Morphine, the proteus of organic molecules
1
2002
... 天然产物是从自然界存在的动物、植物、微生物中分离提取的有机化合物,是生物体在适应环境的漫长进化过程中,为了生存而产生的内源生理活性分子.广泛而多样性的生物,制造出千变万化的天然产物,被认为是大自然赐予人类的瑰宝,也是开发药物活性分子的重要源泉.例如,8000年前人类就已种植罂粟用于观赏和治病,但直到19世纪初才由德国药剂师Friedrich Sertürner首次从罂粟中分离出活性天然产物——吗啡(morphine),这一创举成为人类将纯单体天然化合物用作药物的里程碑性标志(图1)[1-2].人类利用柳树皮镇痛退烧也有数千年的历史,1828年德国药物学家Johann Buchner首次从中提取分离出活性成分水杨苷(salicin),逐渐揭开其神奇功效背后的面纱;1838年意大利化学家Raffaele Piria确定了水杨酸苷的结构,并经水解和氧化制得水杨酸(salicylic acid),但因其对咽喉和胃肠刺激剧烈而无法用于临床治疗;1852年法国化学家Charles Gerhardt将水杨酸钠与乙酰氯进行反应,首次报道了乙酰水杨酸(acetylsalicylic acid)的合成,但对其功效未进行深入研究;在此基础上,1897年德国拜耳公司Arthur Eichengrün和Felix Hoffmann等重新制备了乙酰水杨酸,发现其独特的镇痛退烧功效并申请专利,随后拜耳公司将其命名为阿司匹林(aspirin)推向市场,开创了人类将天然产物类似物(或衍生物)作为药物的先河(图1)[3-4]. ...
The chemical history of Morphine: An 8000-year journey, from resin to de-novo synthesis
1
2017
... 天然产物是从自然界存在的动物、植物、微生物中分离提取的有机化合物,是生物体在适应环境的漫长进化过程中,为了生存而产生的内源生理活性分子.广泛而多样性的生物,制造出千变万化的天然产物,被认为是大自然赐予人类的瑰宝,也是开发药物活性分子的重要源泉.例如,8000年前人类就已种植罂粟用于观赏和治病,但直到19世纪初才由德国药剂师Friedrich Sertürner首次从罂粟中分离出活性天然产物——吗啡(morphine),这一创举成为人类将纯单体天然化合物用作药物的里程碑性标志(图1)[1-2].人类利用柳树皮镇痛退烧也有数千年的历史,1828年德国药物学家Johann Buchner首次从中提取分离出活性成分水杨苷(salicin),逐渐揭开其神奇功效背后的面纱;1838年意大利化学家Raffaele Piria确定了水杨酸苷的结构,并经水解和氧化制得水杨酸(salicylic acid),但因其对咽喉和胃肠刺激剧烈而无法用于临床治疗;1852年法国化学家Charles Gerhardt将水杨酸钠与乙酰氯进行反应,首次报道了乙酰水杨酸(acetylsalicylic acid)的合成,但对其功效未进行深入研究;在此基础上,1897年德国拜耳公司Arthur Eichengrün和Felix Hoffmann等重新制备了乙酰水杨酸,发现其独特的镇痛退烧功效并申请专利,随后拜耳公司将其命名为阿司匹林(aspirin)推向市场,开创了人类将天然产物类似物(或衍生物)作为药物的先河(图1)[3-4]. ...
One hundred years of Aspirin
1
1997
... 天然产物是从自然界存在的动物、植物、微生物中分离提取的有机化合物,是生物体在适应环境的漫长进化过程中,为了生存而产生的内源生理活性分子.广泛而多样性的生物,制造出千变万化的天然产物,被认为是大自然赐予人类的瑰宝,也是开发药物活性分子的重要源泉.例如,8000年前人类就已种植罂粟用于观赏和治病,但直到19世纪初才由德国药剂师Friedrich Sertürner首次从罂粟中分离出活性天然产物——吗啡(morphine),这一创举成为人类将纯单体天然化合物用作药物的里程碑性标志(图1)[1-2].人类利用柳树皮镇痛退烧也有数千年的历史,1828年德国药物学家Johann Buchner首次从中提取分离出活性成分水杨苷(salicin),逐渐揭开其神奇功效背后的面纱;1838年意大利化学家Raffaele Piria确定了水杨酸苷的结构,并经水解和氧化制得水杨酸(salicylic acid),但因其对咽喉和胃肠刺激剧烈而无法用于临床治疗;1852年法国化学家Charles Gerhardt将水杨酸钠与乙酰氯进行反应,首次报道了乙酰水杨酸(acetylsalicylic acid)的合成,但对其功效未进行深入研究;在此基础上,1897年德国拜耳公司Arthur Eichengrün和Felix Hoffmann等重新制备了乙酰水杨酸,发现其独特的镇痛退烧功效并申请专利,随后拜耳公司将其命名为阿司匹林(aspirin)推向市场,开创了人类将天然产物类似物(或衍生物)作为药物的先河(图1)[3-4]. ...
The Aspirin story-from willow to wonder drug
1
2017
... 天然产物是从自然界存在的动物、植物、微生物中分离提取的有机化合物,是生物体在适应环境的漫长进化过程中,为了生存而产生的内源生理活性分子.广泛而多样性的生物,制造出千变万化的天然产物,被认为是大自然赐予人类的瑰宝,也是开发药物活性分子的重要源泉.例如,8000年前人类就已种植罂粟用于观赏和治病,但直到19世纪初才由德国药剂师Friedrich Sertürner首次从罂粟中分离出活性天然产物——吗啡(morphine),这一创举成为人类将纯单体天然化合物用作药物的里程碑性标志(图1)[1-2].人类利用柳树皮镇痛退烧也有数千年的历史,1828年德国药物学家Johann Buchner首次从中提取分离出活性成分水杨苷(salicin),逐渐揭开其神奇功效背后的面纱;1838年意大利化学家Raffaele Piria确定了水杨酸苷的结构,并经水解和氧化制得水杨酸(salicylic acid),但因其对咽喉和胃肠刺激剧烈而无法用于临床治疗;1852年法国化学家Charles Gerhardt将水杨酸钠与乙酰氯进行反应,首次报道了乙酰水杨酸(acetylsalicylic acid)的合成,但对其功效未进行深入研究;在此基础上,1897年德国拜耳公司Arthur Eichengrün和Felix Hoffmann等重新制备了乙酰水杨酸,发现其独特的镇痛退烧功效并申请专利,随后拜耳公司将其命名为阿司匹林(aspirin)推向市场,开创了人类将天然产物类似物(或衍生物)作为药物的先河(图1)[3-4]. ...
1
2004
... 进入20世纪之后,天然产物化学研究获得快速发展,大量天然产物从各类动物、植物、海洋生物和微生物中被提取分离和成功鉴定,很多高生理活性分子被用作治疗疾病的药物[5-6].据统计,目前市场上超过40%的小分子药物来源于天然产物及其类似物[7-9],例如药物化合物库中的明星分子(图2),有源于植物的奎宁(quinine)和青蒿素(artemisinin),是治疗疟疾的特效药物;源于植物的紫杉醇(taxol)、长春碱(vinblastine)及长春新碱(vincristine),是广谱抗癌药物;源于微生物的青霉素(penicillin)、链霉素(streptomycin)、万古霉素(vancomycin)等,是治疗细菌感染类疾病的特效抗生素药物;而源于微生物的洛伐他汀(lovastatin)则能有效控制胆固醇,为高血脂患者带来福音;源于植物的二甲双胍(metformin)及源于哺乳动物的胰岛素(insulin),被用于治疗和控制糖尿病;源于海洋软体动物的软海绵素B(halichondrin B),对一些恶性肿瘤具有神奇疗效(图2).除此之外,很多天然产物及其类似物还被开发出来,已被广泛应用于香料、染料、食品添加剂、农药和兽药等领域.因此天然产物无处不在,与人类生活密切相关. ...
1
2004
... 进入20世纪之后,天然产物化学研究获得快速发展,大量天然产物从各类动物、植物、海洋生物和微生物中被提取分离和成功鉴定,很多高生理活性分子被用作治疗疾病的药物[5-6].据统计,目前市场上超过40%的小分子药物来源于天然产物及其类似物[7-9],例如药物化合物库中的明星分子(图2),有源于植物的奎宁(quinine)和青蒿素(artemisinin),是治疗疟疾的特效药物;源于植物的紫杉醇(taxol)、长春碱(vinblastine)及长春新碱(vincristine),是广谱抗癌药物;源于微生物的青霉素(penicillin)、链霉素(streptomycin)、万古霉素(vancomycin)等,是治疗细菌感染类疾病的特效抗生素药物;而源于微生物的洛伐他汀(lovastatin)则能有效控制胆固醇,为高血脂患者带来福音;源于植物的二甲双胍(metformin)及源于哺乳动物的胰岛素(insulin),被用于治疗和控制糖尿病;源于海洋软体动物的软海绵素B(halichondrin B),对一些恶性肿瘤具有神奇疗效(图2).除此之外,很多天然产物及其类似物还被开发出来,已被广泛应用于香料、染料、食品添加剂、农药和兽药等领域.因此天然产物无处不在,与人类生活密切相关. ...
1
2016
... 进入20世纪之后,天然产物化学研究获得快速发展,大量天然产物从各类动物、植物、海洋生物和微生物中被提取分离和成功鉴定,很多高生理活性分子被用作治疗疾病的药物[5-6].据统计,目前市场上超过40%的小分子药物来源于天然产物及其类似物[7-9],例如药物化合物库中的明星分子(图2),有源于植物的奎宁(quinine)和青蒿素(artemisinin),是治疗疟疾的特效药物;源于植物的紫杉醇(taxol)、长春碱(vinblastine)及长春新碱(vincristine),是广谱抗癌药物;源于微生物的青霉素(penicillin)、链霉素(streptomycin)、万古霉素(vancomycin)等,是治疗细菌感染类疾病的特效抗生素药物;而源于微生物的洛伐他汀(lovastatin)则能有效控制胆固醇,为高血脂患者带来福音;源于植物的二甲双胍(metformin)及源于哺乳动物的胰岛素(insulin),被用于治疗和控制糖尿病;源于海洋软体动物的软海绵素B(halichondrin B),对一些恶性肿瘤具有神奇疗效(图2).除此之外,很多天然产物及其类似物还被开发出来,已被广泛应用于香料、染料、食品添加剂、农药和兽药等领域.因此天然产物无处不在,与人类生活密切相关. ...
Natural products-based drug discovery: some bottlenecks and considerations
1
2009
... 进入20世纪之后,天然产物化学研究获得快速发展,大量天然产物从各类动物、植物、海洋生物和微生物中被提取分离和成功鉴定,很多高生理活性分子被用作治疗疾病的药物[5-6].据统计,目前市场上超过40%的小分子药物来源于天然产物及其类似物[7-9],例如药物化合物库中的明星分子(图2),有源于植物的奎宁(quinine)和青蒿素(artemisinin),是治疗疟疾的特效药物;源于植物的紫杉醇(taxol)、长春碱(vinblastine)及长春新碱(vincristine),是广谱抗癌药物;源于微生物的青霉素(penicillin)、链霉素(streptomycin)、万古霉素(vancomycin)等,是治疗细菌感染类疾病的特效抗生素药物;而源于微生物的洛伐他汀(lovastatin)则能有效控制胆固醇,为高血脂患者带来福音;源于植物的二甲双胍(metformin)及源于哺乳动物的胰岛素(insulin),被用于治疗和控制糖尿病;源于海洋软体动物的软海绵素B(halichondrin B),对一些恶性肿瘤具有神奇疗效(图2).除此之外,很多天然产物及其类似物还被开发出来,已被广泛应用于香料、染料、食品添加剂、农药和兽药等领域.因此天然产物无处不在,与人类生活密切相关. ...
The pharmaceutical industry and natural products: historical status and new trends
0
2015
Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019
1
2020
... 进入20世纪之后,天然产物化学研究获得快速发展,大量天然产物从各类动物、植物、海洋生物和微生物中被提取分离和成功鉴定,很多高生理活性分子被用作治疗疾病的药物[5-6].据统计,目前市场上超过40%的小分子药物来源于天然产物及其类似物[7-9],例如药物化合物库中的明星分子(图2),有源于植物的奎宁(quinine)和青蒿素(artemisinin),是治疗疟疾的特效药物;源于植物的紫杉醇(taxol)、长春碱(vinblastine)及长春新碱(vincristine),是广谱抗癌药物;源于微生物的青霉素(penicillin)、链霉素(streptomycin)、万古霉素(vancomycin)等,是治疗细菌感染类疾病的特效抗生素药物;而源于微生物的洛伐他汀(lovastatin)则能有效控制胆固醇,为高血脂患者带来福音;源于植物的二甲双胍(metformin)及源于哺乳动物的胰岛素(insulin),被用于治疗和控制糖尿病;源于海洋软体动物的软海绵素B(halichondrin B),对一些恶性肿瘤具有神奇疗效(图2).除此之外,很多天然产物及其类似物还被开发出来,已被广泛应用于香料、染料、食品添加剂、农药和兽药等领域.因此天然产物无处不在,与人类生活密切相关. ...
The art and science of total synthesis at the dawn of the twenty-first century
1
2000
... 尽管天然产物具有很好的生物活性,但其在生物体内含量非常低,通过提取分离进行大量制备存在很大难度,从而严重制约其开发和利用.1828年德国化学家Friedrich Wöhler采用无机原料和试剂探索化学反应,在实验室成功制备出尿素(urea),开创了化学方法获取有机分子的新时代[10].1845年Hermann Kolbe用元素碳制备出乙酸(acetic acid),首次提出“合成”(synthesis)这一概念,意指将化学原料通过组装反应制得产物的过程[11];1869年Carl Gräbe和Carl Liebermann以蒽为原料经过氧化反应,合成出天然橙红色染料茜红素(alizarin)[12];1878年Adolf von Baeyer利用邻硝基苯甲醛与丙酮反应,合成出天然产物靛蓝(indigo)[13];而19世纪最引人瞩目的全合成工作,当属1890年Emil Fischer完成的含有5个相连碳立体中心的葡萄糖(glucose)分子[14].这些开拓性研究揭开了天然产物化学全合成的新篇章. ...
