辑完成后对Cas9元件进行去除,实现无标记基因操作,同时结果表明供体DNA同源臂长度仅60 bp就能进行基因缺失编辑.随后,该团队详细介绍了Cas9蛋白与sgRNA组装的RNPs转化产黄青霉原生质体的操作方法[68],该报道为更多丝状真菌实现无痕基因组编辑提供了参考. ...
... Applycations of CRISPR-Cas systems in industrial filamentous fungi
Tab. 1| 丝状真菌 | Cas 类型 | Cas启动子 | sgRNA启动子 | 靶标基因 | 编辑效率 | 参考文献 |
|---|
| 黑曲霉 | Cas9 | P tef1 | P gpdA | albA | 高效 | [52] |
| P tef1 | Af_U6-1p | albA | 高效 | [53] |
| P tef1 | PanU6 | albA | 79% | [54] |
| P gpdA | 5S rRNA | alba, fum5, fum1 | 高达100% | [55] |
| P tef1 | AoU6, AfU6 | fwnA, amyA, glaA | 单基因高达80% | [56-58] |
| P tef1, P gpdA, P glaA | Anp | pyrG, moc, laeA | 高达97.2% | [59] |
| LbCpf1 | P tef1 | Af_U3p | albA | 80% | [60] |
| 米曲霉 | Cas9 | P amyB | 米曲霉U6p | wA, pyrG, yA | 10%~100% | [61] |
| P Aotef1, AMA1-自主复制质粒 | PU6p,AMA1-自主复制质粒 | wA, pyrG, yA | 55.6%~100% | [62] |
| 里氏木霉 | Cas9 | P pdc, P cbh1 | 体外转录合成 | ura5, lae1, vib1, clr2 | 单基因高达93%~100%;多基因同时编辑效率4.2%~45% | [63] |
| 体外转录合成 | 体外转录合成 | cbh-1 | 40% | [64] |
| P pdc | 里氏木霉PU61, PU62 | ura5 | 高效 | [65] |
| 体外转录合成 | 体外转录合成 | Trura5,Trlae1,Trcbh1,Trcbh2, Treg1 | 56.52%~100% | [66] |
| 产黄青霉 | Cas9 | P xyl 或体外 转录合成 | U6, 145 tRNA, Utp25或体外转录合成 | pks17, roqA, lovF,pcbAB, penDE, hcpA | 高达60%~100% | [67, 68] |
| 嗜热毁丝霉 | Cas9 | P tef1 | 嗜热毁丝霉MtU6p | amd S, cre1, res1, alp1, gh1-1, rca1, hcr1, ap3, prk6 | 单基因高达95%;双、三、四基因同时编辑效率22%~70% | [75] |
| SpCas9, FnCpf1, AsCpf1 | P tef1 或体外 转录合成 | U6p或体外 转录合成 | pks4.2, alp1, snc1, ptf1 | 单基因高达100%;多基因同时编辑时单基因效率5%~100% | [76] |
| AsCas12a | P tef1 | MtU6p | cre1, res1, alp1, gh1-1, rca1,neo, bar, hcr1, ap3, prk6 | 单基因高达90%;三、四基因同时编辑效率22%~41% | [77] |
| 棉阿舒囊霉 | SpCas9 | 酿酒酵母P tef1 | P snr52 | ade2, A754, fmp27 | 36%~80% | [78] |
| LbCpf1 | P tsa1 | P snr52 | his3, ade2, trp1, leu2, ura3 | 多基因编辑效率10.4%~77.2% | [79] |
3.3 CRISPR-Cas技术在其他丝状真菌中的应用3.3.1 稻瘟病菌2015年,日本明治大学Shigeru Kuwata教授团队[80]采用共转化载体Cas9表达框和sgRNA表达元件的策略成功在稻瘟病菌(M. oryzae)进行CRISPR-Cas9系统的构建.该团队同时使用稻瘟病菌自身的启动子U6p和构巢曲霉的启动子P trpC 分别转录表达sgRNA,对其编辑效率进行比较评价,成功对基因组中的SDH和SRS2基因进行敲除.2018年,英国埃克塞特大学Nicholas J. Talbot教授团队构建了CRISPR-Cas9 RNPs介导的稻瘟病菌基因组编辑技术体系[81],在不添加模板DNA时,以NHEJ的修复方式对靶标基因alb1进行编辑,并且不引入标记基因,建立了CRISPR-Cas9-RNP介导的无标记基因编辑系统;以HR修复方式时,仅用80 bp单链核苷酸为模板与RNPs共转化就实现了碱基的替换,或者80 bp双链DNA与RNPs共转化就能高效进行双基因同时编辑,并基于带有的苯菌灵抗性的突变菌株(E198A)对乙霉威(diethofencarb)的敏感性,设计了反筛体系,成功去除了标记基因,从而构建了CRISPR-Cas9-RNP介导的无标记基因的编辑系统.2019年,Shigeru Kuwata团队又基于单交叉介导的同源重组修复,改造升级了稻瘟病菌CRISPR-Cas9基因编辑系统,仅使用100 bp同源臂就实现了碱基的替换和GFP基因的敲入[82]. ...
Genome editing in Penicillium chrysogenum using Cas9 ribonucleoprotein particles
2
2018
... 产黄青霉(P. chrysogenum)是青霉素的主要工业生产菌种,作为一种重要的工业丝状真菌,可产生许多重要的次级代谢产物.因此,产黄青霉CRISPR/Cas9编辑系统的构建对提升青霉素以及其他次级代谢物的生产有重要意义.2016年,荷兰格罗宁根大学Nygård教授团队在产黄青霉中构建了3种不同方式的CRISPR/Cas9基因编辑系统[67],包括:含有Cas9和sgRNA表达元件的AMA1自主复制质粒;在细胞内表达Cas9蛋白后,通过转化体外合成的sgRNA;同时在体外转录合成sgRNA和Cas9蛋白,并将Cas9蛋白与sgRNA装配成核糖体核蛋白复合体(RNPs),以核糖体核蛋白复合体的形式通过原生质体转化进入细胞中.AMA1自主复制的质粒会在无抗性压力时丢失,因此该方法能在编辑完成后对Cas9元件进行去除,实现无标记基因操作,同时结果表明供体DNA同源臂长度仅60 bp就能进行基因缺失编辑.随后,该团队详细介绍了Cas9蛋白与sgRNA组装的RNPs转化产黄青霉原生质体的操作方法[68],该报道为更多丝状真菌实现无痕基因组编辑提供了参考. ...
... Applycations of CRISPR-Cas systems in industrial filamentous fungi
Tab. 1| 丝状真菌 | Cas 类型 | Cas启动子 | sgRNA启动子 | 靶标基因 | 编辑效率 | 参考文献 |
|---|
| 黑曲霉 | Cas9 | P tef1 | P gpdA | albA | 高效 | [52] |
| P tef1 | Af_U6-1p | albA | 高效 | [53] |
| P tef1 | PanU6 | albA | 79% | [54] |
| P gpdA | 5S rRNA | alba, fum5, fum1 | 高达100% | [55] |
| P tef1 | AoU6, AfU6 | fwnA, amyA, glaA | 单基因高达80% | [56-58] |
| P tef1, P gpdA, P glaA | Anp | pyrG, moc, laeA | 高达97.2% | [59] |
| LbCpf1 | P tef1 | Af_U3p | albA | 80% | [60] |
| 米曲霉 | Cas9 | P amyB | 米曲霉U6p | wA, pyrG, yA | 10%~100% | [61] |
| P Aotef1, AMA1-自主复制质粒 | PU6p,AMA1-自主复制质粒 | wA, pyrG, yA | 55.6%~100% | [62] |
| 里氏木霉 | Cas9 | P pdc, P cbh1 | 体外转录合成 | ura5, lae1, vib1, clr2 | 单基因高达93%~100%;多基因同时编辑效率4.2%~45% | [63] |
| 体外转录合成 | 体外转录合成 | cbh-1 | 40% | [64] |
| P pdc | 里氏木霉PU61, PU62 | ura5 | 高效 | [65] |
| 体外转录合成 | 体外转录合成 | Trura5,Trlae1,Trcbh1,Trcbh2, Treg1 | 56.52%~100% | [66] |
| 产黄青霉 | Cas9 | P xyl 或体外 转录合成 | U6, 145 tRNA, Utp25或体外转录合成 | pks17, roqA, lovF,pcbAB, penDE, hcpA | 高达60%~100% | [67, 68] |
| 嗜热毁丝霉 | Cas9 | P tef1 | 嗜热毁丝霉MtU6p | amd S, cre1, res1, alp1, gh1-1, rca1, hcr1, ap3, prk6 | 单基因高达95%;双、三、四基因同时编辑效率22%~70% | [75] |
| SpCas9, FnCpf1, AsCpf1 | P tef1 或体外 转录合成 | U6p或体外 转录合成 | pks4.2, alp1, snc1, ptf1 | 单基因高达100%;多基因同时编辑时单基因效率5%~100% | [76] |
| AsCas12a | P tef1 | MtU6p | cre1, res1, alp1, gh1-1, rca1,neo, bar, hcr1, ap3, prk6 | 单基因高达90%;三、四基因同时编辑效率22%~41% | [77] |
| 棉阿舒囊霉 | SpCas9 | 酿酒酵母P tef1 | P snr52 | ade2, A754, fmp27 | 36%~80% | [78] |
| LbCpf1 | P tsa1 | P snr52 | his3, ade2, trp1, leu2, ura3 | 多基因编辑效率10.4%~77.2% | [79] |
3.3 CRISPR-Cas技术在其他丝状真菌中的应用3.3.1 稻瘟病菌2015年,日本明治大学Shigeru Kuwata教授团队[80]采用共转化载体Cas9表达框和sgRNA表达元件的策略成功在稻瘟病菌(M. oryzae)进行CRISPR-Cas9系统的构建.该团队同时使用稻瘟病菌自身的启动子U6p和构巢曲霉的启动子P trpC 分别转录表达sgRNA,对其编辑效率进行比较评价,成功对基因组中的SDH和SRS2基因进行敲除.2018年,英国埃克塞特大学Nicholas J. Talbot教授团队构建了CRISPR-Cas9 RNPs介导的稻瘟病菌基因组编辑技术体系[81],在不添加模板DNA时,以NHEJ的修复方式对靶标基因alb1进行编辑,并且不引入标记基因,建立了CRISPR-Cas9-RNP介导的无标记基因编辑系统;以HR修复方式时,仅用80 bp单链核苷酸为模板与RNPs共转化就实现了碱基的替换,或者80 bp双链DNA与RNPs共转化就能高效进行双基因同时编辑,并基于带有的苯菌灵抗性的突变菌株(E198A)对乙霉威(diethofencarb)的敏感性,设计了反筛体系,成功去除了标记基因,从而构建了CRISPR-Cas9-RNP介导的无标记基因的编辑系统.2019年,Shigeru Kuwata团队又基于单交叉介导的同源重组修复,改造升级了稻瘟病菌CRISPR-Cas9基因编辑系统,仅使用100 bp同源臂就实现了碱基的替换和GFP基因的敲入[82]. ...