1
2008
... 尽管天然产物具有很好的生物活性,但其在生物体内含量非常低,通过提取分离进行大量制备存在很大难度,从而严重制约其开发和利用.1828年德国化学家Friedrich Wöhler采用无机原料和试剂探索化学反应,在实验室成功制备出尿素(urea),开创了化学方法获取有机分子的新时代[10].1845年Hermann Kolbe用元素碳制备出乙酸(acetic acid),首次提出“合成”(synthesis)这一概念,意指将化学原料通过组装反应制得产物的过程[11];1869年Carl Gräbe和Carl Liebermann以蒽为原料经过氧化反应,合成出天然橙红色染料茜红素(alizarin)[12];1878年Adolf von Baeyer利用邻硝基苯甲醛与丙酮反应,合成出天然产物靛蓝(indigo)[13];而19世纪最引人瞩目的全合成工作,当属1890年Emil Fischer完成的含有5个相连碳立体中心的葡萄糖(glucose)分子[14].这些开拓性研究揭开了天然产物化学全合成的新篇章. ...
1
1996
... 尽管天然产物具有很好的生物活性,但其在生物体内含量非常低,通过提取分离进行大量制备存在很大难度,从而严重制约其开发和利用.1828年德国化学家Friedrich Wöhler采用无机原料和试剂探索化学反应,在实验室成功制备出尿素(urea),开创了化学方法获取有机分子的新时代[10].1845年Hermann Kolbe用元素碳制备出乙酸(acetic acid),首次提出“合成”(synthesis)这一概念,意指将化学原料通过组装反应制得产物的过程[11];1869年Carl Gräbe和Carl Liebermann以蒽为原料经过氧化反应,合成出天然橙红色染料茜红素(alizarin)[12];1878年Adolf von Baeyer利用邻硝基苯甲醛与丙酮反应,合成出天然产物靛蓝(indigo)[13];而19世纪最引人瞩目的全合成工作,当属1890年Emil Fischer完成的含有5个相连碳立体中心的葡萄糖(glucose)分子[14].这些开拓性研究揭开了天然产物化学全合成的新篇章. ...
1
2003
... 尽管天然产物具有很好的生物活性,但其在生物体内含量非常低,通过提取分离进行大量制备存在很大难度,从而严重制约其开发和利用.1828年德国化学家Friedrich Wöhler采用无机原料和试剂探索化学反应,在实验室成功制备出尿素(urea),开创了化学方法获取有机分子的新时代[10].1845年Hermann Kolbe用元素碳制备出乙酸(acetic acid),首次提出“合成”(synthesis)这一概念,意指将化学原料通过组装反应制得产物的过程[11];1869年Carl Gräbe和Carl Liebermann以蒽为原料经过氧化反应,合成出天然橙红色染料茜红素(alizarin)[12];1878年Adolf von Baeyer利用邻硝基苯甲醛与丙酮反应,合成出天然产物靛蓝(indigo)[13];而19世纪最引人瞩目的全合成工作,当属1890年Emil Fischer完成的含有5个相连碳立体中心的葡萄糖(glucose)分子[14].这些开拓性研究揭开了天然产物化学全合成的新篇章. ...
1
2011
... 尽管天然产物具有很好的生物活性,但其在生物体内含量非常低,通过提取分离进行大量制备存在很大难度,从而严重制约其开发和利用.1828年德国化学家Friedrich Wöhler采用无机原料和试剂探索化学反应,在实验室成功制备出尿素(urea),开创了化学方法获取有机分子的新时代[10].1845年Hermann Kolbe用元素碳制备出乙酸(acetic acid),首次提出“合成”(synthesis)这一概念,意指将化学原料通过组装反应制得产物的过程[11];1869年Carl Gräbe和Carl Liebermann以蒽为原料经过氧化反应,合成出天然橙红色染料茜红素(alizarin)[12];1878年Adolf von Baeyer利用邻硝基苯甲醛与丙酮反应,合成出天然产物靛蓝(indigo)[13];而19世纪最引人瞩目的全合成工作,当属1890年Emil Fischer完成的含有5个相连碳立体中心的葡萄糖(glucose)分子[14].这些开拓性研究揭开了天然产物化学全合成的新篇章. ...
A synthesis of tropinone
1
1917
... 进入20世纪,天然产物全合成领域涌现一批有机化学大师,他们设计发展了各种各样的化学试剂和有机转化反应,成功实现众多天然产物的化学全合成.例如20世纪初,德国化学家Richard Willstätter和英国化学家Robert Robinson采用不同的工艺路线,完成了托品酮(tropinone)的全合成[15];到了20年代,德国Hans Fischer教授实现了血红素分子(haemin)的全合成[16];30年代美国化学家Werner E. Bachmann对雌甾酮分子(equilenin)的全合成率先获得成功[17].在随后的40多年里,借助于新的分析检测技术,天然产物全合成更取得了突破性发展,例如美国有机合成大师Robert B. Woodward与其合作者,先后完成了奎宁(quinine)[18]、胆固醇(cholesterol)[19]、可的松(cortisone)[20]、马钱子碱(strychnine)[21]、叶绿素α(chlorophyll α)[22]、头孢菌素C(cephalosporin C)[23]及红霉素A(erythromycin A)[24]等明星分子的全合成;并且在全合成维生素B12(vitamin B12)[25]实验过程中,发现电环化协同反应现象,与量子化学家Roald Hoffmann一起建立了分子轨道守恒(the conservation of molecular orbital symmetry)理论[26].另一位有机合成大师Elias J. Corey除了完成烟曲霉素(fumagillin)[27]、红霉内酯B(erythronolide B)[28]、秋水仙碱(colchicine)[29]、喜树碱(camptothecin)[30]、前列腺素F(prostaglandin F)[31]、番红霉素A(saframycin A)[32]、抗疟霉素(aplasmomycin)[33]等众多天然产物的全合成之外,还提出了逆合成分析(retrosynthesis analysis)策略[34],即运用有机反应逻辑,对目标分子进行合理的解构,反推出起始原料和关键反应节点,指导设计出复杂分子的全合成路线. ...
Synthese des h?matoporphyrins, protoporphyrins und h?mins
1
1929
... 进入20世纪,天然产物全合成领域涌现一批有机化学大师,他们设计发展了各种各样的化学试剂和有机转化反应,成功实现众多天然产物的化学全合成.例如20世纪初,德国化学家Richard Willstätter和英国化学家Robert Robinson采用不同的工艺路线,完成了托品酮(tropinone)的全合成[15];到了20年代,德国Hans Fischer教授实现了血红素分子(haemin)的全合成[16];30年代美国化学家Werner E. Bachmann对雌甾酮分子(equilenin)的全合成率先获得成功[17].在随后的40多年里,借助于新的分析检测技术,天然产物全合成更取得了突破性发展,例如美国有机合成大师Robert B. Woodward与其合作者,先后完成了奎宁(quinine)[18]、胆固醇(cholesterol)[19]、可的松(cortisone)[20]、马钱子碱(strychnine)[21]、叶绿素α(chlorophyll α)[22]、头孢菌素C(cephalosporin C)[23]及红霉素A(erythromycin A)[24]等明星分子的全合成;并且在全合成维生素B12(vitamin B12)[25]实验过程中,发现电环化协同反应现象,与量子化学家Roald Hoffmann一起建立了分子轨道守恒(the conservation of molecular orbital symmetry)理论[26].另一位有机合成大师Elias J. Corey除了完成烟曲霉素(fumagillin)[27]、红霉内酯B(erythronolide B)[28]、秋水仙碱(colchicine)[29]、喜树碱(camptothecin)[30]、前列腺素F(prostaglandin F)[31]、番红霉素A(saframycin A)[32]、抗疟霉素(aplasmomycin)[33]等众多天然产物的全合成之外,还提出了逆合成分析(retrosynthesis analysis)策略[34],即运用有机反应逻辑,对目标分子进行合理的解构,反推出起始原料和关键反应节点,指导设计出复杂分子的全合成路线. ...
The total synthesis of the sex hormone equilenin
1
1939
... 进入20世纪,天然产物全合成领域涌现一批有机化学大师,他们设计发展了各种各样的化学试剂和有机转化反应,成功实现众多天然产物的化学全合成.例如20世纪初,德国化学家Richard Willstätter和英国化学家Robert Robinson采用不同的工艺路线,完成了托品酮(tropinone)的全合成[15];到了20年代,德国Hans Fischer教授实现了血红素分子(haemin)的全合成[16];30年代美国化学家Werner E. Bachmann对雌甾酮分子(equilenin)的全合成率先获得成功[17].在随后的40多年里,借助于新的分析检测技术,天然产物全合成更取得了突破性发展,例如美国有机合成大师Robert B. Woodward与其合作者,先后完成了奎宁(quinine)[18]、胆固醇(cholesterol)[19]、可的松(cortisone)[20]、马钱子碱(strychnine)[21]、叶绿素α(chlorophyll α)[22]、头孢菌素C(cephalosporin C)[23]及红霉素A(erythromycin A)[24]等明星分子的全合成;并且在全合成维生素B12(vitamin B12)[25]实验过程中,发现电环化协同反应现象,与量子化学家Roald Hoffmann一起建立了分子轨道守恒(the conservation of molecular orbital symmetry)理论[26].另一位有机合成大师Elias J. Corey除了完成烟曲霉素(fumagillin)[27]、红霉内酯B(erythronolide B)[28]、秋水仙碱(colchicine)[29]、喜树碱(camptothecin)[30]、前列腺素F(prostaglandin F)[31]、番红霉素A(saframycin A)[32]、抗疟霉素(aplasmomycin)[33]等众多天然产物的全合成之外,还提出了逆合成分析(retrosynthesis analysis)策略[34],即运用有机反应逻辑,对目标分子进行合理的解构,反推出起始原料和关键反应节点,指导设计出复杂分子的全合成路线. ...
The total synthesis of quinine
1
1944
... 进入20世纪,天然产物全合成领域涌现一批有机化学大师,他们设计发展了各种各样的化学试剂和有机转化反应,成功实现众多天然产物的化学全合成.例如20世纪初,德国化学家Richard Willstätter和英国化学家Robert Robinson采用不同的工艺路线,完成了托品酮(tropinone)的全合成[15];到了20年代,德国Hans Fischer教授实现了血红素分子(haemin)的全合成[16];30年代美国化学家Werner E. Bachmann对雌甾酮分子(equilenin)的全合成率先获得成功[17].在随后的40多年里,借助于新的分析检测技术,天然产物全合成更取得了突破性发展,例如美国有机合成大师Robert B. Woodward与其合作者,先后完成了奎宁(quinine)[18]、胆固醇(cholesterol)[19]、可的松(cortisone)[20]、马钱子碱(strychnine)[21]、叶绿素α(chlorophyll α)[22]、头孢菌素C(cephalosporin C)[23]及红霉素A(erythromycin A)[24]等明星分子的全合成;并且在全合成维生素B12(vitamin B12)[25]实验过程中,发现电环化协同反应现象,与量子化学家Roald Hoffmann一起建立了分子轨道守恒(the conservation of molecular orbital symmetry)理论[26].另一位有机合成大师Elias J. Corey除了完成烟曲霉素(fumagillin)[27]、红霉内酯B(erythronolide B)[28]、秋水仙碱(colchicine)[29]、喜树碱(camptothecin)[30]、前列腺素F(prostaglandin F)[31]、番红霉素A(saframycin A)[32]、抗疟霉素(aplasmomycin)[33]等众多天然产物的全合成之外,还提出了逆合成分析(retrosynthesis analysis)策略[34],即运用有机反应逻辑,对目标分子进行合理的解构,反推出起始原料和关键反应节点,指导设计出复杂分子的全合成路线. ...
The total synthesis of steroids
1
1952
... 进入20世纪,天然产物全合成领域涌现一批有机化学大师,他们设计发展了各种各样的化学试剂和有机转化反应,成功实现众多天然产物的化学全合成.例如20世纪初,德国化学家Richard Willstätter和英国化学家Robert Robinson采用不同的工艺路线,完成了托品酮(tropinone)的全合成[15];到了20年代,德国Hans Fischer教授实现了血红素分子(haemin)的全合成[16];30年代美国化学家Werner E. Bachmann对雌甾酮分子(equilenin)的全合成率先获得成功[17].在随后的40多年里,借助于新的分析检测技术,天然产物全合成更取得了突破性发展,例如美国有机合成大师Robert B. Woodward与其合作者,先后完成了奎宁(quinine)[18]、胆固醇(cholesterol)[19]、可的松(cortisone)[20]、马钱子碱(strychnine)[21]、叶绿素α(chlorophyll α)[22]、头孢菌素C(cephalosporin C)[23]及红霉素A(erythromycin A)[24]等明星分子的全合成;并且在全合成维生素B12(vitamin B12)[25]实验过程中,发现电环化协同反应现象,与量子化学家Roald Hoffmann一起建立了分子轨道守恒(the conservation of molecular orbital symmetry)理论[26].另一位有机合成大师Elias J. Corey除了完成烟曲霉素(fumagillin)[27]、红霉内酯B(erythronolide B)[28]、秋水仙碱(colchicine)[29]、喜树碱(camptothecin)[30]、前列腺素F(prostaglandin F)[31]、番红霉素A(saframycin A)[32]、抗疟霉素(aplasmomycin)[33]等众多天然产物的全合成之外,还提出了逆合成分析(retrosynthesis analysis)策略[34],即运用有机反应逻辑,对目标分子进行合理的解构,反推出起始原料和关键反应节点,指导设计出复杂分子的全合成路线. ...
The total synthesis of cortisone
1
1951
... 进入20世纪,天然产物全合成领域涌现一批有机化学大师,他们设计发展了各种各样的化学试剂和有机转化反应,成功实现众多天然产物的化学全合成.例如20世纪初,德国化学家Richard Willstätter和英国化学家Robert Robinson采用不同的工艺路线,完成了托品酮(tropinone)的全合成[15];到了20年代,德国Hans Fischer教授实现了血红素分子(haemin)的全合成[16];30年代美国化学家Werner E. Bachmann对雌甾酮分子(equilenin)的全合成率先获得成功[17].在随后的40多年里,借助于新的分析检测技术,天然产物全合成更取得了突破性发展,例如美国有机合成大师Robert B. Woodward与其合作者,先后完成了奎宁(quinine)[18]、胆固醇(cholesterol)[19]、可的松(cortisone)[20]、马钱子碱(strychnine)[21]、叶绿素α(chlorophyll α)[22]、头孢菌素C(cephalosporin C)[23]及红霉素A(erythromycin A)[24]等明星分子的全合成;并且在全合成维生素B12(vitamin B12)[25]实验过程中,发现电环化协同反应现象,与量子化学家Roald Hoffmann一起建立了分子轨道守恒(the conservation of molecular orbital symmetry)理论[26].另一位有机合成大师Elias J. Corey除了完成烟曲霉素(fumagillin)[27]、红霉内酯B(erythronolide B)[28]、秋水仙碱(colchicine)[29]、喜树碱(camptothecin)[30]、前列腺素F(prostaglandin F)[31]、番红霉素A(saframycin A)[32]、抗疟霉素(aplasmomycin)[33]等众多天然产物的全合成之外,还提出了逆合成分析(retrosynthesis analysis)策略[34],即运用有机反应逻辑,对目标分子进行合理的解构,反推出起始原料和关键反应节点,指导设计出复杂分子的全合成路线. ...