Development of a mature fungal technology and production platform for industrial enzymes based on a Myceliophthora thermophila isolate, previously known as Chrysosporium lucknowense C1
1
2011
... 嗜热毁丝霉(M. thermophila)是一种能够快速降解纤维素的嗜热丝状真菌,能够分泌大量的纤维素水解酶,其酶系的种类和数量相当丰富.嗜热毁丝霉具有高温发酵的优势,其所产酶系在高温下有高活力和高稳定性的优点[69].随着2011年嗜热毁丝霉基因组测序的完成和发布[70],极大促进了国内外学者对其的研究和开发,其中多个酶的开发已经取得了显著进展[71].但在2015年之前,嗜热毁丝霉本身的遗传改造研究非常少. ...
Comparative genomic analysis of the thermophilic biomass-degrading fungi Myceliophthora thermophila and Thielavia terrestris
1
2011
... 嗜热毁丝霉(M. thermophila)是一种能够快速降解纤维素的嗜热丝状真菌,能够分泌大量的纤维素水解酶,其酶系的种类和数量相当丰富.嗜热毁丝霉具有高温发酵的优势,其所产酶系在高温下有高活力和高稳定性的优点[69].随着2011年嗜热毁丝霉基因组测序的完成和发布[70],极大促进了国内外学者对其的研究和开发,其中多个酶的开发已经取得了显著进展[71].但在2015年之前,嗜热毁丝霉本身的遗传改造研究非常少. ...
Transcriptome and exoproteome analysis of utilization of plant-derived biomass by Myceliophthora thermophila
1
2014
... 嗜热毁丝霉(M. thermophila)是一种能够快速降解纤维素的嗜热丝状真菌,能够分泌大量的纤维素水解酶,其酶系的种类和数量相当丰富.嗜热毁丝霉具有高温发酵的优势,其所产酶系在高温下有高活力和高稳定性的优点[69].随着2011年嗜热毁丝霉基因组测序的完成和发布[70],极大促进了国内外学者对其的研究和开发,其中多个酶的开发已经取得了显著进展[71].但在2015年之前,嗜热毁丝霉本身的遗传改造研究非常少. ...
Enhancing xylanase production in the thermophilic fungus Myceliophthora thermophila by homologous overexpression of Mtxyr1
1
2015
... 2015年,深圳大学刘刚教授团队在嗜热毁丝霉中发展了过表达[72]和RNA干扰技术[73],成功过表达了转录因子Mtxyr1,将靶标基因Mtcre1进行了沉默,从而提高了纤维素酶和半纤维素酶的产量.与此同时,本文作者所在团队对嗜热毁丝霉的转化系统进行优化[74],利用农杆菌介导的转化技术,结合GFP荧光辅助筛选,能有效对目标基因实施基因过表达和敲除操作,这为后续开展嗜热毁丝霉基因组编辑技术奠定了坚实的基础.2017年,本文作者团队成功构建了CRISPR/Cas9介导的嗜热真菌基因组编辑系统[75],成功对两种嗜热真菌-嗜热毁丝霉和异宗毁丝霉(M. heterothallica)的基因组进行了编辑.该系统包括组成型启动子P tef1 调控Cas9的表达元件和嗜热毁丝霉U6p启动子转录sgRNA的表达元件,通过将Cas9表达框、sgRNA表达框和同源供体共转化导入到宿主细胞中,获得了稳定的遗传性状的基因编辑突变株.毁丝霉的基因组编辑时间缩短到3 d,其基因的同源重组效率显著提高,单基因缺失的同源重组效率高达100%,而且可以同时编辑多个基因位点,其中双基因同时编辑的同源重组效率为61%~69%,3个靶基因同时被敲除的HR效率约为30%,4个靶基因同时被缺失的HR效率仍然约为21%.以纤维素酶分泌途径重要靶点为例,利用该体系对嗜热真菌基因组成功开展了理性重构,并获得一系列蛋白分泌显著提高的工程菌.随后,柏林工业大学Vera Meyer教授团队构建了基于RNPs和质粒体系的嗜热毁丝霉CRISPR-FnCpf1/AsCpf1/SpCas9编辑系统[76],并在Ku80的缺失菌株中进行遗传操作,比较这3种编辑体系的效率,结果表明FnCpf1系统获得了四基因(alp1、pks4.2、snc1和ptf1)突变株,AsCpf1/SpCas9系统获得了三基因突变株,90 bp单链寡核苷酸作为供体DNA实现双基因的编辑.该团队还将转化体积按比例缩小至微量模式,该模式能对菌株进行有效的编辑,有助于将来构建嗜热毁丝霉高通量筛选平台.与此同时,2019年笔者团队成功升级了嗜热真菌CRISPR-Cas基因组编辑系统[77],构建了crRNA串联表达介导的嗜热毁丝霉CRISPR-AsCas12a基因组编辑系统,该系统能够高效、简易实现基因组多基因的编辑,拓展了嗜热真菌CRISPR-Cas基因组编辑系统,使其不再局限于Cas9.基于Cas12a和Cas9系统,团队建立了标记基因可循环利用的基因组编辑系统(CRISPR-Cas-assisted marker recycling technology),将其命名为Camr技术,该系统成功解决了目前丝状真菌多基因(多于10个)的编辑困难,以及基因组被编辑后的工程菌由于缺乏合适的抗性基因无法进行多次编辑的问题,可以对丝状真菌基因组进行快速、高效、多轮的可循环操作编辑.利用该体系以嗜热毁丝霉纤维素酶分泌途径中9个关键调控基因为靶点,通过交替使用2个标记基因进行三轮操作,共编辑了11个位点,人工重构了工业丝状真菌纤维素酶合成分泌途径,并获得了纤维素酶产量显著提高的9M突变体菌株.该研究对其他真菌的基因组编辑和代谢工程改造有一定参考意义. ...
Enhancing cellulase production in thermophilic fungus Myceliophthora thermophila ATCC42464 by RNA interference of cre1 gene expression
1
2015
... 2015年,深圳大学刘刚教授团队在嗜热毁丝霉中发展了过表达[72]和RNA干扰技术[73],成功过表达了转录因子Mtxyr1,将靶标基因Mtcre1进行了沉默,从而提高了纤维素酶和半纤维素酶的产量.与此同时,本文作者所在团队对嗜热毁丝霉的转化系统进行优化[74],利用农杆菌介导的转化技术,结合GFP荧光辅助筛选,能有效对目标基因实施基因过表达和敲除操作,这为后续开展嗜热毁丝霉基因组编辑技术奠定了坚实的基础.2017年,本文作者团队成功构建了CRISPR/Cas9介导的嗜热真菌基因组编辑系统[75],成功对两种嗜热真菌-嗜热毁丝霉和异宗毁丝霉(M. heterothallica)的基因组进行了编辑.该系统包括组成型启动子P tef1 调控Cas9的表达元件和嗜热毁丝霉U6p启动子转录sgRNA的表达元件,通过将Cas9表达框、sgRNA表达框和同源供体共转化导入到宿主细胞中,获得了稳定的遗传性状的基因编辑突变株.毁丝霉的基因组编辑时间缩短到3 d,其基因的同源重组效率显著提高,单基因缺失的同源重组效率高达100%,而且可以同时编辑多个基因位点,其中双基因同时编辑的同源重组效率为61%~69%,3个靶基因同时被敲除的HR效率约为30%,4个靶基因同时被缺失的HR效率仍然约为21%.以纤维素酶分泌途径重要靶点为例,利用该体系对嗜热真菌基因组成功开展了理性重构,并获得一系列蛋白分泌显著提高的工程菌.随后,柏林工业大学Vera Meyer教授团队构建了基于RNPs和质粒体系的嗜热毁丝霉CRISPR-FnCpf1/AsCpf1/SpCas9编辑系统[76],并在Ku80的缺失菌株中进行遗传操作,比较这3种编辑体系的效率,结果表明FnCpf1系统获得了四基因(alp1、pks4.2、snc1和ptf1)突变株,AsCpf1/SpCas9系统获得了三基因突变株,90 bp单链寡核苷酸作为供体DNA实现双基因的编辑.该团队还将转化体积按比例缩小至微量模式,该模式能对菌株进行有效的编辑,有助于将来构建嗜热毁丝霉高通量筛选平台.与此同时,2019年笔者团队成功升级了嗜热真菌CRISPR-Cas基因组编辑系统[77],构建了crRNA串联表达介导的嗜热毁丝霉CRISPR-AsCas12a基因组编辑系统,该系统能够高效、简易实现基因组多基因的编辑,拓展了嗜热真菌CRISPR-Cas基因组编辑系统,使其不再局限于Cas9.基于Cas12a和Cas9系统,团队建立了标记基因可循环利用的基因组编辑系统(CRISPR-Cas-assisted marker recycling technology),将其命名为Camr技术,该系统成功解决了目前丝状真菌多基因(多于10个)的编辑困难,以及基因组被编辑后的工程菌由于缺乏合适的抗性基因无法进行多次编辑的问题,可以对丝状真菌基因组进行快速、高效、多轮的可循环操作编辑.利用该体系以嗜热毁丝霉纤维素酶分泌途径中9个关键调控基因为靶点,通过交替使用2个标记基因进行三轮操作,共编辑了11个位点,人工重构了工业丝状真菌纤维素酶合成分泌途径,并获得了纤维素酶产量显著提高的9M突变体菌株.该研究对其他真菌的基因组编辑和代谢工程改造有一定参考意义. ...
Development of genetic tools for Myceliophthora thermophila
1
2015
... 2015年,深圳大学刘刚教授团队在嗜热毁丝霉中发展了过表达[72]和RNA干扰技术[73],成功过表达了转录因子Mtxyr1,将靶标基因Mtcre1进行了沉默,从而提高了纤维素酶和半纤维素酶的产量.与此同时,本文作者所在团队对嗜热毁丝霉的转化系统进行优化[74],利用农杆菌介导的转化技术,结合GFP荧光辅助筛选,能有效对目标基因实施基因过表达和敲除操作,这为后续开展嗜热毁丝霉基因组编辑技术奠定了坚实的基础.2017年,本文作者团队成功构建了CRISPR/Cas9介导的嗜热真菌基因组编辑系统[75],成功对两种嗜热真菌-嗜热毁丝霉和异宗毁丝霉(M. heterothallica)的基因组进行了编辑.该系统包括组成型启动子P tef1 调控Cas9的表达元件和嗜热毁丝霉U6p启动子转录sgRNA的表达元件,通过将Cas9表达框、sgRNA表达框和同源供体共转化导入到宿主细胞中,获得了稳定的遗传性状的基因编辑突变株.毁丝霉的基因组编辑时间缩短到3 d,其基因的同源重组效率显著提高,单基因缺失的同源重组效率高达100%,而且可以同时编辑多个基因位点,其中双基因同时编辑的同源重组效率为61%~69%,3个靶基因同时被敲除的HR效率约为30%,4个靶基因同时被缺失的HR效率仍然约为21%.以纤维素酶分泌途径重要靶点为例,利用该体系对嗜热真菌基因组成功开展了理性重构,并获得一系列蛋白分泌显著提高的工程菌.随后,柏林工业大学Vera Meyer教授团队构建了基于RNPs和质粒体系的嗜热毁丝霉CRISPR-FnCpf1/AsCpf1/SpCas9编辑系统[76],并在Ku80的缺失菌株中进行遗传操作,比较这3种编辑体系的效率,结果表明FnCpf1系统获得了四基因(alp1、pks4.2、snc1和ptf1)突变株,AsCpf1/SpCas9系统获得了三基因突变株,90 bp单链寡核苷酸作为供体DNA实现双基因的编辑.该团队还将转化体积按比例缩小至微量模式,该模式能对菌株进行有效的编辑,有助于将来构建嗜热毁丝霉高通量筛选平台.与此同时,2019年笔者团队成功升级了嗜热真菌CRISPR-Cas基因组编辑系统[77],构建了crRNA串联表达介导的嗜热毁丝霉CRISPR-AsCas12a基因组编辑系统,该系统能够高效、简易实现基因组多基因的编辑,拓展了嗜热真菌CRISPR-Cas基因组编辑系统,使其不再局限于Cas9.基于Cas12a和Cas9系统,团队建立了标记基因可循环利用的基因组编辑系统(CRISPR-Cas-assisted marker recycling technology),将其命名为Camr技术,该系统成功解决了目前丝状真菌多基因(多于10个)的编辑困难,以及基因组被编辑后的工程菌由于缺乏合适的抗性基因无法进行多次编辑的问题,可以对丝状真菌基因组进行快速、高效、多轮的可循环操作编辑.利用该体系以嗜热毁丝霉纤维素酶分泌途径中9个关键调控基因为靶点,通过交替使用2个标记基因进行三轮操作,共编辑了11个位点,人工重构了工业丝状真菌纤维素酶合成分泌途径,并获得了纤维素酶产量显著提高的9M突变体菌株.该研究对其他真菌的基因组编辑和代谢工程改造有一定参考意义. ...
Development of a genome-editing CRISPR/Cas9 system in thermophilic fungal Myceliophthora species and its application to hyper-cellulase production strain engineering
2
2017
... 2015年,深圳大学刘刚教授团队在嗜热毁丝霉中发展了过表达[72]和RNA干扰技术[73],成功过表达了转录因子Mtxyr1,将靶标基因Mtcre1进行了沉默,从而提高了纤维素酶和半纤维素酶的产量.与此同时,本文作者所在团队对嗜热毁丝霉的转化系统进行优化[74],利用农杆菌介导的转化技术,结合GFP荧光辅助筛选,能有效对目标基因实施基因过表达和敲除操作,这为后续开展嗜热毁丝霉基因组编辑技术奠定了坚实的基础.2017年,本文作者团队成功构建了CRISPR/Cas9介导的嗜热真菌基因组编辑系统[75],成功对两种嗜热真菌-嗜热毁丝霉和异宗毁丝霉(M. heterothallica)的基因组进行了编辑.该系统包括组成型启动子P tef1 调控Cas9的表达元件和嗜热毁丝霉U6p启动子转录sgRNA的表达元件,通过将Cas9表达框、sgRNA表达框和同源供体共转化导入到宿主细胞中,获得了稳定的遗传性状的基因编辑突变株.毁丝霉的基因组编辑时间缩短到3 d,其基因的同源重组效率显著提高,单基因缺失的同源重组效率高达100%,而且可以同时编辑多个基因位点,其中双基因同时编辑的同源重组效率为61%~69%,3个靶基因同时被敲除的HR效率约为30%,4个靶基因同时被缺失的HR效率仍然约为21%.以纤维素酶分泌途径重要靶点为例,利用该体系对嗜热真菌基因组成功开展了理性重构,并获得一系列蛋白分泌显著提高的工程菌.随后,柏林工业大学Vera Meyer教授团队构建了基于RNPs和质粒体系的嗜热毁丝霉CRISPR-FnCpf1/AsCpf1/SpCas9编辑系统[76],并在Ku80的缺失菌株中进行遗传操作,比较这3种编辑体系的效率,结果表明FnCpf1系统获得了四基因(alp1、pks4.2、snc1和ptf1)突变株,AsCpf1/SpCas9系统获得了三基因突变株,90 bp单链寡核苷酸作为供体DNA实现双基因的编辑.该团队还将转化体积按比例缩小至微量模式,该模式能对菌株进行有效的编辑,有助于将来构建嗜热毁丝霉高通量筛选平台.与此同时,2019年笔者团队成功升级了嗜热真菌CRISPR-Cas基因组编辑系统[77],构建了crRNA串联表达介导的嗜热毁丝霉CRISPR-AsCas12a基因组编辑系统,该系统能够高效、简易实现基因组多基因的编辑,拓展了嗜热真菌CRISPR-Cas基因组编辑系统,使其不再局限于Cas9.基于Cas12a和Cas9系统,团队建立了标记基因可循环利用的基因组编辑系统(CRISPR-Cas-assisted marker recycling technology),将其命名为Camr技术,该系统成功解决了目前丝状真菌多基因(多于10个)的编辑困难,以及基因组被编辑后的工程菌由于缺乏合适的抗性基因无法进行多次编辑的问题,可以对丝状真菌基因组进行快速、高效、多轮的可循环操作编辑.利用该体系以嗜热毁丝霉纤维素酶分泌途径中9个关键调控基因为靶点,通过交替使用2个标记基因进行三轮操作,共编辑了11个位点,人工重构了工业丝状真菌纤维素酶合成分泌途径,并获得了纤维素酶产量显著提高的9M突变体菌株.该研究对其他真菌的基因组编辑和代谢工程改造有一定参考意义. ...