The total synthesis of strychnine
1
1954
... 进入20世纪,天然产物全合成领域涌现一批有机化学大师,他们设计发展了各种各样的化学试剂和有机转化反应,成功实现众多天然产物的化学全合成.例如20世纪初,德国化学家Richard Willstätter和英国化学家Robert Robinson采用不同的工艺路线,完成了托品酮(tropinone)的全合成[15];到了20年代,德国Hans Fischer教授实现了血红素分子(haemin)的全合成[16];30年代美国化学家Werner E. Bachmann对雌甾酮分子(equilenin)的全合成率先获得成功[17].在随后的40多年里,借助于新的分析检测技术,天然产物全合成更取得了突破性发展,例如美国有机合成大师Robert B. Woodward与其合作者,先后完成了奎宁(quinine)[18]、胆固醇(cholesterol)[19]、可的松(cortisone)[20]、马钱子碱(strychnine)[21]、叶绿素α(chlorophyll α)[22]、头孢菌素C(cephalosporin C)[23]及红霉素A(erythromycin A)[24]等明星分子的全合成;并且在全合成维生素B12(vitamin B12)[25]实验过程中,发现电环化协同反应现象,与量子化学家Roald Hoffmann一起建立了分子轨道守恒(the conservation of molecular orbital symmetry)理论[26].另一位有机合成大师Elias J. Corey除了完成烟曲霉素(fumagillin)[27]、红霉内酯B(erythronolide B)[28]、秋水仙碱(colchicine)[29]、喜树碱(camptothecin)[30]、前列腺素F(prostaglandin F)[31]、番红霉素A(saframycin A)[32]、抗疟霉素(aplasmomycin)[33]等众多天然产物的全合成之外,还提出了逆合成分析(retrosynthesis analysis)策略[34],即运用有机反应逻辑,对目标分子进行合理的解构,反推出起始原料和关键反应节点,指导设计出复杂分子的全合成路线. ...
The total synthesis of Chlorophyll
1
1960
... 进入20世纪,天然产物全合成领域涌现一批有机化学大师,他们设计发展了各种各样的化学试剂和有机转化反应,成功实现众多天然产物的化学全合成.例如20世纪初,德国化学家Richard Willstätter和英国化学家Robert Robinson采用不同的工艺路线,完成了托品酮(tropinone)的全合成[15];到了20年代,德国Hans Fischer教授实现了血红素分子(haemin)的全合成[16];30年代美国化学家Werner E. Bachmann对雌甾酮分子(equilenin)的全合成率先获得成功[17].在随后的40多年里,借助于新的分析检测技术,天然产物全合成更取得了突破性发展,例如美国有机合成大师Robert B. Woodward与其合作者,先后完成了奎宁(quinine)[18]、胆固醇(cholesterol)[19]、可的松(cortisone)[20]、马钱子碱(strychnine)[21]、叶绿素α(chlorophyll α)[22]、头孢菌素C(cephalosporin C)[23]及红霉素A(erythromycin A)[24]等明星分子的全合成;并且在全合成维生素B12(vitamin B12)[25]实验过程中,发现电环化协同反应现象,与量子化学家Roald Hoffmann一起建立了分子轨道守恒(the conservation of molecular orbital symmetry)理论[26].另一位有机合成大师Elias J. Corey除了完成烟曲霉素(fumagillin)[27]、红霉内酯B(erythronolide B)[28]、秋水仙碱(colchicine)[29]、喜树碱(camptothecin)[30]、前列腺素F(prostaglandin F)[31]、番红霉素A(saframycin A)[32]、抗疟霉素(aplasmomycin)[33]等众多天然产物的全合成之外,还提出了逆合成分析(retrosynthesis analysis)策略[34],即运用有机反应逻辑,对目标分子进行合理的解构,反推出起始原料和关键反应节点,指导设计出复杂分子的全合成路线. ...
The total synthesis of cephalosporin C
1
1966
... 进入20世纪,天然产物全合成领域涌现一批有机化学大师,他们设计发展了各种各样的化学试剂和有机转化反应,成功实现众多天然产物的化学全合成.例如20世纪初,德国化学家Richard Willstätter和英国化学家Robert Robinson采用不同的工艺路线,完成了托品酮(tropinone)的全合成[15];到了20年代,德国Hans Fischer教授实现了血红素分子(haemin)的全合成[16];30年代美国化学家Werner E. Bachmann对雌甾酮分子(equilenin)的全合成率先获得成功[17].在随后的40多年里,借助于新的分析检测技术,天然产物全合成更取得了突破性发展,例如美国有机合成大师Robert B. Woodward与其合作者,先后完成了奎宁(quinine)[18]、胆固醇(cholesterol)[19]、可的松(cortisone)[20]、马钱子碱(strychnine)[21]、叶绿素α(chlorophyll α)[22]、头孢菌素C(cephalosporin C)[23]及红霉素A(erythromycin A)[24]等明星分子的全合成;并且在全合成维生素B12(vitamin B12)[25]实验过程中,发现电环化协同反应现象,与量子化学家Roald Hoffmann一起建立了分子轨道守恒(the conservation of molecular orbital symmetry)理论[26].另一位有机合成大师Elias J. Corey除了完成烟曲霉素(fumagillin)[27]、红霉内酯B(erythronolide B)[28]、秋水仙碱(colchicine)[29]、喜树碱(camptothecin)[30]、前列腺素F(prostaglandin F)[31]、番红霉素A(saframycin A)[32]、抗疟霉素(aplasmomycin)[33]等众多天然产物的全合成之外,还提出了逆合成分析(retrosynthesis analysis)策略[34],即运用有机反应逻辑,对目标分子进行合理的解构,反推出起始原料和关键反应节点,指导设计出复杂分子的全合成路线. ...
Asymmetric total synthesis of erythromycin. 2. Synthesis of an erythronolide A lactone system
1
1981
... 进入20世纪,天然产物全合成领域涌现一批有机化学大师,他们设计发展了各种各样的化学试剂和有机转化反应,成功实现众多天然产物的化学全合成.例如20世纪初,德国化学家Richard Willstätter和英国化学家Robert Robinson采用不同的工艺路线,完成了托品酮(tropinone)的全合成[15];到了20年代,德国Hans Fischer教授实现了血红素分子(haemin)的全合成[16];30年代美国化学家Werner E. Bachmann对雌甾酮分子(equilenin)的全合成率先获得成功[17].在随后的40多年里,借助于新的分析检测技术,天然产物全合成更取得了突破性发展,例如美国有机合成大师Robert B. Woodward与其合作者,先后完成了奎宁(quinine)[18]、胆固醇(cholesterol)[19]、可的松(cortisone)[20]、马钱子碱(strychnine)[21]、叶绿素α(chlorophyll α)[22]、头孢菌素C(cephalosporin C)[23]及红霉素A(erythromycin A)[24]等明星分子的全合成;并且在全合成维生素B12(vitamin B12)[25]实验过程中,发现电环化协同反应现象,与量子化学家Roald Hoffmann一起建立了分子轨道守恒(the conservation of molecular orbital symmetry)理论[26].另一位有机合成大师Elias J. Corey除了完成烟曲霉素(fumagillin)[27]、红霉内酯B(erythronolide B)[28]、秋水仙碱(colchicine)[29]、喜树碱(camptothecin)[30]、前列腺素F(prostaglandin F)[31]、番红霉素A(saframycin A)[32]、抗疟霉素(aplasmomycin)[33]等众多天然产物的全合成之外,还提出了逆合成分析(retrosynthesis analysis)策略[34],即运用有机反应逻辑,对目标分子进行合理的解构,反推出起始原料和关键反应节点,指导设计出复杂分子的全合成路线. ...
Recent advances in the chemistry of natural products
1
1968
... 进入20世纪,天然产物全合成领域涌现一批有机化学大师,他们设计发展了各种各样的化学试剂和有机转化反应,成功实现众多天然产物的化学全合成.例如20世纪初,德国化学家Richard Willstätter和英国化学家Robert Robinson采用不同的工艺路线,完成了托品酮(tropinone)的全合成[15];到了20年代,德国Hans Fischer教授实现了血红素分子(haemin)的全合成[16];30年代美国化学家Werner E. Bachmann对雌甾酮分子(equilenin)的全合成率先获得成功[17].在随后的40多年里,借助于新的分析检测技术,天然产物全合成更取得了突破性发展,例如美国有机合成大师Robert B. Woodward与其合作者,先后完成了奎宁(quinine)[18]、胆固醇(cholesterol)[19]、可的松(cortisone)[20]、马钱子碱(strychnine)[21]、叶绿素α(chlorophyll α)[22]、头孢菌素C(cephalosporin C)[23]及红霉素A(erythromycin A)[24]等明星分子的全合成;并且在全合成维生素B12(vitamin B12)[25]实验过程中,发现电环化协同反应现象,与量子化学家Roald Hoffmann一起建立了分子轨道守恒(the conservation of molecular orbital symmetry)理论[26].另一位有机合成大师Elias J. Corey除了完成烟曲霉素(fumagillin)[27]、红霉内酯B(erythronolide B)[28]、秋水仙碱(colchicine)[29]、喜树碱(camptothecin)[30]、前列腺素F(prostaglandin F)[31]、番红霉素A(saframycin A)[32]、抗疟霉素(aplasmomycin)[33]等众多天然产物的全合成之外,还提出了逆合成分析(retrosynthesis analysis)策略[34],即运用有机反应逻辑,对目标分子进行合理的解构,反推出起始原料和关键反应节点,指导设计出复杂分子的全合成路线. ...
Structure of electrocyclic Reactions
1
1966
... 进入20世纪,天然产物全合成领域涌现一批有机化学大师,他们设计发展了各种各样的化学试剂和有机转化反应,成功实现众多天然产物的化学全合成.例如20世纪初,德国化学家Richard Willstätter和英国化学家Robert Robinson采用不同的工艺路线,完成了托品酮(tropinone)的全合成[15];到了20年代,德国Hans Fischer教授实现了血红素分子(haemin)的全合成[16];30年代美国化学家Werner E. Bachmann对雌甾酮分子(equilenin)的全合成率先获得成功[17].在随后的40多年里,借助于新的分析检测技术,天然产物全合成更取得了突破性发展,例如美国有机合成大师Robert B. Woodward与其合作者,先后完成了奎宁(quinine)[18]、胆固醇(cholesterol)[19]、可的松(cortisone)[20]、马钱子碱(strychnine)[21]、叶绿素α(chlorophyll α)[22]、头孢菌素C(cephalosporin C)[23]及红霉素A(erythromycin A)[24]等明星分子的全合成;并且在全合成维生素B12(vitamin B12)[25]实验过程中,发现电环化协同反应现象,与量子化学家Roald Hoffmann一起建立了分子轨道守恒(the conservation of molecular orbital symmetry)理论[26].另一位有机合成大师Elias J. Corey除了完成烟曲霉素(fumagillin)[27]、红霉内酯B(erythronolide B)[28]、秋水仙碱(colchicine)[29]、喜树碱(camptothecin)[30]、前列腺素F(prostaglandin F)[31]、番红霉素A(saframycin A)[32]、抗疟霉素(aplasmomycin)[33]等众多天然产物的全合成之外,还提出了逆合成分析(retrosynthesis analysis)策略[34],即运用有机反应逻辑,对目标分子进行合理的解构,反推出起始原料和关键反应节点,指导设计出复杂分子的全合成路线. ...
Total synthesis of (+ -)-fumagillin
1
1972
... 进入20世纪,天然产物全合成领域涌现一批有机化学大师,他们设计发展了各种各样的化学试剂和有机转化反应,成功实现众多天然产物的化学全合成.例如20世纪初,德国化学家Richard Willstätter和英国化学家Robert Robinson采用不同的工艺路线,完成了托品酮(tropinone)的全合成[15];到了20年代,德国Hans Fischer教授实现了血红素分子(haemin)的全合成[16];30年代美国化学家Werner E. Bachmann对雌甾酮分子(equilenin)的全合成率先获得成功[17].在随后的40多年里,借助于新的分析检测技术,天然产物全合成更取得了突破性发展,例如美国有机合成大师Robert B. Woodward与其合作者,先后完成了奎宁(quinine)[18]、胆固醇(cholesterol)[19]、可的松(cortisone)[20]、马钱子碱(strychnine)[21]、叶绿素α(chlorophyll α)[22]、头孢菌素C(cephalosporin C)[23]及红霉素A(erythromycin A)[24]等明星分子的全合成;并且在全合成维生素B12(vitamin B12)[25]实验过程中,发现电环化协同反应现象,与量子化学家Roald Hoffmann一起建立了分子轨道守恒(the conservation of molecular orbital symmetry)理论[26].另一位有机合成大师Elias J. Corey除了完成烟曲霉素(fumagillin)[27]、红霉内酯B(erythronolide B)[28]、秋水仙碱(colchicine)[29]、喜树碱(camptothecin)[30]、前列腺素F(prostaglandin F)[31]、番红霉素A(saframycin A)[32]、抗疟霉素(aplasmomycin)[33]等众多天然产物的全合成之外,还提出了逆合成分析(retrosynthesis analysis)策略[34],即运用有机反应逻辑,对目标分子进行合理的解构,反推出起始原料和关键反应节点,指导设计出复杂分子的全合成路线. ...