... Applycations of CRISPR-Cas systems in industrial filamentous fungi
Tab. 1| 丝状真菌 | Cas 类型 | Cas启动子 | sgRNA启动子 | 靶标基因 | 编辑效率 | 参考文献 |
|---|
| 黑曲霉 | Cas9 | P tef1 | P gpdA | albA | 高效 | [52] |
| P tef1 | Af_U6-1p | albA | 高效 | [53] |
| P tef1 | PanU6 | albA | 79% | [54] |
| P gpdA | 5S rRNA | alba, fum5, fum1 | 高达100% | [55] |
| P tef1 | AoU6, AfU6 | fwnA, amyA, glaA | 单基因高达80% | [56-58] |
| P tef1, P gpdA, P glaA | Anp | pyrG, moc, laeA | 高达97.2% | [59] |
| LbCpf1 | P tef1 | Af_U3p | albA | 80% | [60] |
| 米曲霉 | Cas9 | P amyB | 米曲霉U6p | wA, pyrG, yA | 10%~100% | [61] |
| P Aotef1, AMA1-自主复制质粒 | PU6p,AMA1-自主复制质粒 | wA, pyrG, yA | 55.6%~100% | [62] |
| 里氏木霉 | Cas9 | P pdc, P cbh1 | 体外转录合成 | ura5, lae1, vib1, clr2 | 单基因高达93%~100%;多基因同时编辑效率4.2%~45% | [63] |
| 体外转录合成 | 体外转录合成 | cbh-1 | 40% | [64] |
| P pdc | 里氏木霉PU61, PU62 | ura5 | 高效 | [65] |
| 体外转录合成 | 体外转录合成 | Trura5,Trlae1,Trcbh1,Trcbh2, Treg1 | 56.52%~100% | [66] |
| 产黄青霉 | Cas9 | P xyl 或体外 转录合成 | U6, 145 tRNA, Utp25或体外转录合成 | pks17, roqA, lovF,pcbAB, penDE, hcpA | 高达60%~100% | [67, 68] |
| 嗜热毁丝霉 | Cas9 | P tef1 | 嗜热毁丝霉MtU6p | amd S, cre1, res1, alp1, gh1-1, rca1, hcr1, ap3, prk6 | 单基因高达95%;双、三、四基因同时编辑效率22%~70% | [75] |
| SpCas9, FnCpf1, AsCpf1 | P tef1 或体外 转录合成 | U6p或体外 转录合成 | pks4.2, alp1, snc1, ptf1 | 单基因高达100%;多基因同时编辑时单基因效率5%~100% | [76] |
| AsCas12a | P tef1 | MtU6p | cre1, res1, alp1, gh1-1, rca1,neo, bar, hcr1, ap3, prk6 | 单基因高达90%;三、四基因同时编辑效率22%~41% | [77] |
| 棉阿舒囊霉 | SpCas9 | 酿酒酵母P tef1 | P snr52 | ade2, A754, fmp27 | 36%~80% | [78] |
| LbCpf1 | P tsa1 | P snr52 | his3, ade2, trp1, leu2, ura3 | 多基因编辑效率10.4%~77.2% | [79] |
3.3 CRISPR-Cas技术在其他丝状真菌中的应用3.3.1 稻瘟病菌2015年,日本明治大学Shigeru Kuwata教授团队[80]采用共转化载体Cas9表达框和sgRNA表达元件的策略成功在稻瘟病菌(M. oryzae)进行CRISPR-Cas9系统的构建.该团队同时使用稻瘟病菌自身的启动子U6p和构巢曲霉的启动子P trpC 分别转录表达sgRNA,对其编辑效率进行比较评价,成功对基因组中的SDH和SRS2基因进行敲除.2018年,英国埃克塞特大学Nicholas J. Talbot教授团队构建了CRISPR-Cas9 RNPs介导的稻瘟病菌基因组编辑技术体系[81],在不添加模板DNA时,以NHEJ的修复方式对靶标基因alb1进行编辑,并且不引入标记基因,建立了CRISPR-Cas9-RNP介导的无标记基因编辑系统;以HR修复方式时,仅用80 bp单链核苷酸为模板与RNPs共转化就实现了碱基的替换,或者80 bp双链DNA与RNPs共转化就能高效进行双基因同时编辑,并基于带有的苯菌灵抗性的突变菌株(E198A)对乙霉威(diethofencarb)的敏感性,设计了反筛体系,成功去除了标记基因,从而构建了CRISPR-Cas9-RNP介导的无标记基因的编辑系统.2019年,Shigeru Kuwata团队又基于单交叉介导的同源重组修复,改造升级了稻瘟病菌CRISPR-Cas9基因编辑系统,仅使用100 bp同源臂就实现了碱基的替换和GFP基因的敲入[82]. ...
Practical guidance for the implementation of the CRISPR genome editing tool in filamentous fungi
2
2019
... 2015年,深圳大学刘刚教授团队在嗜热毁丝霉中发展了过表达[72]和RNA干扰技术[73],成功过表达了转录因子Mtxyr1,将靶标基因Mtcre1进行了沉默,从而提高了纤维素酶和半纤维素酶的产量.与此同时,本文作者所在团队对嗜热毁丝霉的转化系统进行优化[74],利用农杆菌介导的转化技术,结合GFP荧光辅助筛选,能有效对目标基因实施基因过表达和敲除操作,这为后续开展嗜热毁丝霉基因组编辑技术奠定了坚实的基础.2017年,本文作者团队成功构建了CRISPR/Cas9介导的嗜热真菌基因组编辑系统[75],成功对两种嗜热真菌-嗜热毁丝霉和异宗毁丝霉(M. heterothallica)的基因组进行了编辑.该系统包括组成型启动子P tef1 调控Cas9的表达元件和嗜热毁丝霉U6p启动子转录sgRNA的表达元件,通过将Cas9表达框、sgRNA表达框和同源供体共转化导入到宿主细胞中,获得了稳定的遗传性状的基因编辑突变株.毁丝霉的基因组编辑时间缩短到3 d,其基因的同源重组效率显著提高,单基因缺失的同源重组效率高达100%,而且可以同时编辑多个基因位点,其中双基因同时编辑的同源重组效率为61%~69%,3个靶基因同时被敲除的HR效率约为30%,4个靶基因同时被缺失的HR效率仍然约为21%.以纤维素酶分泌途径重要靶点为例,利用该体系对嗜热真菌基因组成功开展了理性重构,并获得一系列蛋白分泌显著提高的工程菌.随后,柏林工业大学Vera Meyer教授团队构建了基于RNPs和质粒体系的嗜热毁丝霉CRISPR-FnCpf1/AsCpf1/SpCas9编辑系统[76],并在Ku80的缺失菌株中进行遗传操作,比较这3种编辑体系的效率,结果表明FnCpf1系统获得了四基因(alp1、pks4.2、snc1和ptf1)突变株,AsCpf1/SpCas9系统获得了三基因突变株,90 bp单链寡核苷酸作为供体DNA实现双基因的编辑.该团队还将转化体积按比例缩小至微量模式,该模式能对菌株进行有效的编辑,有助于将来构建嗜热毁丝霉高通量筛选平台.与此同时,2019年笔者团队成功升级了嗜热真菌CRISPR-Cas基因组编辑系统[77],构建了crRNA串联表达介导的嗜热毁丝霉CRISPR-AsCas12a基因组编辑系统,该系统能够高效、简易实现基因组多基因的编辑,拓展了嗜热真菌CRISPR-Cas基因组编辑系统,使其不再局限于Cas9.基于Cas12a和Cas9系统,团队建立了标记基因可循环利用的基因组编辑系统(CRISPR-Cas-assisted marker recycling technology),将其命名为Camr技术,该系统成功解决了目前丝状真菌多基因(多于10个)的编辑困难,以及基因组被编辑后的工程菌由于缺乏合适的抗性基因无法进行多次编辑的问题,可以对丝状真菌基因组进行快速、高效、多轮的可循环操作编辑.利用该体系以嗜热毁丝霉纤维素酶分泌途径中9个关键调控基因为靶点,通过交替使用2个标记基因进行三轮操作,共编辑了11个位点,人工重构了工业丝状真菌纤维素酶合成分泌途径,并获得了纤维素酶产量显著提高的9M突变体菌株.该研究对其他真菌的基因组编辑和代谢工程改造有一定参考意义. ...
... Applycations of CRISPR-Cas systems in industrial filamentous fungi
Tab. 1| 丝状真菌 | Cas 类型 | Cas启动子 | sgRNA启动子 | 靶标基因 | 编辑效率 | 参考文献 |
|---|
| 黑曲霉 | Cas9 | P tef1 | P gpdA | albA | 高效 | [52] |
| P tef1 | Af_U6-1p | albA | 高效 | [53] |
| P tef1 | PanU6 | albA | 79% | [54] |
| P gpdA | 5S rRNA | alba, fum5, fum1 | 高达100% | [55] |
| P tef1 | AoU6, AfU6 | fwnA, amyA, glaA | 单基因高达80% | [56-58] |
| P tef1, P gpdA, P glaA | Anp | pyrG, moc, laeA | 高达97.2% | [59] |
| LbCpf1 | P tef1 | Af_U3p | albA | 80% | [60] |
| 米曲霉 | Cas9 | P amyB | 米曲霉U6p | wA, pyrG, yA | 10%~100% | [61] |
| P Aotef1, AMA1-自主复制质粒 | PU6p,AMA1-自主复制质粒 | wA, pyrG, yA | 55.6%~100% | [62] |
| 里氏木霉 | Cas9 | P pdc, P cbh1 | 体外转录合成 | ura5, lae1, vib1, clr2 | 单基因高达93%~100%;多基因同时编辑效率4.2%~45% | [63] |
| 体外转录合成 | 体外转录合成 | cbh-1 | 40% | [64] |
| P pdc | 里氏木霉PU61, PU62 | ura5 | 高效 | [65] |
| 体外转录合成 | 体外转录合成 | Trura5,Trlae1,Trcbh1,Trcbh2, Treg1 | 56.52%~100% | [66] |
| 产黄青霉 | Cas9 | P xyl 或体外 转录合成 | U6, 145 tRNA, Utp25或体外转录合成 | pks17, roqA, lovF,pcbAB, penDE, hcpA | 高达60%~100% | [67, 68] |
| 嗜热毁丝霉 | Cas9 | P tef1 | 嗜热毁丝霉MtU6p | amd S, cre1, res1, alp1, gh1-1, rca1, hcr1, ap3, prk6 | 单基因高达95%;双、三、四基因同时编辑效率22%~70% | [75] |
| SpCas9, FnCpf1, AsCpf1 | P tef1 或体外 转录合成 | U6p或体外 转录合成 | pks4.2, alp1, snc1, ptf1 | 单基因高达100%;多基因同时编辑时单基因效率5%~100% | [76] |
| AsCas12a | P tef1 | MtU6p | cre1, res1, alp1, gh1-1, rca1,neo, bar, hcr1, ap3, prk6 | 单基因高达90%;三、四基因同时编辑效率22%~41% | [77] |
| 棉阿舒囊霉 | SpCas9 | 酿酒酵母P tef1 | P snr52 | ade2, A754, fmp27 | 36%~80% | [78] |
| LbCpf1 | P tsa1 | P snr52 | his3, ade2, trp1, leu2, ura3 | 多基因编辑效率10.4%~77.2% | [79] |
3.3 CRISPR-Cas技术在其他丝状真菌中的应用3.3.1 稻瘟病菌2015年,日本明治大学Shigeru Kuwata教授团队[80]采用共转化载体Cas9表达框和sgRNA表达元件的策略成功在稻瘟病菌(M. oryzae)进行CRISPR-Cas9系统的构建.该团队同时使用稻瘟病菌自身的启动子U6p和构巢曲霉的启动子P trpC 分别转录表达sgRNA,对其编辑效率进行比较评价,成功对基因组中的SDH和SRS2基因进行敲除.2018年,英国埃克塞特大学Nicholas J. Talbot教授团队构建了CRISPR-Cas9 RNPs介导的稻瘟病菌基因组编辑技术体系[81],在不添加模板DNA时,以NHEJ的修复方式对靶标基因alb1进行编辑,并且不引入标记基因,建立了CRISPR-Cas9-RNP介导的无标记基因编辑系统;以HR修复方式时,仅用80 bp单链核苷酸为模板与RNPs共转化就实现了碱基的替换,或者80 bp双链DNA与RNPs共转化就能高效进行双基因同时编辑,并基于带有的苯菌灵抗性的突变菌株(E198A)对乙霉威(diethofencarb)的敏感性,设计了反筛体系,成功去除了标记基因,从而构建了CRISPR-Cas9-RNP介导的无标记基因的编辑系统.2019年,Shigeru Kuwata团队又基于单交叉介导的同源重组修复,改造升级了稻瘟病菌CRISPR-Cas9基因编辑系统,仅使用100 bp同源臂就实现了碱基的替换和GFP基因的敲入[82]. ...