Total synthesis of erythromycins. 3. Stereoselective routes to intermediates corresponding to C(1) to C(9) and C(10) to C(13) fragments of erythronolide B
1
1978
... 进入20世纪,天然产物全合成领域涌现一批有机化学大师,他们设计发展了各种各样的化学试剂和有机转化反应,成功实现众多天然产物的化学全合成.例如20世纪初,德国化学家Richard Willstätter和英国化学家Robert Robinson采用不同的工艺路线,完成了托品酮(tropinone)的全合成[15];到了20年代,德国Hans Fischer教授实现了血红素分子(haemin)的全合成[16];30年代美国化学家Werner E. Bachmann对雌甾酮分子(equilenin)的全合成率先获得成功[17].在随后的40多年里,借助于新的分析检测技术,天然产物全合成更取得了突破性发展,例如美国有机合成大师Robert B. Woodward与其合作者,先后完成了奎宁(quinine)[18]、胆固醇(cholesterol)[19]、可的松(cortisone)[20]、马钱子碱(strychnine)[21]、叶绿素α(chlorophyll α)[22]、头孢菌素C(cephalosporin C)[23]及红霉素A(erythromycin A)[24]等明星分子的全合成;并且在全合成维生素B12(vitamin B12)[25]实验过程中,发现电环化协同反应现象,与量子化学家Roald Hoffmann一起建立了分子轨道守恒(the conservation of molecular orbital symmetry)理论[26].另一位有机合成大师Elias J. Corey除了完成烟曲霉素(fumagillin)[27]、红霉内酯B(erythronolide B)[28]、秋水仙碱(colchicine)[29]、喜树碱(camptothecin)[30]、前列腺素F(prostaglandin F)[31]、番红霉素A(saframycin A)[32]、抗疟霉素(aplasmomycin)[33]等众多天然产物的全合成之外,还提出了逆合成分析(retrosynthesis analysis)策略[34],即运用有机反应逻辑,对目标分子进行合理的解构,反推出起始原料和关键反应节点,指导设计出复杂分子的全合成路线. ...
The total synthesis of colchicine
1
1959
... 进入20世纪,天然产物全合成领域涌现一批有机化学大师,他们设计发展了各种各样的化学试剂和有机转化反应,成功实现众多天然产物的化学全合成.例如20世纪初,德国化学家Richard Willstätter和英国化学家Robert Robinson采用不同的工艺路线,完成了托品酮(tropinone)的全合成[15];到了20年代,德国Hans Fischer教授实现了血红素分子(haemin)的全合成[16];30年代美国化学家Werner E. Bachmann对雌甾酮分子(equilenin)的全合成率先获得成功[17].在随后的40多年里,借助于新的分析检测技术,天然产物全合成更取得了突破性发展,例如美国有机合成大师Robert B. Woodward与其合作者,先后完成了奎宁(quinine)[18]、胆固醇(cholesterol)[19]、可的松(cortisone)[20]、马钱子碱(strychnine)[21]、叶绿素α(chlorophyll α)[22]、头孢菌素C(cephalosporin C)[23]及红霉素A(erythromycin A)[24]等明星分子的全合成;并且在全合成维生素B12(vitamin B12)[25]实验过程中,发现电环化协同反应现象,与量子化学家Roald Hoffmann一起建立了分子轨道守恒(the conservation of molecular orbital symmetry)理论[26].另一位有机合成大师Elias J. Corey除了完成烟曲霉素(fumagillin)[27]、红霉内酯B(erythronolide B)[28]、秋水仙碱(colchicine)[29]、喜树碱(camptothecin)[30]、前列腺素F(prostaglandin F)[31]、番红霉素A(saframycin A)[32]、抗疟霉素(aplasmomycin)[33]等众多天然产物的全合成之外,还提出了逆合成分析(retrosynthesis analysis)策略[34],即运用有机反应逻辑,对目标分子进行合理的解构,反推出起始原料和关键反应节点,指导设计出复杂分子的全合成路线. ...
Total synthesis of natural 20(S)-camptothecin
1
1975
... 进入20世纪,天然产物全合成领域涌现一批有机化学大师,他们设计发展了各种各样的化学试剂和有机转化反应,成功实现众多天然产物的化学全合成.例如20世纪初,德国化学家Richard Willstätter和英国化学家Robert Robinson采用不同的工艺路线,完成了托品酮(tropinone)的全合成[15];到了20年代,德国Hans Fischer教授实现了血红素分子(haemin)的全合成[16];30年代美国化学家Werner E. Bachmann对雌甾酮分子(equilenin)的全合成率先获得成功[17].在随后的40多年里,借助于新的分析检测技术,天然产物全合成更取得了突破性发展,例如美国有机合成大师Robert B. Woodward与其合作者,先后完成了奎宁(quinine)[18]、胆固醇(cholesterol)[19]、可的松(cortisone)[20]、马钱子碱(strychnine)[21]、叶绿素α(chlorophyll α)[22]、头孢菌素C(cephalosporin C)[23]及红霉素A(erythromycin A)[24]等明星分子的全合成;并且在全合成维生素B12(vitamin B12)[25]实验过程中,发现电环化协同反应现象,与量子化学家Roald Hoffmann一起建立了分子轨道守恒(the conservation of molecular orbital symmetry)理论[26].另一位有机合成大师Elias J. Corey除了完成烟曲霉素(fumagillin)[27]、红霉内酯B(erythronolide B)[28]、秋水仙碱(colchicine)[29]、喜树碱(camptothecin)[30]、前列腺素F(prostaglandin F)[31]、番红霉素A(saframycin A)[32]、抗疟霉素(aplasmomycin)[33]等众多天然产物的全合成之外,还提出了逆合成分析(retrosynthesis analysis)策略[34],即运用有机反应逻辑,对目标分子进行合理的解构,反推出起始原料和关键反应节点,指导设计出复杂分子的全合成路线. ...
Total synthesis of Prostaglandins F2α and E2 as the naturally occuring forms
1
1970
... 进入20世纪,天然产物全合成领域涌现一批有机化学大师,他们设计发展了各种各样的化学试剂和有机转化反应,成功实现众多天然产物的化学全合成.例如20世纪初,德国化学家Richard Willstätter和英国化学家Robert Robinson采用不同的工艺路线,完成了托品酮(tropinone)的全合成[15];到了20年代,德国Hans Fischer教授实现了血红素分子(haemin)的全合成[16];30年代美国化学家Werner E. Bachmann对雌甾酮分子(equilenin)的全合成率先获得成功[17].在随后的40多年里,借助于新的分析检测技术,天然产物全合成更取得了突破性发展,例如美国有机合成大师Robert B. Woodward与其合作者,先后完成了奎宁(quinine)[18]、胆固醇(cholesterol)[19]、可的松(cortisone)[20]、马钱子碱(strychnine)[21]、叶绿素α(chlorophyll α)[22]、头孢菌素C(cephalosporin C)[23]及红霉素A(erythromycin A)[24]等明星分子的全合成;并且在全合成维生素B12(vitamin B12)[25]实验过程中,发现电环化协同反应现象,与量子化学家Roald Hoffmann一起建立了分子轨道守恒(the conservation of molecular orbital symmetry)理论[26].另一位有机合成大师Elias J. Corey除了完成烟曲霉素(fumagillin)[27]、红霉内酯B(erythronolide B)[28]、秋水仙碱(colchicine)[29]、喜树碱(camptothecin)[30]、前列腺素F(prostaglandin F)[31]、番红霉素A(saframycin A)[32]、抗疟霉素(aplasmomycin)[33]等众多天然产物的全合成之外,还提出了逆合成分析(retrosynthesis analysis)策略[34],即运用有机反应逻辑,对目标分子进行合理的解构,反推出起始原料和关键反应节点,指导设计出复杂分子的全合成路线. ...
Total synthesis of (.+ -.)-saframycin A
1
1990
... 进入20世纪,天然产物全合成领域涌现一批有机化学大师,他们设计发展了各种各样的化学试剂和有机转化反应,成功实现众多天然产物的化学全合成.例如20世纪初,德国化学家Richard Willstätter和英国化学家Robert Robinson采用不同的工艺路线,完成了托品酮(tropinone)的全合成[15];到了20年代,德国Hans Fischer教授实现了血红素分子(haemin)的全合成[16];30年代美国化学家Werner E. Bachmann对雌甾酮分子(equilenin)的全合成率先获得成功[17].在随后的40多年里,借助于新的分析检测技术,天然产物全合成更取得了突破性发展,例如美国有机合成大师Robert B. Woodward与其合作者,先后完成了奎宁(quinine)[18]、胆固醇(cholesterol)[19]、可的松(cortisone)[20]、马钱子碱(strychnine)[21]、叶绿素α(chlorophyll α)[22]、头孢菌素C(cephalosporin C)[23]及红霉素A(erythromycin A)[24]等明星分子的全合成;并且在全合成维生素B12(vitamin B12)[25]实验过程中,发现电环化协同反应现象,与量子化学家Roald Hoffmann一起建立了分子轨道守恒(the conservation of molecular orbital symmetry)理论[26].另一位有机合成大师Elias J. Corey除了完成烟曲霉素(fumagillin)[27]、红霉内酯B(erythronolide B)[28]、秋水仙碱(colchicine)[29]、喜树碱(camptothecin)[30]、前列腺素F(prostaglandin F)[31]、番红霉素A(saframycin A)[32]、抗疟霉素(aplasmomycin)[33]等众多天然产物的全合成之外,还提出了逆合成分析(retrosynthesis analysis)策略[34],即运用有机反应逻辑,对目标分子进行合理的解构,反推出起始原料和关键反应节点,指导设计出复杂分子的全合成路线. ...
Total synthesis of aplasmomycin
1
1982
... 进入20世纪,天然产物全合成领域涌现一批有机化学大师,他们设计发展了各种各样的化学试剂和有机转化反应,成功实现众多天然产物的化学全合成.例如20世纪初,德国化学家Richard Willstätter和英国化学家Robert Robinson采用不同的工艺路线,完成了托品酮(tropinone)的全合成[15];到了20年代,德国Hans Fischer教授实现了血红素分子(haemin)的全合成[16];30年代美国化学家Werner E. Bachmann对雌甾酮分子(equilenin)的全合成率先获得成功[17].在随后的40多年里,借助于新的分析检测技术,天然产物全合成更取得了突破性发展,例如美国有机合成大师Robert B. Woodward与其合作者,先后完成了奎宁(quinine)[18]、胆固醇(cholesterol)[19]、可的松(cortisone)[20]、马钱子碱(strychnine)[21]、叶绿素α(chlorophyll α)[22]、头孢菌素C(cephalosporin C)[23]及红霉素A(erythromycin A)[24]等明星分子的全合成;并且在全合成维生素B12(vitamin B12)[25]实验过程中,发现电环化协同反应现象,与量子化学家Roald Hoffmann一起建立了分子轨道守恒(the conservation of molecular orbital symmetry)理论[26].另一位有机合成大师Elias J. Corey除了完成烟曲霉素(fumagillin)[27]、红霉内酯B(erythronolide B)[28]、秋水仙碱(colchicine)[29]、喜树碱(camptothecin)[30]、前列腺素F(prostaglandin F)[31]、番红霉素A(saframycin A)[32]、抗疟霉素(aplasmomycin)[33]等众多天然产物的全合成之外,还提出了逆合成分析(retrosynthesis analysis)策略[34],即运用有机反应逻辑,对目标分子进行合理的解构,反推出起始原料和关键反应节点,指导设计出复杂分子的全合成路线. ...
1
1989
... 进入20世纪,天然产物全合成领域涌现一批有机化学大师,他们设计发展了各种各样的化学试剂和有机转化反应,成功实现众多天然产物的化学全合成.例如20世纪初,德国化学家Richard Willstätter和英国化学家Robert Robinson采用不同的工艺路线,完成了托品酮(tropinone)的全合成[15];到了20年代,德国Hans Fischer教授实现了血红素分子(haemin)的全合成[16];30年代美国化学家Werner E. Bachmann对雌甾酮分子(equilenin)的全合成率先获得成功[17].在随后的40多年里,借助于新的分析检测技术,天然产物全合成更取得了突破性发展,例如美国有机合成大师Robert B. Woodward与其合作者,先后完成了奎宁(quinine)[18]、胆固醇(cholesterol)[19]、可的松(cortisone)[20]、马钱子碱(strychnine)[21]、叶绿素α(chlorophyll α)[22]、头孢菌素C(cephalosporin C)[23]及红霉素A(erythromycin A)[24]等明星分子的全合成;并且在全合成维生素B12(vitamin B12)[25]实验过程中,发现电环化协同反应现象,与量子化学家Roald Hoffmann一起建立了分子轨道守恒(the conservation of molecular orbital symmetry)理论[26].另一位有机合成大师Elias J. Corey除了完成烟曲霉素(fumagillin)[27]、红霉内酯B(erythronolide B)[28]、秋水仙碱(colchicine)[29]、喜树碱(camptothecin)[30]、前列腺素F(prostaglandin F)[31]、番红霉素A(saframycin A)[32]、抗疟霉素(aplasmomycin)[33]等众多天然产物的全合成之外,还提出了逆合成分析(retrosynthesis analysis)策略[34],即运用有机反应逻辑,对目标分子进行合理的解构,反推出起始原料和关键反应节点,指导设计出复杂分子的全合成路线. ...
Total synthesis of crystalline bovine insulin
1
1965
... 在这期间我国化学家的全合成工作同样引人瞩目:1965年中国科学院上海生物化学研究所和上海有机化学研究所与北京大学化学系历时7年完成了牛胰岛素(insulin bovine)的全合成,获得晶体并确定了其分子结构,这是世界上首次以氨基酸为原料、通过化学反应人工制备出蛋白质;经过生物活性测试,合成的结晶牛胰岛素和天然牛胰岛素具有同样的生物活性[35-37].1981年中国科学院的4个研究所(上海生物化学研究所、上海细胞生物学研究所、上海有机化学研究所、生物物理研究所)和北京大学等单位,利用化学和酶促相结合的方法,首次人工合成了76个核苷酸的整分子酵母丙氨酸转移核糖核酸(yeast alanine transfer RNA),其结构与天然分子完全相同,并具有较高的丙氨酸接受和转移活性[38-39].1983—1984年中国科学院上海有机化学研究所周维善院士团队以手性香茅醛为原料,经过20步有机反应,实现了抗疟药物青蒿素(artemisinin)分子的全合成[40-41]. ...
Total synthesis of crystalline insulin
0
1966
Total synthesis of insulin in red China
1
1966
... 在这期间我国化学家的全合成工作同样引人瞩目:1965年中国科学院上海生物化学研究所和上海有机化学研究所与北京大学化学系历时7年完成了牛胰岛素(insulin bovine)的全合成,获得晶体并确定了其分子结构,这是世界上首次以氨基酸为原料、通过化学反应人工制备出蛋白质;经过生物活性测试,合成的结晶牛胰岛素和天然牛胰岛素具有同样的生物活性[35-37].1981年中国科学院的4个研究所(上海生物化学研究所、上海细胞生物学研究所、上海有机化学研究所、生物物理研究所)和北京大学等单位,利用化学和酶促相结合的方法,首次人工合成了76个核苷酸的整分子酵母丙氨酸转移核糖核酸(yeast alanine transfer RNA),其结构与天然分子完全相同,并具有较高的丙氨酸接受和转移活性[38-39].1983—1984年中国科学院上海有机化学研究所周维善院士团队以手性香茅醛为原料,经过20步有机反应,实现了抗疟药物青蒿素(artemisinin)分子的全合成[40-41]. ...
酵母丙氨酸转移核糖核酸的全合成
1
1982
... 在这期间我国化学家的全合成工作同样引人瞩目:1965年中国科学院上海生物化学研究所和上海有机化学研究所与北京大学化学系历时7年完成了牛胰岛素(insulin bovine)的全合成,获得晶体并确定了其分子结构,这是世界上首次以氨基酸为原料、通过化学反应人工制备出蛋白质;经过生物活性测试,合成的结晶牛胰岛素和天然牛胰岛素具有同样的生物活性[35-37].1981年中国科学院的4个研究所(上海生物化学研究所、上海细胞生物学研究所、上海有机化学研究所、生物物理研究所)和北京大学等单位,利用化学和酶促相结合的方法,首次人工合成了76个核苷酸的整分子酵母丙氨酸转移核糖核酸(yeast alanine transfer RNA),其结构与天然分子完全相同,并具有较高的丙氨酸接受和转移活性[38-39].1983—1984年中国科学院上海有机化学研究所周维善院士团队以手性香茅醛为原料,经过20步有机反应,实现了抗疟药物青蒿素(artemisinin)分子的全合成[40-41]. ...
The total synthesis of yeast alanine transfer RNA
1
1982
... 在这期间我国化学家的全合成工作同样引人瞩目:1965年中国科学院上海生物化学研究所和上海有机化学研究所与北京大学化学系历时7年完成了牛胰岛素(insulin bovine)的全合成,获得晶体并确定了其分子结构,这是世界上首次以氨基酸为原料、通过化学反应人工制备出蛋白质;经过生物活性测试,合成的结晶牛胰岛素和天然牛胰岛素具有同样的生物活性[35-37].1981年中国科学院的4个研究所(上海生物化学研究所、上海细胞生物学研究所、上海有机化学研究所、生物物理研究所)和北京大学等单位,利用化学和酶促相结合的方法,首次人工合成了76个核苷酸的整分子酵母丙氨酸转移核糖核酸(yeast alanine transfer RNA),其结构与天然分子完全相同,并具有较高的丙氨酸接受和转移活性[38-39].1983—1984年中国科学院上海有机化学研究所周维善院士团队以手性香茅醛为原料,经过20步有机反应,实现了抗疟药物青蒿素(artemisinin)分子的全合成[40-41]. ...
酵母丙氨酸转移核糖核酸(酵母丙氨酸t-RNA)人工全合成
1
1983
... 在这期间我国化学家的全合成工作同样引人瞩目:1965年中国科学院上海生物化学研究所和上海有机化学研究所与北京大学化学系历时7年完成了牛胰岛素(insulin bovine)的全合成,获得晶体并确定了其分子结构,这是世界上首次以氨基酸为原料、通过化学反应人工制备出蛋白质;经过生物活性测试,合成的结晶牛胰岛素和天然牛胰岛素具有同样的生物活性[35-37].1981年中国科学院的4个研究所(上海生物化学研究所、上海细胞生物学研究所、上海有机化学研究所、生物物理研究所)和北京大学等单位,利用化学和酶促相结合的方法,首次人工合成了76个核苷酸的整分子酵母丙氨酸转移核糖核酸(yeast alanine transfer RNA),其结构与天然分子完全相同,并具有较高的丙氨酸接受和转移活性[38-39].1983—1984年中国科学院上海有机化学研究所周维善院士团队以手性香茅醛为原料,经过20步有机反应,实现了抗疟药物青蒿素(artemisinin)分子的全合成[40-41]. ...
The man-total synthesis of yeast alanine transfer RNA
1
1983
... 在这期间我国化学家的全合成工作同样引人瞩目:1965年中国科学院上海生物化学研究所和上海有机化学研究所与北京大学化学系历时7年完成了牛胰岛素(insulin bovine)的全合成,获得晶体并确定了其分子结构,这是世界上首次以氨基酸为原料、通过化学反应人工制备出蛋白质;经过生物活性测试,合成的结晶牛胰岛素和天然牛胰岛素具有同样的生物活性[35-37].1981年中国科学院的4个研究所(上海生物化学研究所、上海细胞生物学研究所、上海有机化学研究所、生物物理研究所)和北京大学等单位,利用化学和酶促相结合的方法,首次人工合成了76个核苷酸的整分子酵母丙氨酸转移核糖核酸(yeast alanine transfer RNA),其结构与天然分子完全相同,并具有较高的丙氨酸接受和转移活性[38-39].1983—1984年中国科学院上海有机化学研究所周维善院士团队以手性香茅醛为原料,经过20步有机反应,实现了抗疟药物青蒿素(artemisinin)分子的全合成[40-41]. ...
青蒿素及其一类物结构和合成的研究X: 从青蒿酸立体控制合成青蒿素和脱氧青蒿素
1
1983
... 在这期间我国化学家的全合成工作同样引人瞩目:1965年中国科学院上海生物化学研究所和上海有机化学研究所与北京大学化学系历时7年完成了牛胰岛素(insulin bovine)的全合成,获得晶体并确定了其分子结构,这是世界上首次以氨基酸为原料、通过化学反应人工制备出蛋白质;经过生物活性测试,合成的结晶牛胰岛素和天然牛胰岛素具有同样的生物活性[35-37].1981年中国科学院的4个研究所(上海生物化学研究所、上海细胞生物学研究所、上海有机化学研究所、生物物理研究所)和北京大学等单位,利用化学和酶促相结合的方法,首次人工合成了76个核苷酸的整分子酵母丙氨酸转移核糖核酸(yeast alanine transfer RNA),其结构与天然分子完全相同,并具有较高的丙氨酸接受和转移活性[38-39].1983—1984年中国科学院上海有机化学研究所周维善院士团队以手性香茅醛为原料,经过20步有机反应,实现了抗疟药物青蒿素(artemisinin)分子的全合成[40-41]. ...
Studies on structure and syntheses of arteannuin and related compound XVII: The stereocontrolled synthesis of arteannuin and deoxyarteannuin from arteannuic acid
1
1983
... 在这期间我国化学家的全合成工作同样引人瞩目:1965年中国科学院上海生物化学研究所和上海有机化学研究所与北京大学化学系历时7年完成了牛胰岛素(insulin bovine)的全合成,获得晶体并确定了其分子结构,这是世界上首次以氨基酸为原料、通过化学反应人工制备出蛋白质;经过生物活性测试,合成的结晶牛胰岛素和天然牛胰岛素具有同样的生物活性[35-37].1981年中国科学院的4个研究所(上海生物化学研究所、上海细胞生物学研究所、上海有机化学研究所、生物物理研究所)和北京大学等单位,利用化学和酶促相结合的方法,首次人工合成了76个核苷酸的整分子酵母丙氨酸转移核糖核酸(yeast alanine transfer RNA),其结构与天然分子完全相同,并具有较高的丙氨酸接受和转移活性[38-39].1983—1984年中国科学院上海有机化学研究所周维善院士团队以手性香茅醛为原料,经过20步有机反应,实现了抗疟药物青蒿素(artemisinin)分子的全合成[40-41]. ...
青蒿素及其一类物结构和合成的研究ⅩⅦ: 双氢青蒿酸甲酯的立体控制性全合成——青蒿素全合成
1
1984
... 在这期间我国化学家的全合成工作同样引人瞩目:1965年中国科学院上海生物化学研究所和上海有机化学研究所与北京大学化学系历时7年完成了牛胰岛素(insulin bovine)的全合成,获得晶体并确定了其分子结构,这是世界上首次以氨基酸为原料、通过化学反应人工制备出蛋白质;经过生物活性测试,合成的结晶牛胰岛素和天然牛胰岛素具有同样的生物活性[35-37].1981年中国科学院的4个研究所(上海生物化学研究所、上海细胞生物学研究所、上海有机化学研究所、生物物理研究所)和北京大学等单位,利用化学和酶促相结合的方法,首次人工合成了76个核苷酸的整分子酵母丙氨酸转移核糖核酸(yeast alanine transfer RNA),其结构与天然分子完全相同,并具有较高的丙氨酸接受和转移活性[38-39].1983—1984年中国科学院上海有机化学研究所周维善院士团队以手性香茅醛为原料,经过20步有机反应,实现了抗疟药物青蒿素(artemisinin)分子的全合成[40-41]. ...
Studies on structure and syntheses of arteannuin and related compound ⅩⅦ: The stereocontrolled total synthesis of methyl dihydroarteannuate─the total synthesis of arteannuin
1
1984
... 在这期间我国化学家的全合成工作同样引人瞩目:1965年中国科学院上海生物化学研究所和上海有机化学研究所与北京大学化学系历时7年完成了牛胰岛素(insulin bovine)的全合成,获得晶体并确定了其分子结构,这是世界上首次以氨基酸为原料、通过化学反应人工制备出蛋白质;经过生物活性测试,合成的结晶牛胰岛素和天然牛胰岛素具有同样的生物活性[35-37].1981年中国科学院的4个研究所(上海生物化学研究所、上海细胞生物学研究所、上海有机化学研究所、生物物理研究所)和北京大学等单位,利用化学和酶促相结合的方法,首次人工合成了76个核苷酸的整分子酵母丙氨酸转移核糖核酸(yeast alanine transfer RNA),其结构与天然分子完全相同,并具有较高的丙氨酸接受和转移活性[38-39].1983—1984年中国科学院上海有机化学研究所周维善院士团队以手性香茅醛为原料,经过20步有机反应,实现了抗疟药物青蒿素(artemisinin)分子的全合成[40-41]. ...
Total synthesis of taxol
1
1994
... 20世纪90年代至今,有机化学家进一步完成了许多高药物活性天然产物的全合成.例如1994年美国化学家Kyriacos Nicolau和Robert Holton两个团队同年攻克抗癌明星药物紫杉醇分子的全合成(图3)[42-48],其中Robert Holton以天然氧化绿叶烯为原料,采用线性策略,经过41步反应,以不到3%总收率得到紫杉醇;除此之外,化学家采用其他原料和策略还发展了多条不同的全合成路线,但都因为合成步骤太多,工艺路线过长,反应条件苛刻,总收率均低于1%,而无法实现规模化放大生产.1981年法国科学家Pierre Potier从紫杉茎叶中分离得到一种高含量化合物——10-脱乙酰基巴卡丁-Ⅲ(10-DAB),具有与紫杉醇相似的四并环骨架.随后开发出四步合成转化法,将10-脱乙酰基巴卡丁-Ⅲ以80%总收率转化为紫杉醇,并在美国施贵宝公司实现了工业化生产.1999年Kyriacos Nicolau、David Evans和Dale Boger三个课题组几乎同时报道了万古霉素的全合成工作[49-51];进入21世纪之后,英国剑桥大学Steven Ley和其合作者率先报道了雷帕霉素(rapamycin)的全合成[52];美籍日裔化学家Yoshito Kishi先后完成了海洋天然产物海葵毒素(palytoxin)[53]、河豚毒素(tetrodotoxin)[54]、软海绵素B(halichondrin B)[55]等的全合成;2006年日本卫材(Eisa)公司化学家基于软海绵素B结构改造,经62步有机反应合成出了含有19个碳手性中心的抗癌药物——甲磺酸艾日布林(eribulin),该药物在2010年获美国FDA批准,是第1个衍生自天然产物而用于转移性乳腺癌患者获得总生存期改善的化疗单药[56-58]. ...
First total synthesis of taxol: 1. Functionalization of the B ring
0
1994
First total synthesis of taxol: 2. Completion of the C and D rings
0
1994
Structure and biochemistry of products isolated from Taxus baccata
0
1981
Highly efficient, practical approach to natural taxol
0
1988
Taxol and taxotere: discovery, chemistry, and structure-activity relationships
0
1993
A new semisynthesis of paclitaxel from baccatinⅢ
1
1999
... 20世纪90年代至今,有机化学家进一步完成了许多高药物活性天然产物的全合成.例如1994年美国化学家Kyriacos Nicolau和Robert Holton两个团队同年攻克抗癌明星药物紫杉醇分子的全合成(图3)[42-48],其中Robert Holton以天然氧化绿叶烯为原料,采用线性策略,经过41步反应,以不到3%总收率得到紫杉醇;除此之外,化学家采用其他原料和策略还发展了多条不同的全合成路线,但都因为合成步骤太多,工艺路线过长,反应条件苛刻,总收率均低于1%,而无法实现规模化放大生产.1981年法国科学家Pierre Potier从紫杉茎叶中分离得到一种高含量化合物——10-脱乙酰基巴卡丁-Ⅲ(10-DAB),具有与紫杉醇相似的四并环骨架.随后开发出四步合成转化法,将10-脱乙酰基巴卡丁-Ⅲ以80%总收率转化为紫杉醇,并在美国施贵宝公司实现了工业化生产.1999年Kyriacos Nicolau、David Evans和Dale Boger三个课题组几乎同时报道了万古霉素的全合成工作[49-51];进入21世纪之后,英国剑桥大学Steven Ley和其合作者率先报道了雷帕霉素(rapamycin)的全合成[52];美籍日裔化学家Yoshito Kishi先后完成了海洋天然产物海葵毒素(palytoxin)[53]、河豚毒素(tetrodotoxin)[54]、软海绵素B(halichondrin B)[55]等的全合成;2006年日本卫材(Eisa)公司化学家基于软海绵素B结构改造,经62步有机反应合成出了含有19个碳手性中心的抗癌药物——甲磺酸艾日布林(eribulin),该药物在2010年获美国FDA批准,是第1个衍生自天然产物而用于转移性乳腺癌患者获得总生存期改善的化疗单药[56-58]. ...