Upgrading of efficient and scalable CRISPR-Cas-mediated technology for genetic engineering in thermophilic fungus Myceliophthora thermophila
2
2019
... 2015年,深圳大学刘刚教授团队在嗜热毁丝霉中发展了过表达[72]和RNA干扰技术[73],成功过表达了转录因子Mtxyr1,将靶标基因Mtcre1进行了沉默,从而提高了纤维素酶和半纤维素酶的产量.与此同时,本文作者所在团队对嗜热毁丝霉的转化系统进行优化[74],利用农杆菌介导的转化技术,结合GFP荧光辅助筛选,能有效对目标基因实施基因过表达和敲除操作,这为后续开展嗜热毁丝霉基因组编辑技术奠定了坚实的基础.2017年,本文作者团队成功构建了CRISPR/Cas9介导的嗜热真菌基因组编辑系统[75],成功对两种嗜热真菌-嗜热毁丝霉和异宗毁丝霉(M. heterothallica)的基因组进行了编辑.该系统包括组成型启动子P tef1 调控Cas9的表达元件和嗜热毁丝霉U6p启动子转录sgRNA的表达元件,通过将Cas9表达框、sgRNA表达框和同源供体共转化导入到宿主细胞中,获得了稳定的遗传性状的基因编辑突变株.毁丝霉的基因组编辑时间缩短到3 d,其基因的同源重组效率显著提高,单基因缺失的同源重组效率高达100%,而且可以同时编辑多个基因位点,其中双基因同时编辑的同源重组效率为61%~69%,3个靶基因同时被敲除的HR效率约为30%,4个靶基因同时被缺失的HR效率仍然约为21%.以纤维素酶分泌途径重要靶点为例,利用该体系对嗜热真菌基因组成功开展了理性重构,并获得一系列蛋白分泌显著提高的工程菌.随后,柏林工业大学Vera Meyer教授团队构建了基于RNPs和质粒体系的嗜热毁丝霉CRISPR-FnCpf1/AsCpf1/SpCas9编辑系统[76],并在Ku80的缺失菌株中进行遗传操作,比较这3种编辑体系的效率,结果表明FnCpf1系统获得了四基因(alp1、pks4.2、snc1和ptf1)突变株,AsCpf1/SpCas9系统获得了三基因突变株,90 bp单链寡核苷酸作为供体DNA实现双基因的编辑.该团队还将转化体积按比例缩小至微量模式,该模式能对菌株进行有效的编辑,有助于将来构建嗜热毁丝霉高通量筛选平台.与此同时,2019年笔者团队成功升级了嗜热真菌CRISPR-Cas基因组编辑系统[77],构建了crRNA串联表达介导的嗜热毁丝霉CRISPR-AsCas12a基因组编辑系统,该系统能够高效、简易实现基因组多基因的编辑,拓展了嗜热真菌CRISPR-Cas基因组编辑系统,使其不再局限于Cas9.基于Cas12a和Cas9系统,团队建立了标记基因可循环利用的基因组编辑系统(CRISPR-Cas-assisted marker recycling technology),将其命名为Camr技术,该系统成功解决了目前丝状真菌多基因(多于10个)的编辑困难,以及基因组被编辑后的工程菌由于缺乏合适的抗性基因无法进行多次编辑的问题,可以对丝状真菌基因组进行快速、高效、多轮的可循环操作编辑.利用该体系以嗜热毁丝霉纤维素酶分泌途径中9个关键调控基因为靶点,通过交替使用2个标记基因进行三轮操作,共编辑了11个位点,人工重构了工业丝状真菌纤维素酶合成分泌途径,并获得了纤维素酶产量显著提高的9M突变体菌株.该研究对其他真菌的基因组编辑和代谢工程改造有一定参考意义. ...
... Applycations of CRISPR-Cas systems in industrial filamentous fungi
Tab. 1| 丝状真菌 | Cas 类型 | Cas启动子 | sgRNA启动子 | 靶标基因 | 编辑效率 | 参考文献 |
|---|
| 黑曲霉 | Cas9 | P tef1 | P gpdA | albA | 高效 | [52] |
| P tef1 | Af_U6-1p | albA | 高效 | [53] |
| P tef1 | PanU6 | albA | 79% | [54] |
| P gpdA | 5S rRNA | alba, fum5, fum1 | 高达100% | [55] |
| P tef1 | AoU6, AfU6 | fwnA, amyA, glaA | 单基因高达80% | [56-58] |
| P tef1, P gpdA, P glaA | Anp | pyrG, moc, laeA | 高达97.2% | [59] |
| LbCpf1 | P tef1 | Af_U3p | albA | 80% | [60] |
| 米曲霉 | Cas9 | P amyB | 米曲霉U6p | wA, pyrG, yA | 10%~100% | [61] |
| P Aotef1, AMA1-自主复制质粒 | PU6p,AMA1-自主复制质粒 | wA, pyrG, yA | 55.6%~100% | [62] |
| 里氏木霉 | Cas9 | P pdc, P cbh1 | 体外转录合成 | ura5, lae1, vib1, clr2 | 单基因高达93%~100%;多基因同时编辑效率4.2%~45% | [63] |
| 体外转录合成 | 体外转录合成 | cbh-1 | 40% | [64] |
| P pdc | 里氏木霉PU61, PU62 | ura5 | 高效 | [65] |
| 体外转录合成 | 体外转录合成 | Trura5,Trlae1,Trcbh1,Trcbh2, Treg1 | 56.52%~100% | [66] |
| 产黄青霉 | Cas9 | P xyl 或体外 转录合成 | U6, 145 tRNA, Utp25或体外转录合成 | pks17, roqA, lovF,pcbAB, penDE, hcpA | 高达60%~100% | [67, 68] |
| 嗜热毁丝霉 | Cas9 | P tef1 | 嗜热毁丝霉MtU6p | amd S, cre1, res1, alp1, gh1-1, rca1, hcr1, ap3, prk6 | 单基因高达95%;双、三、四基因同时编辑效率22%~70% | [75] |
| SpCas9, FnCpf1, AsCpf1 | P tef1 或体外 转录合成 | U6p或体外 转录合成 | pks4.2, alp1, snc1, ptf1 | 单基因高达100%;多基因同时编辑时单基因效率5%~100% | [76] |
| AsCas12a | P tef1 | MtU6p | cre1, res1, alp1, gh1-1, rca1,neo, bar, hcr1, ap3, prk6 | 单基因高达90%;三、四基因同时编辑效率22%~41% | [77] |
| 棉阿舒囊霉 | SpCas9 | 酿酒酵母P tef1 | P snr52 | ade2, A754, fmp27 | 36%~80% | [78] |
| LbCpf1 | P tsa1 | P snr52 | his3, ade2, trp1, leu2, ura3 | 多基因编辑效率10.4%~77.2% | [79] |
3.3 CRISPR-Cas技术在其他丝状真菌中的应用3.3.1 稻瘟病菌2015年,日本明治大学Shigeru Kuwata教授团队[80]采用共转化载体Cas9表达框和sgRNA表达元件的策略成功在稻瘟病菌(M. oryzae)进行CRISPR-Cas9系统的构建.该团队同时使用稻瘟病菌自身的启动子U6p和构巢曲霉的启动子P trpC 分别转录表达sgRNA,对其编辑效率进行比较评价,成功对基因组中的SDH和SRS2基因进行敲除.2018年,英国埃克塞特大学Nicholas J. Talbot教授团队构建了CRISPR-Cas9 RNPs介导的稻瘟病菌基因组编辑技术体系[81],在不添加模板DNA时,以NHEJ的修复方式对靶标基因alb1进行编辑,并且不引入标记基因,建立了CRISPR-Cas9-RNP介导的无标记基因编辑系统;以HR修复方式时,仅用80 bp单链核苷酸为模板与RNPs共转化就实现了碱基的替换,或者80 bp双链DNA与RNPs共转化就能高效进行双基因同时编辑,并基于带有的苯菌灵抗性的突变菌株(E198A)对乙霉威(diethofencarb)的敏感性,设计了反筛体系,成功去除了标记基因,从而构建了CRISPR-Cas9-RNP介导的无标记基因的编辑系统.2019年,Shigeru Kuwata团队又基于单交叉介导的同源重组修复,改造升级了稻瘟病菌CRISPR-Cas9基因编辑系统,仅使用100 bp同源臂就实现了碱基的替换和GFP基因的敲入[82]. ...
One-vector CRISPR/Cas9 genome engineering of the industrial fungus Ashbya gossypii
2
2019
... 棉阿舒囊霉(Ashbya gossypii)是一种天然具有生产核黄素能力的工业丝状真菌,有望被发展成为一类新型的细胞工厂底盘,用于生产叶酸、生物脂质、核苷和重组蛋白等.2019年,西班牙萨拉曼卡大学José Luis Revuelta教授团队构建了棉阿舒囊霉CRISPR-Cas9基因组编辑系统[78].该系统使用菌株自身的SNR52启动子转录sgRNA,并将Cas9、sgRNA和donor-DNA这3个元件放置于同一个质粒系统中,同时结合loxP技术将标记基因和CRISPR-Cas9系统进行去除,实现无痕操作.利用该系统高效实现了目标基因ade2的敲除失活、定点插入和突变,以及目标基因FMP27的点突变.随后,2020年该团队基于LbCpf1核酸酶构建了棉阿舒囊霉CRISPR/Cpf1多基因编辑技术[79],该体系通过crRNA和donor DNA分别串联表达(crRNA Array和dDNA Array)成功对5个营养缺陷型基因(his3,ade2,trp1,leu2和ura3)进行了同时敲除编辑,并获得了三基因和四基因的突变菌株.这两种CRISPR/Cas编辑技术的成功构建将极大地促进该菌在工业生产中的代谢改造研究. ...
... Applycations of CRISPR-Cas systems in industrial filamentous fungi
Tab. 1| 丝状真菌 | Cas 类型 | Cas启动子 | sgRNA启动子 | 靶标基因 | 编辑效率 | 参考文献 |
|---|
| 黑曲霉 | Cas9 | P tef1 | P gpdA | albA | 高效 | [52] |
| P tef1 | Af_U6-1p | albA | 高效 | [53] |
| P tef1 | PanU6 | albA | 79% | [54] |
| P gpdA | 5S rRNA | alba, fum5, fum1 | 高达100% | [55] |
| P tef1 | AoU6, AfU6 | fwnA, amyA, glaA | 单基因高达80% | [56-58] |
| P tef1, P gpdA, P glaA | Anp | pyrG, moc, laeA | 高达97.2% | [59] |
| LbCpf1 | P tef1 | Af_U3p | albA | 80% | [60] |
| 米曲霉 | Cas9 | P amyB | 米曲霉U6p | wA, pyrG, yA | 10%~100% | [61] |
| P Aotef1, AMA1-自主复制质粒 | PU6p,AMA1-自主复制质粒 | wA, pyrG, yA | 55.6%~100% | [62] |
| 里氏木霉 | Cas9 | P pdc, P cbh1 | 体外转录合成 | ura5, lae1, vib1, clr2 | 单基因高达93%~100%;多基因同时编辑效率4.2%~45% | [63] |
| 体外转录合成 | 体外转录合成 | cbh-1 | 40% | [64] |
| P pdc | 里氏木霉PU61, PU62 | ura5 | 高效 | [65] |
| 体外转录合成 | 体外转录合成 | Trura5,Trlae1,Trcbh1,Trcbh2, Treg1 | 56.52%~100% | [66] |
| 产黄青霉 | Cas9 | P xyl 或体外 转录合成 | U6, 145 tRNA, Utp25或体外转录合成 | pks17, roqA, lovF,pcbAB, penDE, hcpA | 高达60%~100% | [67, 68] |
| 嗜热毁丝霉 | Cas9 | P tef1 | 嗜热毁丝霉MtU6p | amd S, cre1, res1, alp1, gh1-1, rca1, hcr1, ap3, prk6 | 单基因高达95%;双、三、四基因同时编辑效率22%~70% | [75] |
| SpCas9, FnCpf1, AsCpf1 | P tef1 或体外 转录合成 | U6p或体外 转录合成 | pks4.2, alp1, snc1, ptf1 | 单基因高达100%;多基因同时编辑时单基因效率5%~100% | [76] |
| AsCas12a | P tef1 | MtU6p | cre1, res1, alp1, gh1-1, rca1,neo, bar, hcr1, ap3, prk6 | 单基因高达90%;三、四基因同时编辑效率22%~41% | [77] |
| 棉阿舒囊霉 | SpCas9 | 酿酒酵母P tef1 | P snr52 | ade2, A754, fmp27 | 36%~80% | [78] |
| LbCpf1 | P tsa1 | P snr52 | his3, ade2, trp1, leu2, ura3 | 多基因编辑效率10.4%~77.2% | [79] |
3.3 CRISPR-Cas技术在其他丝状真菌中的应用3.3.1 稻瘟病菌2015年,日本明治大学Shigeru Kuwata教授团队[80]采用共转化载体Cas9表达框和sgRNA表达元件的策略成功在稻瘟病菌(M. oryzae)进行CRISPR-Cas9系统的构建.该团队同时使用稻瘟病菌自身的启动子U6p和构巢曲霉的启动子P trpC 分别转录表达sgRNA,对其编辑效率进行比较评价,成功对基因组中的SDH和SRS2基因进行敲除.2018年,英国埃克塞特大学Nicholas J. Talbot教授团队构建了CRISPR-Cas9 RNPs介导的稻瘟病菌基因组编辑技术体系[81],在不添加模板DNA时,以NHEJ的修复方式对靶标基因alb1进行编辑,并且不引入标记基因,建立了CRISPR-Cas9-RNP介导的无标记基因编辑系统;以HR修复方式时,仅用80 bp单链核苷酸为模板与RNPs共转化就实现了碱基的替换,或者80 bp双链DNA与RNPs共转化就能高效进行双基因同时编辑,并基于带有的苯菌灵抗性的突变菌株(E198A)对乙霉威(diethofencarb)的敏感性,设计了反筛体系,成功去除了标记基因,从而构建了CRISPR-Cas9-RNP介导的无标记基因的编辑系统.2019年,Shigeru Kuwata团队又基于单交叉介导的同源重组修复,改造升级了稻瘟病菌CRISPR-Cas9基因编辑系统,仅使用100 bp同源臂就实现了碱基的替换和GFP基因的敲入[82]. ...
Multiplex genome editing in Ashbya gossypii using CRISPR-Cpf1
2
2020
... 棉阿舒囊霉(Ashbya gossypii)是一种天然具有生产核黄素能力的工业丝状真菌,有望被发展成为一类新型的细胞工厂底盘,用于生产叶酸、生物脂质、核苷和重组蛋白等.2019年,西班牙萨拉曼卡大学José Luis Revuelta教授团队构建了棉阿舒囊霉CRISPR-Cas9基因组编辑系统[78].该系统使用菌株自身的SNR52启动子转录sgRNA,并将Cas9、sgRNA和donor-DNA这3个元件放置于同一个质粒系统中,同时结合loxP技术将标记基因和CRISPR-Cas9系统进行去除,实现无痕操作.利用该系统高效实现了目标基因ade2的敲除失活、定点插入和突变,以及目标基因FMP27的点突变.随后,2020年该团队基于LbCpf1核酸酶构建了棉阿舒囊霉CRISPR/Cpf1多基因编辑技术[79],该体系通过crRNA和donor DNA分别串联表达(crRNA Array和dDNA Array)成功对5个营养缺陷型基因(his3,ade2,trp1,leu2和ura3)进行了同时敲除编辑,并获得了三基因和四基因的突变菌株.这两种CRISPR/Cas编辑技术的成功构建将极大地促进该菌在工业生产中的代谢改造研究. ...