Total synthesis of vancomycin
1
1999
... 20世纪90年代至今,有机化学家进一步完成了许多高药物活性天然产物的全合成.例如1994年美国化学家Kyriacos Nicolau和Robert Holton两个团队同年攻克抗癌明星药物紫杉醇分子的全合成(图3)[42-48],其中Robert Holton以天然氧化绿叶烯为原料,采用线性策略,经过41步反应,以不到3%总收率得到紫杉醇;除此之外,化学家采用其他原料和策略还发展了多条不同的全合成路线,但都因为合成步骤太多,工艺路线过长,反应条件苛刻,总收率均低于1%,而无法实现规模化放大生产.1981年法国科学家Pierre Potier从紫杉茎叶中分离得到一种高含量化合物——10-脱乙酰基巴卡丁-Ⅲ(10-DAB),具有与紫杉醇相似的四并环骨架.随后开发出四步合成转化法,将10-脱乙酰基巴卡丁-Ⅲ以80%总收率转化为紫杉醇,并在美国施贵宝公司实现了工业化生产.1999年Kyriacos Nicolau、David Evans和Dale Boger三个课题组几乎同时报道了万古霉素的全合成工作[49-51];进入21世纪之后,英国剑桥大学Steven Ley和其合作者率先报道了雷帕霉素(rapamycin)的全合成[52];美籍日裔化学家Yoshito Kishi先后完成了海洋天然产物海葵毒素(palytoxin)[53]、河豚毒素(tetrodotoxin)[54]、软海绵素B(halichondrin B)[55]等的全合成;2006年日本卫材(Eisa)公司化学家基于软海绵素B结构改造,经62步有机反应合成出了含有19个碳手性中心的抗癌药物——甲磺酸艾日布林(eribulin),该药物在2010年获美国FDA批准,是第1个衍生自天然产物而用于转移性乳腺癌患者获得总生存期改善的化疗单药[56-58]. ...
Total syntheses of vancomycin and eremomycin aglycons
0
1998
Total synthesis of the vancomycin aglycon
1
1999
... 20世纪90年代至今,有机化学家进一步完成了许多高药物活性天然产物的全合成.例如1994年美国化学家Kyriacos Nicolau和Robert Holton两个团队同年攻克抗癌明星药物紫杉醇分子的全合成(图3)[42-48],其中Robert Holton以天然氧化绿叶烯为原料,采用线性策略,经过41步反应,以不到3%总收率得到紫杉醇;除此之外,化学家采用其他原料和策略还发展了多条不同的全合成路线,但都因为合成步骤太多,工艺路线过长,反应条件苛刻,总收率均低于1%,而无法实现规模化放大生产.1981年法国科学家Pierre Potier从紫杉茎叶中分离得到一种高含量化合物——10-脱乙酰基巴卡丁-Ⅲ(10-DAB),具有与紫杉醇相似的四并环骨架.随后开发出四步合成转化法,将10-脱乙酰基巴卡丁-Ⅲ以80%总收率转化为紫杉醇,并在美国施贵宝公司实现了工业化生产.1999年Kyriacos Nicolau、David Evans和Dale Boger三个课题组几乎同时报道了万古霉素的全合成工作[49-51];进入21世纪之后,英国剑桥大学Steven Ley和其合作者率先报道了雷帕霉素(rapamycin)的全合成[52];美籍日裔化学家Yoshito Kishi先后完成了海洋天然产物海葵毒素(palytoxin)[53]、河豚毒素(tetrodotoxin)[54]、软海绵素B(halichondrin B)[55]等的全合成;2006年日本卫材(Eisa)公司化学家基于软海绵素B结构改造,经62步有机反应合成出了含有19个碳手性中心的抗癌药物——甲磺酸艾日布林(eribulin),该药物在2010年获美国FDA批准,是第1个衍生自天然产物而用于转移性乳腺癌患者获得总生存期改善的化疗单药[56-58]. ...
Total synthesis of rapamycin
1
2007
... 20世纪90年代至今,有机化学家进一步完成了许多高药物活性天然产物的全合成.例如1994年美国化学家Kyriacos Nicolau和Robert Holton两个团队同年攻克抗癌明星药物紫杉醇分子的全合成(图3)[42-48],其中Robert Holton以天然氧化绿叶烯为原料,采用线性策略,经过41步反应,以不到3%总收率得到紫杉醇;除此之外,化学家采用其他原料和策略还发展了多条不同的全合成路线,但都因为合成步骤太多,工艺路线过长,反应条件苛刻,总收率均低于1%,而无法实现规模化放大生产.1981年法国科学家Pierre Potier从紫杉茎叶中分离得到一种高含量化合物——10-脱乙酰基巴卡丁-Ⅲ(10-DAB),具有与紫杉醇相似的四并环骨架.随后开发出四步合成转化法,将10-脱乙酰基巴卡丁-Ⅲ以80%总收率转化为紫杉醇,并在美国施贵宝公司实现了工业化生产.1999年Kyriacos Nicolau、David Evans和Dale Boger三个课题组几乎同时报道了万古霉素的全合成工作[49-51];进入21世纪之后,英国剑桥大学Steven Ley和其合作者率先报道了雷帕霉素(rapamycin)的全合成[52];美籍日裔化学家Yoshito Kishi先后完成了海洋天然产物海葵毒素(palytoxin)[53]、河豚毒素(tetrodotoxin)[54]、软海绵素B(halichondrin B)[55]等的全合成;2006年日本卫材(Eisa)公司化学家基于软海绵素B结构改造,经62步有机反应合成出了含有19个碳手性中心的抗癌药物——甲磺酸艾日布林(eribulin),该药物在2010年获美国FDA批准,是第1个衍生自天然产物而用于转移性乳腺癌患者获得总生存期改善的化疗单药[56-58]. ...
Synthesis of palytoxin from palytoxin carboxylic acid
1
1994
... 20世纪90年代至今,有机化学家进一步完成了许多高药物活性天然产物的全合成.例如1994年美国化学家Kyriacos Nicolau和Robert Holton两个团队同年攻克抗癌明星药物紫杉醇分子的全合成(图3)[42-48],其中Robert Holton以天然氧化绿叶烯为原料,采用线性策略,经过41步反应,以不到3%总收率得到紫杉醇;除此之外,化学家采用其他原料和策略还发展了多条不同的全合成路线,但都因为合成步骤太多,工艺路线过长,反应条件苛刻,总收率均低于1%,而无法实现规模化放大生产.1981年法国科学家Pierre Potier从紫杉茎叶中分离得到一种高含量化合物——10-脱乙酰基巴卡丁-Ⅲ(10-DAB),具有与紫杉醇相似的四并环骨架.随后开发出四步合成转化法,将10-脱乙酰基巴卡丁-Ⅲ以80%总收率转化为紫杉醇,并在美国施贵宝公司实现了工业化生产.1999年Kyriacos Nicolau、David Evans和Dale Boger三个课题组几乎同时报道了万古霉素的全合成工作[49-51];进入21世纪之后,英国剑桥大学Steven Ley和其合作者率先报道了雷帕霉素(rapamycin)的全合成[52];美籍日裔化学家Yoshito Kishi先后完成了海洋天然产物海葵毒素(palytoxin)[53]、河豚毒素(tetrodotoxin)[54]、软海绵素B(halichondrin B)[55]等的全合成;2006年日本卫材(Eisa)公司化学家基于软海绵素B结构改造,经62步有机反应合成出了含有19个碳手性中心的抗癌药物——甲磺酸艾日布林(eribulin),该药物在2010年获美国FDA批准,是第1个衍生自天然产物而用于转移性乳腺癌患者获得总生存期改善的化疗单药[56-58]. ...
Synthetic studies on tetrodotoxin and related compounds.Ⅲ. Stereospecific synthesis of an equivalent of acetylated tetrodamine
1
1972
... 20世纪90年代至今,有机化学家进一步完成了许多高药物活性天然产物的全合成.例如1994年美国化学家Kyriacos Nicolau和Robert Holton两个团队同年攻克抗癌明星药物紫杉醇分子的全合成(图3)[42-48],其中Robert Holton以天然氧化绿叶烯为原料,采用线性策略,经过41步反应,以不到3%总收率得到紫杉醇;除此之外,化学家采用其他原料和策略还发展了多条不同的全合成路线,但都因为合成步骤太多,工艺路线过长,反应条件苛刻,总收率均低于1%,而无法实现规模化放大生产.1981年法国科学家Pierre Potier从紫杉茎叶中分离得到一种高含量化合物——10-脱乙酰基巴卡丁-Ⅲ(10-DAB),具有与紫杉醇相似的四并环骨架.随后开发出四步合成转化法,将10-脱乙酰基巴卡丁-Ⅲ以80%总收率转化为紫杉醇,并在美国施贵宝公司实现了工业化生产.1999年Kyriacos Nicolau、David Evans和Dale Boger三个课题组几乎同时报道了万古霉素的全合成工作[49-51];进入21世纪之后,英国剑桥大学Steven Ley和其合作者率先报道了雷帕霉素(rapamycin)的全合成[52];美籍日裔化学家Yoshito Kishi先后完成了海洋天然产物海葵毒素(palytoxin)[53]、河豚毒素(tetrodotoxin)[54]、软海绵素B(halichondrin B)[55]等的全合成;2006年日本卫材(Eisa)公司化学家基于软海绵素B结构改造,经62步有机反应合成出了含有19个碳手性中心的抗癌药物——甲磺酸艾日布林(eribulin),该药物在2010年获美国FDA批准,是第1个衍生自天然产物而用于转移性乳腺癌患者获得总生存期改善的化疗单药[56-58]. ...
Total synthesis of halichondrin B and norhalichondrin B
1
1992
... 20世纪90年代至今,有机化学家进一步完成了许多高药物活性天然产物的全合成.例如1994年美国化学家Kyriacos Nicolau和Robert Holton两个团队同年攻克抗癌明星药物紫杉醇分子的全合成(图3)[42-48],其中Robert Holton以天然氧化绿叶烯为原料,采用线性策略,经过41步反应,以不到3%总收率得到紫杉醇;除此之外,化学家采用其他原料和策略还发展了多条不同的全合成路线,但都因为合成步骤太多,工艺路线过长,反应条件苛刻,总收率均低于1%,而无法实现规模化放大生产.1981年法国科学家Pierre Potier从紫杉茎叶中分离得到一种高含量化合物——10-脱乙酰基巴卡丁-Ⅲ(10-DAB),具有与紫杉醇相似的四并环骨架.随后开发出四步合成转化法,将10-脱乙酰基巴卡丁-Ⅲ以80%总收率转化为紫杉醇,并在美国施贵宝公司实现了工业化生产.1999年Kyriacos Nicolau、David Evans和Dale Boger三个课题组几乎同时报道了万古霉素的全合成工作[49-51];进入21世纪之后,英国剑桥大学Steven Ley和其合作者率先报道了雷帕霉素(rapamycin)的全合成[52];美籍日裔化学家Yoshito Kishi先后完成了海洋天然产物海葵毒素(palytoxin)[53]、河豚毒素(tetrodotoxin)[54]、软海绵素B(halichondrin B)[55]等的全合成;2006年日本卫材(Eisa)公司化学家基于软海绵素B结构改造,经62步有机反应合成出了含有19个碳手性中心的抗癌药物——甲磺酸艾日布林(eribulin),该药物在2010年获美国FDA批准,是第1个衍生自天然产物而用于转移性乳腺癌患者获得总生存期改善的化疗单药[56-58]. ...
Macrocyclic ketone analogues of halichondrin B
1
2004
... 20世纪90年代至今,有机化学家进一步完成了许多高药物活性天然产物的全合成.例如1994年美国化学家Kyriacos Nicolau和Robert Holton两个团队同年攻克抗癌明星药物紫杉醇分子的全合成(图3)[42-48],其中Robert Holton以天然氧化绿叶烯为原料,采用线性策略,经过41步反应,以不到3%总收率得到紫杉醇;除此之外,化学家采用其他原料和策略还发展了多条不同的全合成路线,但都因为合成步骤太多,工艺路线过长,反应条件苛刻,总收率均低于1%,而无法实现规模化放大生产.1981年法国科学家Pierre Potier从紫杉茎叶中分离得到一种高含量化合物——10-脱乙酰基巴卡丁-Ⅲ(10-DAB),具有与紫杉醇相似的四并环骨架.随后开发出四步合成转化法,将10-脱乙酰基巴卡丁-Ⅲ以80%总收率转化为紫杉醇,并在美国施贵宝公司实现了工业化生产.1999年Kyriacos Nicolau、David Evans和Dale Boger三个课题组几乎同时报道了万古霉素的全合成工作[49-51];进入21世纪之后,英国剑桥大学Steven Ley和其合作者率先报道了雷帕霉素(rapamycin)的全合成[52];美籍日裔化学家Yoshito Kishi先后完成了海洋天然产物海葵毒素(palytoxin)[53]、河豚毒素(tetrodotoxin)[54]、软海绵素B(halichondrin B)[55]等的全合成;2006年日本卫材(Eisa)公司化学家基于软海绵素B结构改造,经62步有机反应合成出了含有19个碳手性中心的抗癌药物——甲磺酸艾日布林(eribulin),该药物在2010年获美国FDA批准,是第1个衍生自天然产物而用于转移性乳腺癌患者获得总生存期改善的化疗单药[56-58]. ...
Case history: discovery of eribulin (HALAVEN (TM)), a halichondrin B analogue that prolongs overall survival in patients with metastatic breast cancer
0
2011
From micrograms to grams: scale-up synthesis of eribulin mesylate
1
2013
... 20世纪90年代至今,有机化学家进一步完成了许多高药物活性天然产物的全合成.例如1994年美国化学家Kyriacos Nicolau和Robert Holton两个团队同年攻克抗癌明星药物紫杉醇分子的全合成(图3)[42-48],其中Robert Holton以天然氧化绿叶烯为原料,采用线性策略,经过41步反应,以不到3%总收率得到紫杉醇;除此之外,化学家采用其他原料和策略还发展了多条不同的全合成路线,但都因为合成步骤太多,工艺路线过长,反应条件苛刻,总收率均低于1%,而无法实现规模化放大生产.1981年法国科学家Pierre Potier从紫杉茎叶中分离得到一种高含量化合物——10-脱乙酰基巴卡丁-Ⅲ(10-DAB),具有与紫杉醇相似的四并环骨架.随后开发出四步合成转化法,将10-脱乙酰基巴卡丁-Ⅲ以80%总收率转化为紫杉醇,并在美国施贵宝公司实现了工业化生产.1999年Kyriacos Nicolau、David Evans和Dale Boger三个课题组几乎同时报道了万古霉素的全合成工作[49-51];进入21世纪之后,英国剑桥大学Steven Ley和其合作者率先报道了雷帕霉素(rapamycin)的全合成[52];美籍日裔化学家Yoshito Kishi先后完成了海洋天然产物海葵毒素(palytoxin)[53]、河豚毒素(tetrodotoxin)[54]、软海绵素B(halichondrin B)[55]等的全合成;2006年日本卫材(Eisa)公司化学家基于软海绵素B结构改造,经62步有机反应合成出了含有19个碳手性中心的抗癌药物——甲磺酸艾日布林(eribulin),该药物在2010年获美国FDA批准,是第1个衍生自天然产物而用于转移性乳腺癌患者获得总生存期改善的化疗单药[56-58]. ...
合成生物学与天然产物开发
1
2011
... 天然产物的化学提取分离与结构鉴定,促进了生物合成研究的发展.早期科学家研究微生物次级代谢天然产物的策略较为单一,主要通过大量筛选寻找能够分泌具有药理活性的微生物菌种,然后进行培养和优化研究,并对最佳菌种开展大规模工业化发酵,最后通过提取和精制,完成天然产物生物全合成制备.如今,随着基因组学、转录组学、蛋白组学、代谢组学和生物信息学等多学科交叉领域研究的不断深入,合成生物学家通过物质/能量代谢及其调控通路的重构,可以在同源或异源微生物细胞中实现天然产物的生物全合成[59-64].生物制造给天然产物的全合成,带来了一场影响深远的变革.在此将通过几个典型的同源和异源微生物细胞合成途径重构的例子,分析天然药物分子的生物全合成. ...
Synthetic biology and natural products development
1
2011
... 天然产物的化学提取分离与结构鉴定,促进了生物合成研究的发展.早期科学家研究微生物次级代谢天然产物的策略较为单一,主要通过大量筛选寻找能够分泌具有药理活性的微生物菌种,然后进行培养和优化研究,并对最佳菌种开展大规模工业化发酵,最后通过提取和精制,完成天然产物生物全合成制备.如今,随着基因组学、转录组学、蛋白组学、代谢组学和生物信息学等多学科交叉领域研究的不断深入,合成生物学家通过物质/能量代谢及其调控通路的重构,可以在同源或异源微生物细胞中实现天然产物的生物全合成[59-64].生物制造给天然产物的全合成,带来了一场影响深远的变革.在此将通过几个典型的同源和异源微生物细胞合成途径重构的例子,分析天然药物分子的生物全合成. ...
天然产物生物合成与抗肿瘤药物合成生物学研究
0
2015
Biosynthesis of natural products and synthetic biology of antitumor drugs
0
2015
Role of biocatalysis in sustainable chemistry
0
2018
多酶催化串联策略在复杂天然产物合成中的应用
0
2020
Applications of the multienzyme-catalyzed tandem strategy in the synthesis of complex natural products
0
2020
Synthetic biology for the directed evolution of protein biocatalysts: navigating sequence space intelligently
0
2015
Synthetic biology strategies for microbial biosynthesis of plant natural products
1
2019
... 天然产物的化学提取分离与结构鉴定,促进了生物合成研究的发展.早期科学家研究微生物次级代谢天然产物的策略较为单一,主要通过大量筛选寻找能够分泌具有药理活性的微生物菌种,然后进行培养和优化研究,并对最佳菌种开展大规模工业化发酵,最后通过提取和精制,完成天然产物生物全合成制备.如今,随着基因组学、转录组学、蛋白组学、代谢组学和生物信息学等多学科交叉领域研究的不断深入,合成生物学家通过物质/能量代谢及其调控通路的重构,可以在同源或异源微生物细胞中实现天然产物的生物全合成[59-64].生物制造给天然产物的全合成,带来了一场影响深远的变革.在此将通过几个典型的同源和异源微生物细胞合成途径重构的例子,分析天然药物分子的生物全合成. ...
微生物药物的合成生物学研究进展
1
2020
... 自然界微生物的种类多样、生长速度快、容易培养和优化等特点.在此基础上,对微生物次级代谢产物发现及利用促进了天然产物生物全合成领域的逐渐成长.随着科学和技术的不断发展,进一步通过对同源微生物细胞的遗传操作,可以实现次级代谢天然产物的高效调控生物全合成[65-66]. ...
Recent progress of synthetic biology applications in microbial pharmaceuticals research
1
2020
... 自然界微生物的种类多样、生长速度快、容易培养和优化等特点.在此基础上,对微生物次级代谢产物发现及利用促进了天然产物生物全合成领域的逐渐成长.随着科学和技术的不断发展,进一步通过对同源微生物细胞的遗传操作,可以实现次级代谢天然产物的高效调控生物全合成[65-66]. ...
Synthetic biology and metabolic engineering of actinomycetes for natural product discovery
1
2019
... 自然界微生物的种类多样、生长速度快、容易培养和优化等特点.在此基础上,对微生物次级代谢产物发现及利用促进了天然产物生物全合成领域的逐渐成长.随着科学和技术的不断发展,进一步通过对同源微生物细胞的遗传操作,可以实现次级代谢天然产物的高效调控生物全合成[65-66]. ...
The X-ray analysis of the structure of penicillin
1
1949
... 1928年英国微生物学家Alexander Fleming最早发现了青霉菌分泌物——青霉素,但由于当时技术条件的局限,他并没有分离纯化出青霉素.10年后德国生物化学家Ernst Chain阅读了Fleming的研究报道,开始尝试提纯实验;1941年他与英国牛津大学病理学家Howard Florey一起实现了青霉素的分离与纯化,并发现其对链球菌、白喉杆菌等多种细菌感染疾病的疗效.随后,Florey又在腐烂的甜瓜上发现了一种可供大量提取青霉素的菌种,并使用玉米粉配制出相应的培养液.在这些研究成果推动下,美国制药企业于1942年开始对青霉素进行大批量生产;1945年英国化学家Dorothy Hodgkin用X射线衍射法确定了青霉素的分子结构[67].但直到20世纪80年代,青霉素的生物合成途径才获得解析[68-69].如图4所示青霉素G的生物合成途径:葡萄糖经过代谢后合成3种重要的前体氨基酸,分别为L-缬氨酸(L-Val)、L-半胱氨酸(L-Cys)和L-氨基己二酸(L-AAA),然后经ACV三肽合成酶缩合形成ACV三肽,通过异青霉素N合成酶(IPNS)催化环化合成出异青霉素N(IPN),再经酰基转移酶AT通过侧链转换得到青霉素G.发展至今,青霉素的工业生物全合成已经非常成熟,主要包括三个流程:首先将青霉菌接种到固体培养基上,室温培养制取青霉菌孢子培养物;随后将孢子悬浮液接种到带有灭菌培养基的种子罐中,搅拌发酵培养;最后将发酵液过滤,提取和精制.继青霉素之后,链霉素、氯霉素、土霉素、四环素、金霉素、万古霉素等抗生素不断被发现,也都是通过类似流程,实现了大规模工业化生产. ...
Development of a transformation system in Penicillium chrysogenum: cloning of genes involved in penicillin biosynthesis
1
1987
... 1928年英国微生物学家Alexander Fleming最早发现了青霉菌分泌物——青霉素,但由于当时技术条件的局限,他并没有分离纯化出青霉素.10年后德国生物化学家Ernst Chain阅读了Fleming的研究报道,开始尝试提纯实验;1941年他与英国牛津大学病理学家Howard Florey一起实现了青霉素的分离与纯化,并发现其对链球菌、白喉杆菌等多种细菌感染疾病的疗效.随后,Florey又在腐烂的甜瓜上发现了一种可供大量提取青霉素的菌种,并使用玉米粉配制出相应的培养液.在这些研究成果推动下,美国制药企业于1942年开始对青霉素进行大批量生产;1945年英国化学家Dorothy Hodgkin用X射线衍射法确定了青霉素的分子结构[67].但直到20世纪80年代,青霉素的生物合成途径才获得解析[68-69].如图4所示青霉素G的生物合成途径:葡萄糖经过代谢后合成3种重要的前体氨基酸,分别为L-缬氨酸(L-Val)、L-半胱氨酸(L-Cys)和L-氨基己二酸(L-AAA),然后经ACV三肽合成酶缩合形成ACV三肽,通过异青霉素N合成酶(IPNS)催化环化合成出异青霉素N(IPN),再经酰基转移酶AT通过侧链转换得到青霉素G.发展至今,青霉素的工业生物全合成已经非常成熟,主要包括三个流程:首先将青霉菌接种到固体培养基上,室温培养制取青霉菌孢子培养物;随后将孢子悬浮液接种到带有灭菌培养基的种子罐中,搅拌发酵培养;最后将发酵液过滤,提取和精制.继青霉素之后,链霉素、氯霉素、土霉素、四环素、金霉素、万古霉素等抗生素不断被发现,也都是通过类似流程,实现了大规模工业化生产. ...
Purification to homogeneity and characterization of acyl coenzyme A: 6-aminopenicillanic acid acyltransferase of Penicillium chrysogenum
1
1987
... 1928年英国微生物学家Alexander Fleming最早发现了青霉菌分泌物——青霉素,但由于当时技术条件的局限,他并没有分离纯化出青霉素.10年后德国生物化学家Ernst Chain阅读了Fleming的研究报道,开始尝试提纯实验;1941年他与英国牛津大学病理学家Howard Florey一起实现了青霉素的分离与纯化,并发现其对链球菌、白喉杆菌等多种细菌感染疾病的疗效.随后,Florey又在腐烂的甜瓜上发现了一种可供大量提取青霉素的菌种,并使用玉米粉配制出相应的培养液.在这些研究成果推动下,美国制药企业于1942年开始对青霉素进行大批量生产;1945年英国化学家Dorothy Hodgkin用X射线衍射法确定了青霉素的分子结构[67].但直到20世纪80年代,青霉素的生物合成途径才获得解析[68-69].如图4所示青霉素G的生物合成途径:葡萄糖经过代谢后合成3种重要的前体氨基酸,分别为L-缬氨酸(L-Val)、L-半胱氨酸(L-Cys)和L-氨基己二酸(L-AAA),然后经ACV三肽合成酶缩合形成ACV三肽,通过异青霉素N合成酶(IPNS)催化环化合成出异青霉素N(IPN),再经酰基转移酶AT通过侧链转换得到青霉素G.发展至今,青霉素的工业生物全合成已经非常成熟,主要包括三个流程:首先将青霉菌接种到固体培养基上,室温培养制取青霉菌孢子培养物;随后将孢子悬浮液接种到带有灭菌培养基的种子罐中,搅拌发酵培养;最后将发酵液过滤,提取和精制.继青霉素之后,链霉素、氯霉素、土霉素、四环素、金霉素、万古霉素等抗生素不断被发现,也都是通过类似流程,实现了大规模工业化生产. ...
Ilotycin, a new antibiotic
1
1952
... 作为放线菌来源的聚酮类天然产物,红霉素属于大环内酯类抗生素,可用来治疗革兰氏阳性细菌感染.1952年James McGire等首次从红色糖多孢菌(Saccharopolyspora erythraea)的发酵产物中分离得到红霉素[70].目前红霉素的生物体内合成途径已被探明[71]:首先通过Ⅰ型聚酮合酶(polyketide synthase-Ⅰ, PKS-Ⅰ)多步延伸模块催化1分子丙酰辅酶A(propionyl-CoA)和6分子甲基丙二酸单酰辅酶A[(2S)-methylmalonyl-CoA],合成十四元环中间产物6-脱氧红霉内酯(6-deoxy-erythronolide B,6-dEB);此后,6-dEB经历多步修饰,包括P450羟化酶(EryF)在其大环骨架C6位点进行羟化等反应,生成红霉内酯(erythronolide B);再通过两步糖基转移反应,即糖基转移酶EryBV在C3羟基位点连接一个L-碳霉糖,形成3-O-碳霉糖基红霉内酯(3-O-mycarosyl erythronolide B),以及在此基础上经糖基转移酶EryCⅢ在C5羟基加上一个D-德胺糖,进而形成红霉素生物合成途径中第1个具有生物活性的中间产物红霉素D(Er-D).接下来,羟化酶EryK催化红霉素D的C12位点羟化,形成红霉素C(Er-C),最后经甲基化酶EryG在碳霉糖糖基的C3位点进行甲基化修饰,得到最终产物红霉素A(Er-A)(图5). ...
Biosynthesis and combinatorial biosynthesis of erythromycin
1
2012
... 作为放线菌来源的聚酮类天然产物,红霉素属于大环内酯类抗生素,可用来治疗革兰氏阳性细菌感染.1952年James McGire等首次从红色糖多孢菌(Saccharopolyspora erythraea)的发酵产物中分离得到红霉素[70].目前红霉素的生物体内合成途径已被探明[71]:首先通过Ⅰ型聚酮合酶(polyketide synthase-Ⅰ, PKS-Ⅰ)多步延伸模块催化1分子丙酰辅酶A(propionyl-CoA)和6分子甲基丙二酸单酰辅酶A[(2S)-methylmalonyl-CoA],合成十四元环中间产物6-脱氧红霉内酯(6-deoxy-erythronolide B,6-dEB);此后,6-dEB经历多步修饰,包括P450羟化酶(EryF)在其大环骨架C6位点进行羟化等反应,生成红霉内酯(erythronolide B);再通过两步糖基转移反应,即糖基转移酶EryBV在C3羟基位点连接一个L-碳霉糖,形成3-O-碳霉糖基红霉内酯(3-O-mycarosyl erythronolide B),以及在此基础上经糖基转移酶EryCⅢ在C5羟基加上一个D-德胺糖,进而形成红霉素生物合成途径中第1个具有生物活性的中间产物红霉素D(Er-D).接下来,羟化酶EryK催化红霉素D的C12位点羟化,形成红霉素C(Er-C),最后经甲基化酶EryG在碳霉糖糖基的C3位点进行甲基化修饰,得到最终产物红霉素A(Er-A)(图5). ...