... Applycations of CRISPR-Cas systems in industrial filamentous fungi
Tab. 1| 丝状真菌 | Cas 类型 | Cas启动子 | sgRNA启动子 | 靶标基因 | 编辑效率 | 参考文献 |
|---|
| 黑曲霉 | Cas9 | P tef1 | P gpdA | albA | 高效 | [52] |
| P tef1 | Af_U6-1p | albA | 高效 | [53] |
| P tef1 | PanU6 | albA | 79% | [54] |
| P gpdA | 5S rRNA | alba, fum5, fum1 | 高达100% | [55] |
| P tef1 | AoU6, AfU6 | fwnA, amyA, glaA | 单基因高达80% | [56-58] |
| P tef1, P gpdA, P glaA | Anp | pyrG, moc, laeA | 高达97.2% | [59] |
| LbCpf1 | P tef1 | Af_U3p | albA | 80% | [60] |
| 米曲霉 | Cas9 | P amyB | 米曲霉U6p | wA, pyrG, yA | 10%~100% | [61] |
| P Aotef1, AMA1-自主复制质粒 | PU6p,AMA1-自主复制质粒 | wA, pyrG, yA | 55.6%~100% | [62] |
| 里氏木霉 | Cas9 | P pdc, P cbh1 | 体外转录合成 | ura5, lae1, vib1, clr2 | 单基因高达93%~100%;多基因同时编辑效率4.2%~45% | [63] |
| 体外转录合成 | 体外转录合成 | cbh-1 | 40% | [64] |
| P pdc | 里氏木霉PU61, PU62 | ura5 | 高效 | [65] |
| 体外转录合成 | 体外转录合成 | Trura5,Trlae1,Trcbh1,Trcbh2, Treg1 | 56.52%~100% | [66] |
| 产黄青霉 | Cas9 | P xyl 或体外 转录合成 | U6, 145 tRNA, Utp25或体外转录合成 | pks17, roqA, lovF,pcbAB, penDE, hcpA | 高达60%~100% | [67, 68] |
| 嗜热毁丝霉 | Cas9 | P tef1 | 嗜热毁丝霉MtU6p | amd S, cre1, res1, alp1, gh1-1, rca1, hcr1, ap3, prk6 | 单基因高达95%;双、三、四基因同时编辑效率22%~70% | [75] |
| SpCas9, FnCpf1, AsCpf1 | P tef1 或体外 转录合成 | U6p或体外 转录合成 | pks4.2, alp1, snc1, ptf1 | 单基因高达100%;多基因同时编辑时单基因效率5%~100% | [76] |
| AsCas12a | P tef1 | MtU6p | cre1, res1, alp1, gh1-1, rca1,neo, bar, hcr1, ap3, prk6 | 单基因高达90%;三、四基因同时编辑效率22%~41% | [77] |
| 棉阿舒囊霉 | SpCas9 | 酿酒酵母P tef1 | P snr52 | ade2, A754, fmp27 | 36%~80% | [78] |
| LbCpf1 | P tsa1 | P snr52 | his3, ade2, trp1, leu2, ura3 | 多基因编辑效率10.4%~77.2% | [79] |
3.3 CRISPR-Cas技术在其他丝状真菌中的应用3.3.1 稻瘟病菌2015年,日本明治大学Shigeru Kuwata教授团队[80]采用共转化载体Cas9表达框和sgRNA表达元件的策略成功在稻瘟病菌(M. oryzae)进行CRISPR-Cas9系统的构建.该团队同时使用稻瘟病菌自身的启动子U6p和构巢曲霉的启动子P trpC 分别转录表达sgRNA,对其编辑效率进行比较评价,成功对基因组中的SDH和SRS2基因进行敲除.2018年,英国埃克塞特大学Nicholas J. Talbot教授团队构建了CRISPR-Cas9 RNPs介导的稻瘟病菌基因组编辑技术体系[81],在不添加模板DNA时,以NHEJ的修复方式对靶标基因alb1进行编辑,并且不引入标记基因,建立了CRISPR-Cas9-RNP介导的无标记基因编辑系统;以HR修复方式时,仅用80 bp单链核苷酸为模板与RNPs共转化就实现了碱基的替换,或者80 bp双链DNA与RNPs共转化就能高效进行双基因同时编辑,并基于带有的苯菌灵抗性的突变菌株(E198A)对乙霉威(diethofencarb)的敏感性,设计了反筛体系,成功去除了标记基因,从而构建了CRISPR-Cas9-RNP介导的无标记基因的编辑系统.2019年,Shigeru Kuwata团队又基于单交叉介导的同源重组修复,改造升级了稻瘟病菌CRISPR-Cas9基因编辑系统,仅使用100 bp同源臂就实现了碱基的替换和GFP基因的敲入[82]. ...
Tailor-made CRISPR/Cas system for highly efficient targeted gene replacement in the rice blast fungus
1
2015
... 2015年,日本明治大学Shigeru Kuwata教授团队[80]采用共转化载体Cas9表达框和sgRNA表达元件的策略成功在稻瘟病菌(M. oryzae)进行CRISPR-Cas9系统的构建.该团队同时使用稻瘟病菌自身的启动子U6p和构巢曲霉的启动子P trpC 分别转录表达sgRNA,对其编辑效率进行比较评价,成功对基因组中的SDH和SRS2基因进行敲除.2018年,英国埃克塞特大学Nicholas J. Talbot教授团队构建了CRISPR-Cas9 RNPs介导的稻瘟病菌基因组编辑技术体系[81],在不添加模板DNA时,以NHEJ的修复方式对靶标基因alb1进行编辑,并且不引入标记基因,建立了CRISPR-Cas9-RNP介导的无标记基因编辑系统;以HR修复方式时,仅用80 bp单链核苷酸为模板与RNPs共转化就实现了碱基的替换,或者80 bp双链DNA与RNPs共转化就能高效进行双基因同时编辑,并基于带有的苯菌灵抗性的突变菌株(E198A)对乙霉威(diethofencarb)的敏感性,设计了反筛体系,成功去除了标记基因,从而构建了CRISPR-Cas9-RNP介导的无标记基因的编辑系统.2019年,Shigeru Kuwata团队又基于单交叉介导的同源重组修复,改造升级了稻瘟病菌CRISPR-Cas9基因编辑系统,仅使用100 bp同源臂就实现了碱基的替换和GFP基因的敲入[82]. ...
CRISPR-Cas9 ribonucleoprotein-mediated co-editing and counterselection in the rice blast fungus
1
2018
... 2015年,日本明治大学Shigeru Kuwata教授团队[80]采用共转化载体Cas9表达框和sgRNA表达元件的策略成功在稻瘟病菌(M. oryzae)进行CRISPR-Cas9系统的构建.该团队同时使用稻瘟病菌自身的启动子U6p和构巢曲霉的启动子P trpC 分别转录表达sgRNA,对其编辑效率进行比较评价,成功对基因组中的SDH和SRS2基因进行敲除.2018年,英国埃克塞特大学Nicholas J. Talbot教授团队构建了CRISPR-Cas9 RNPs介导的稻瘟病菌基因组编辑技术体系[81],在不添加模板DNA时,以NHEJ的修复方式对靶标基因alb1进行编辑,并且不引入标记基因,建立了CRISPR-Cas9-RNP介导的无标记基因编辑系统;以HR修复方式时,仅用80 bp单链核苷酸为模板与RNPs共转化就实现了碱基的替换,或者80 bp双链DNA与RNPs共转化就能高效进行双基因同时编辑,并基于带有的苯菌灵抗性的突变菌株(E198A)对乙霉威(diethofencarb)的敏感性,设计了反筛体系,成功去除了标记基因,从而构建了CRISPR-Cas9-RNP介导的无标记基因的编辑系统.2019年,Shigeru Kuwata团队又基于单交叉介导的同源重组修复,改造升级了稻瘟病菌CRISPR-Cas9基因编辑系统,仅使用100 bp同源臂就实现了碱基的替换和GFP基因的敲入[82]. ...
Single crossover-mediated targeted nucleotide substitution and knock-in strategies with CRISPR/Cas9 system in the rice blast fungus
1
2019
... 2015年,日本明治大学Shigeru Kuwata教授团队[80]采用共转化载体Cas9表达框和sgRNA表达元件的策略成功在稻瘟病菌(M. oryzae)进行CRISPR-Cas9系统的构建.该团队同时使用稻瘟病菌自身的启动子U6p和构巢曲霉的启动子P trpC 分别转录表达sgRNA,对其编辑效率进行比较评价,成功对基因组中的SDH和SRS2基因进行敲除.2018年,英国埃克塞特大学Nicholas J. Talbot教授团队构建了CRISPR-Cas9 RNPs介导的稻瘟病菌基因组编辑技术体系[81],在不添加模板DNA时,以NHEJ的修复方式对靶标基因alb1进行编辑,并且不引入标记基因,建立了CRISPR-Cas9-RNP介导的无标记基因编辑系统;以HR修复方式时,仅用80 bp单链核苷酸为模板与RNPs共转化就实现了碱基的替换,或者80 bp双链DNA与RNPs共转化就能高效进行双基因同时编辑,并基于带有的苯菌灵抗性的突变菌株(E198A)对乙霉威(diethofencarb)的敏感性,设计了反筛体系,成功去除了标记基因,从而构建了CRISPR-Cas9-RNP介导的无标记基因的编辑系统.2019年,Shigeru Kuwata团队又基于单交叉介导的同源重组修复,改造升级了稻瘟病菌CRISPR-Cas9基因编辑系统,仅使用100 bp同源臂就实现了碱基的替换和GFP基因的敲入[82]. ...
Development of the CRISPR/Cas9 system for targeted gene disruption in Aspergillus fumigatus
1
2015
... 2015年,美国达特茅斯学院Jay C. Dunlap教授团队[83]对烟曲霉(A. fumigatus)成功开展了CRISPR/Cas9系统的研发,该研究发现持续性表达Cas9基因不会影响烟曲霉的正常生长和致病性,该系统使用酿酒酵母SNR52启动子调控sgRNA的转录表达,对靶标基因pksP的编辑效率为53%.随后,南京师范大学陆玲教授团队[84]对烟曲霉成功开展了高效的CRISPR/Cas9基因组编辑系统的研发,该系统利用烟曲霉自身的U6p启动子来转录表达sgRNA,通过微同源修复机制(microhomology-mediated end joining,MMEJ)显著减少了同源臂序列的长度,简化了donor DNA的构建过程,缩短了时间,极大提高了烟曲霉CRISPR-Cas9系统在靶标位点的编辑效率.在标记基因hph两侧加入仅28 bp的同源序列就能高效实现基因cnaA的敲除,敲除效率高达97%.在基因敲入方面,仅通过35 bp的同源序列就能将外源GFP定点插入到cnaA位点,基因编辑效率接近100%.2017年,德国Axel A. Brakhage教授团队通过使用Mortensen构建的II型RNA聚合酶转录HH-sgRNA-HDV复合体获得sgRNA,Cas9基因的表达由多西环素依赖性系统(Tet-ON)诱导调控,显著提高了CRISPR/Cas9系统在烟曲霉的编辑效率,同时也鉴定出合成代谢产物trypacidin的关键基因tynC,这也是在丝状真菌中将次级代谢产物与基因遗传表型来源联系起来的重要研究[85]. ...
Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening
0
2017
Inducible and multiplex gene regulation using CRISPR-Cpf1-based transcription factors
0
2017
Reprogrammable CRISPR/dCas9-based recruitment of DNMT1 for site-specific DNA demethylation and gene regulation
0
2019
CRISPR-assisted multi-dimensional regulation for fine-tuning gene expression in Bacillus subtilis
1
2019
... 随着CRISPR技术的发展,相信CRISPR-Cas9系统在诸多方面可以进一步优化改进,包括降低脱靶效应、解除Cas蛋白的PAM序列的限制性、提升精确定点编辑的效率等.例如近期研究表明可以通过人工改造Cas9蛋白(xCas9、SpCas9-non-G PAMs等)[92-95]或者是寻找并鉴定更多的Cas同源蛋白(Cas12b、Cas14等)[96-98],使其识别更多的PAM位点.与此同时,基于dCas9或者nCas9的CRISPR相关衍生技术在工业真菌中有待开发,例如CRISPRa和CRISPRi介导的基因表达调控技术[99-103]在丝状真菌中尚未报道,多功能CRISPR介导的组合调控技术还没有在丝状真菌中研发和建立.未来建立不局限于某个真菌物种的通用编辑技术体系,以及将CRISPR系统与高通量技术相结合进行高通量编辑改造等都值得期待.除前文提到的在黑曲霉中建立了胞嘧啶碱基编辑器(cytosine base editor,CBE)以外,在其他的丝状真菌中还没有构建起来,并且腺嘌呤碱基编辑器(adenine base editor,ABE)也尚未在丝状真菌中研发报道.随着CRISPR基因组编辑及相关技术的创新发展,我们相信CRISPR系统将在真菌基因功能研究、代谢途径重构、精确表达调控、蛋白定向进化以及高性能底盘构建等方面得到快速发展和技术革新,从而促进真菌合成生物学的快速发展. ...
Highly efficient CRISPR mutagenesis by microhomology-mediated end joining in Aspergillus fumigatus
1
2016
... 2015年,美国达特茅斯学院Jay C. Dunlap教授团队[83]对烟曲霉(A. fumigatus)成功开展了CRISPR/Cas9系统的研发,该研究发现持续性表达Cas9基因不会影响烟曲霉的正常生长和致病性,该系统使用酿酒酵母SNR52启动子调控sgRNA的转录表达,对靶标基因pksP的编辑效率为53%.随后,南京师范大学陆玲教授团队[84]对烟曲霉成功开展了高效的CRISPR/Cas9基因组编辑系统的研发,该系统利用烟曲霉自身的U6p启动子来转录表达sgRNA,通过微同源修复机制(microhomology-mediated end joining,MMEJ)显著减少了同源臂序列的长度,简化了donor DNA的构建过程,缩短了时间,极大提高了烟曲霉CRISPR-Cas9系统在靶标位点的编辑效率.在标记基因hph两侧加入仅28 bp的同源序列就能高效实现基因cnaA的敲除,敲除效率高达97%.在基因敲入方面,仅通过35 bp的同源序列就能将外源GFP定点插入到cnaA位点,基因编辑效率接近100%.2017年,德国Axel A. Brakhage教授团队通过使用Mortensen构建的II型RNA聚合酶转录HH-sgRNA-HDV复合体获得sgRNA,Cas9基因的表达由多西环素依赖性系统(Tet-ON)诱导调控,显著提高了CRISPR/Cas9系统在烟曲霉的编辑效率,同时也鉴定出合成代谢产物trypacidin的关键基因tynC,这也是在丝状真菌中将次级代谢产物与基因遗传表型来源联系起来的重要研究[85]. ...
Functional reconstitution of a fungal natural product gene cluster by advanced genome editing
1
2017
... 2015年,美国达特茅斯学院Jay C. Dunlap教授团队[83]对烟曲霉(A. fumigatus)成功开展了CRISPR/Cas9系统的研发,该研究发现持续性表达Cas9基因不会影响烟曲霉的正常生长和致病性,该系统使用酿酒酵母SNR52启动子调控sgRNA的转录表达,对靶标基因pksP的编辑效率为53%.随后,南京师范大学陆玲教授团队[84]对烟曲霉成功开展了高效的CRISPR/Cas9基因组编辑系统的研发,该系统利用烟曲霉自身的U6p启动子来转录表达sgRNA,通过微同源修复机制(microhomology-mediated end joining,MMEJ)显著减少了同源臂序列的长度,简化了donor DNA的构建过程,缩短了时间,极大提高了烟曲霉CRISPR-Cas9系统在靶标位点的编辑效率.在标记基因hph两侧加入仅28 bp的同源序列就能高效实现基因cnaA的敲除,敲除效率高达97%.在基因敲入方面,仅通过35 bp的同源序列就能将外源GFP定点插入到cnaA位点,基因编辑效率接近100%.2017年,德国Axel A. Brakhage教授团队通过使用Mortensen构建的II型RNA聚合酶转录HH-sgRNA-HDV复合体获得sgRNA,Cas9基因的表达由多西环素依赖性系统(Tet-ON)诱导调控,显著提高了CRISPR/Cas9系统在烟曲霉的编辑效率,同时也鉴定出合成代谢产物trypacidin的关键基因tynC,这也是在丝状真菌中将次级代谢产物与基因遗传表型来源联系起来的重要研究[85]. ...
Genome editing in Ustilago maydis using the CRISPR-Cas system
1
2016
... 2016年,德国马普学会陆生微生物研究所Regine Kahmann教授团队对另一种植物病原真菌玉米黑粉菌(U. maydis)成功开展CRISPR/Cas9系统的构建[86],使用玉米黑粉菌自身U6启动子转录sgRNA的表达,密码子优化的Cas9基因N端和C端都添加核定位信号(NLS)并置于持续性强启动子P otef 下进行表达,将Cas9和sgRNA两个表达元件一起装载在自我复制的质粒pMS7上,并且过表达Cas9不会对玉米黑粉菌的细胞产生毒性,该体系通过NHEJ修复方式成功对两个靶基因bE1 和bW2实现了编辑,编辑效率高达70%~100%. ...
CRISPR/Cas9-mediated efficient genome editing via blastospore-based transformation in entomopathogenic fungus Beauveria bassiana
1
2017
... 中国科学院上海生命科学研究院王四宝研究员团队成功对昆虫病原真菌球孢白僵菌(B. bassiana)开展了CRISPR/Cas9系统的研发[87].该系统利用强启动子P gpdA 转录表达密码子优化的Cas9-NLS蛋白,使用体外转录sgRNA的方式,通过芽生孢子转化体系将其共转化进入球孢白僵菌细胞中,成功对基因ura5进行NHEJ修复编辑;并通过同源重组修复机制,再次利用内源ura5作为标记基因,对egfp、Bbmp1和Bbrgs1成功进行了单、双和三基因的同时敲除编辑. ...



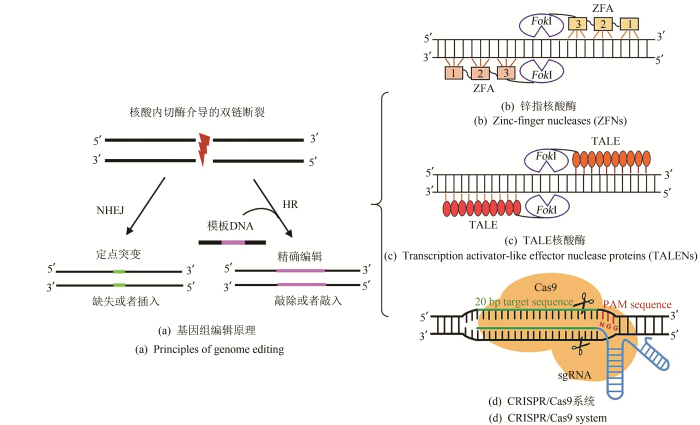
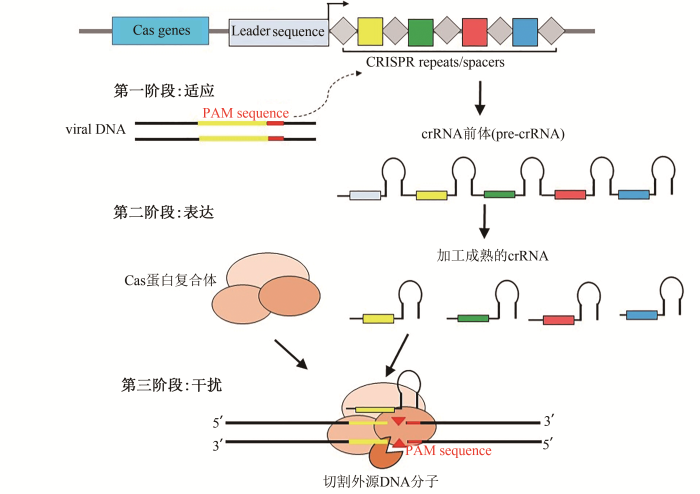


{kind=link}
{kind=link}
{kind=link}
{kind=link}