以生物合成为基础的红霉素A的产量提高和结构改造
1
2015
... 尽管对于红霉素在生物体内合成路线研究得比较清楚,但如何提高产量仍具有很大挑战.中国科学院上海有机化学研究所刘文团队,运用组合生物合成技术对红霉素工业用高产菌株进行了针对性的遗传改良,例如:倍增了红色糖多孢菌体内PKS编码基因,使红霉素产量提高50%,并使发酵周期缩短1/3[72];通过优化整合到染色体上关键羟化酶基因eryK和甲基化酶基因eryG的拷贝数,可以消除副产物B和C,同时红霉素A的产量可以提高30%[73],为红霉素的工业化生产奠定了基础. ...
Biosynthesis-based production improvement and structure modification of erythromycin A
1
2015
... 尽管对于红霉素在生物体内合成路线研究得比较清楚,但如何提高产量仍具有很大挑战.中国科学院上海有机化学研究所刘文团队,运用组合生物合成技术对红霉素工业用高产菌株进行了针对性的遗传改良,例如:倍增了红色糖多孢菌体内PKS编码基因,使红霉素产量提高50%,并使发酵周期缩短1/3[72];通过优化整合到染色体上关键羟化酶基因eryK和甲基化酶基因eryG的拷贝数,可以消除副产物B和C,同时红霉素A的产量可以提高30%[73],为红霉素的工业化生产奠定了基础. ...
Genetic modulation of the overexpression of tailoring genes eryK and eryG leading to the improvement of erythromycin a purity and production in Saccharopolyspora erythraea fermentation
1
2008
... 尽管对于红霉素在生物体内合成路线研究得比较清楚,但如何提高产量仍具有很大挑战.中国科学院上海有机化学研究所刘文团队,运用组合生物合成技术对红霉素工业用高产菌株进行了针对性的遗传改良,例如:倍增了红色糖多孢菌体内PKS编码基因,使红霉素产量提高50%,并使发酵周期缩短1/3[72];通过优化整合到染色体上关键羟化酶基因eryK和甲基化酶基因eryG的拷贝数,可以消除副产物B和C,同时红霉素A的产量可以提高30%[73],为红霉素的工业化生产奠定了基础. ...
History of avermectin and ivermectin, with notes on the history of other macrocyclic lactone antiparasitic agents
1
2012
... 作为农作物保护的高效杀虫剂,阿维菌素是一种具有十六元环结构的聚酮大环内酯类抗生素,工业上主要采用除虫链霉菌(Streptomyces avermitilis)发酵分离获得[74].21世纪初,阿维菌素的生物合成基因模块已被科学家全部探明[75-76],其生物体内合成途径主要包括3个阶段(图6):①起始单元合成,由L-异亮氨酸(L-Ile)及L-缬氨酸(L-Val)两条路径出发,经多步酶促反应分别生成2-甲基丁酰辅酶A(2-methylbutyryl CoA)及异丁酰辅酶A(isobutyryl CoA);②大环内酯骨架合成,在多模块聚酮合酶PKS作用下,这两种起始单元转化为阿维菌素大环内酯骨架结构;③阿维菌素合成,经多步聚酮后修饰反应,包括环氧化、糖基化、还原及甲基化等,大环内酯骨架转化为系列阿维菌素化合物[77].根据其骨架C5、C22、C23以及C26位点结构上的差别,阿维菌素由8个组分构成,即4个主要组分(A1a、A2a、B1a、B2a)以及4个次要组分(A1b、A2b、B1b、B2b).其中B1a毒性最低、生物活性最强,也是市售阿维菌素农药的主要杀虫成分. ...
Organization of the biosynthetic gene cluster for the polyketide anthelmintic macrolide avermectin in Streptomyces avermitilis
1
1999
... 作为农作物保护的高效杀虫剂,阿维菌素是一种具有十六元环结构的聚酮大环内酯类抗生素,工业上主要采用除虫链霉菌(Streptomyces avermitilis)发酵分离获得[74].21世纪初,阿维菌素的生物合成基因模块已被科学家全部探明[75-76],其生物体内合成途径主要包括3个阶段(图6):①起始单元合成,由L-异亮氨酸(L-Ile)及L-缬氨酸(L-Val)两条路径出发,经多步酶促反应分别生成2-甲基丁酰辅酶A(2-methylbutyryl CoA)及异丁酰辅酶A(isobutyryl CoA);②大环内酯骨架合成,在多模块聚酮合酶PKS作用下,这两种起始单元转化为阿维菌素大环内酯骨架结构;③阿维菌素合成,经多步聚酮后修饰反应,包括环氧化、糖基化、还原及甲基化等,大环内酯骨架转化为系列阿维菌素化合物[77].根据其骨架C5、C22、C23以及C26位点结构上的差别,阿维菌素由8个组分构成,即4个主要组分(A1a、A2a、B1a、B2a)以及4个次要组分(A1b、A2b、B1b、B2b).其中B1a毒性最低、生物活性最强,也是市售阿维菌素农药的主要杀虫成分. ...
Genome sequence of an industrial microorganism Streptomyces avermitilis: deducing the ability of producing secondary metabolites
1
2001
... 作为农作物保护的高效杀虫剂,阿维菌素是一种具有十六元环结构的聚酮大环内酯类抗生素,工业上主要采用除虫链霉菌(Streptomyces avermitilis)发酵分离获得[74].21世纪初,阿维菌素的生物合成基因模块已被科学家全部探明[75-76],其生物体内合成途径主要包括3个阶段(图6):①起始单元合成,由L-异亮氨酸(L-Ile)及L-缬氨酸(L-Val)两条路径出发,经多步酶促反应分别生成2-甲基丁酰辅酶A(2-methylbutyryl CoA)及异丁酰辅酶A(isobutyryl CoA);②大环内酯骨架合成,在多模块聚酮合酶PKS作用下,这两种起始单元转化为阿维菌素大环内酯骨架结构;③阿维菌素合成,经多步聚酮后修饰反应,包括环氧化、糖基化、还原及甲基化等,大环内酯骨架转化为系列阿维菌素化合物[77].根据其骨架C5、C22、C23以及C26位点结构上的差别,阿维菌素由8个组分构成,即4个主要组分(A1a、A2a、B1a、B2a)以及4个次要组分(A1b、A2b、B1b、B2b).其中B1a毒性最低、生物活性最强,也是市售阿维菌素农药的主要杀虫成分. ...
Avermectin: biochemical and molecular basis of its biosynthesis and regulation
1
2004
... 作为农作物保护的高效杀虫剂,阿维菌素是一种具有十六元环结构的聚酮大环内酯类抗生素,工业上主要采用除虫链霉菌(Streptomyces avermitilis)发酵分离获得[74].21世纪初,阿维菌素的生物合成基因模块已被科学家全部探明[75-76],其生物体内合成途径主要包括3个阶段(图6):①起始单元合成,由L-异亮氨酸(L-Ile)及L-缬氨酸(L-Val)两条路径出发,经多步酶促反应分别生成2-甲基丁酰辅酶A(2-methylbutyryl CoA)及异丁酰辅酶A(isobutyryl CoA);②大环内酯骨架合成,在多模块聚酮合酶PKS作用下,这两种起始单元转化为阿维菌素大环内酯骨架结构;③阿维菌素合成,经多步聚酮后修饰反应,包括环氧化、糖基化、还原及甲基化等,大环内酯骨架转化为系列阿维菌素化合物[77].根据其骨架C5、C22、C23以及C26位点结构上的差别,阿维菌素由8个组分构成,即4个主要组分(A1a、A2a、B1a、B2a)以及4个次要组分(A1b、A2b、B1b、B2b).其中B1a毒性最低、生物活性最强,也是市售阿维菌素农药的主要杀虫成分. ...
Reverse biological engineering of hrdB to enhance the production of avermectins in an industrial strain of Streptomyces avermitilis
1
2010
... 我国原有阿维菌素生产菌株的效价较低,且能耗高、污染大.中国科学院微生物研究所张立新团队经基因组学分析,发现并解析了阿维菌素的高产机制,通过精准调控转录因子σHrdB的基因表达等遗传操作,获得系列阿维菌素的高产突变菌株,同时优化了发酵工艺,实现了阿维菌素的高效生物合成.相较传统工艺,在120~550 m3发酵罐中阿维菌素B1a产量提高1000倍,达到9 g/L以上,生产效率较国际上阿维菌素发酵水平最高的美国默克公司提高了60%[78]. ...
Plant synthetic biology
1
2015
... 自然界动物和植物的多样性,是众多天然产物的重要源泉.但由于动植物的结构复杂、代谢更新周期长以及资源局限性等问题,大量获取这类天然产物难度高.近年来,科学家通过解析天然产物的结构,探索其生化反应机制和合成路线,将相关基因在异源微生物细胞里进行表达和调控,从而完成这类天然产物的生物全合成[79-81]. ...
From plant metabolic engineering to plant synthetic biology: the evolution of the design/build/test/learn cycle
0
2018
植物合成生物学研究进展
1
2020
... 自然界动物和植物的多样性,是众多天然产物的重要源泉.但由于动植物的结构复杂、代谢更新周期长以及资源局限性等问题,大量获取这类天然产物难度高.近年来,科学家通过解析天然产物的结构,探索其生化反应机制和合成路线,将相关基因在异源微生物细胞里进行表达和调控,从而完成这类天然产物的生物全合成[79-81]. ...
Recent advances in plant synthetic biology
1
2020
... 自然界动物和植物的多样性,是众多天然产物的重要源泉.但由于动植物的结构复杂、代谢更新周期长以及资源局限性等问题,大量获取这类天然产物难度高.近年来,科学家通过解析天然产物的结构,探索其生化反应机制和合成路线,将相关基因在异源微生物细胞里进行表达和调控,从而完成这类天然产物的生物全合成[79-81]. ...
The total synthesis of vitamin B12
1
1973
... 维生素B12是一种含有三价钴的多环有机分子,又叫钴胺素,也是唯一含有金属元素的维生素.可参与制造骨髓红细胞,防止恶性贫血,保护大脑神经系统不受破坏.只能从动物的内脏中经人工提取和精制得到,价格极其昂贵.美国哈佛大学Robert Woodward教授与苏黎世联邦理工学院(ETH) Albert Eschenmoser教授联合组织了14个国家的上百位有机化学家,历时12年协同攻关,在1973年成功实现维生素B12的化学全合成[82-83];但由于有机反应步骤多,合成路线太长,根本无法进行大量制备.2018年,中国科学院天津工业生物技术研究所张大伟团队,在大肠杆菌中实现了维生素B12的从头合成(图7).首先解析维生素B12好氧合成路径中钴螯合与腺苷钴啉醇酰胺磷酸的合成机理,将来源于5种细菌中的28个基因在大肠杆菌细胞中成功异源表达,并按其人工合成途径划分为5个模块进行人工途径组装.在克服多基因适配等难题后,最终实现了维生素B12的从头合成,通过途径优化和发酵过程调控,产量达到307.00 µg/g干细胞菌体,合成菌种发酵周期仅为目前工业生产菌株的1/10,极具工业应用前景[84]. ...
Natural product synthesis and vitamin B12
1
1977
... 维生素B12是一种含有三价钴的多环有机分子,又叫钴胺素,也是唯一含有金属元素的维生素.可参与制造骨髓红细胞,防止恶性贫血,保护大脑神经系统不受破坏.只能从动物的内脏中经人工提取和精制得到,价格极其昂贵.美国哈佛大学Robert Woodward教授与苏黎世联邦理工学院(ETH) Albert Eschenmoser教授联合组织了14个国家的上百位有机化学家,历时12年协同攻关,在1973年成功实现维生素B12的化学全合成[82-83];但由于有机反应步骤多,合成路线太长,根本无法进行大量制备.2018年,中国科学院天津工业生物技术研究所张大伟团队,在大肠杆菌中实现了维生素B12的从头合成(图7).首先解析维生素B12好氧合成路径中钴螯合与腺苷钴啉醇酰胺磷酸的合成机理,将来源于5种细菌中的28个基因在大肠杆菌细胞中成功异源表达,并按其人工合成途径划分为5个模块进行人工途径组装.在克服多基因适配等难题后,最终实现了维生素B12的从头合成,通过途径优化和发酵过程调控,产量达到307.00 µg/g干细胞菌体,合成菌种发酵周期仅为目前工业生产菌株的1/10,极具工业应用前景[84]. ...
Metabolic engineering of Escherichia coli for de novo biosynthesis of vitamin B12
1
2018
... 维生素B12是一种含有三价钴的多环有机分子,又叫钴胺素,也是唯一含有金属元素的维生素.可参与制造骨髓红细胞,防止恶性贫血,保护大脑神经系统不受破坏.只能从动物的内脏中经人工提取和精制得到,价格极其昂贵.美国哈佛大学Robert Woodward教授与苏黎世联邦理工学院(ETH) Albert Eschenmoser教授联合组织了14个国家的上百位有机化学家,历时12年协同攻关,在1973年成功实现维生素B12的化学全合成[82-83];但由于有机反应步骤多,合成路线太长,根本无法进行大量制备.2018年,中国科学院天津工业生物技术研究所张大伟团队,在大肠杆菌中实现了维生素B12的从头合成(图7).首先解析维生素B12好氧合成路径中钴螯合与腺苷钴啉醇酰胺磷酸的合成机理,将来源于5种细菌中的28个基因在大肠杆菌细胞中成功异源表达,并按其人工合成途径划分为5个模块进行人工途径组装.在克服多基因适配等难题后,最终实现了维生素B12的从头合成,通过途径优化和发酵过程调控,产量达到307.00 µg/g干细胞菌体,合成菌种发酵周期仅为目前工业生产菌株的1/10,极具工业应用前景[84]. ...
Tropane alkaloid biosynthesis: a centennial review
1
2021
... 来自茄科植物的莨菪烷类生物碱是神经递质抑制剂,可用于治疗神经肌肉疾病,已经被世界卫生组织列为基本药物.从天然植物获取这类药物,受到气候环境等因素的严重制约,而化学全合成路线较长、副产物较多,因此该类化合物的生物合成策略逐渐受到关注[85].莨菪烷类生物碱的生物合成途径共涉及13个酶[图8(a)]:从鸟氨酸开始,经历鸟氨酸脱羧酶(ODC)→N-甲基转移酶(PMT)→N-甲基腐胺氧化酶(MPO)→自发环化→Ⅲ型聚酮合酶(PYKS)→托品酮合成酶(CYP82M3)→托品酮还原酶(TRI)历程形成托品碱;另一方面从苯丙氨酸出发,经历氨基转移酶(ArAT4)→苯丙酮酸还原酶(PPAR)→糖基转移酶(UGT1)形成糖基化苯乳酸,随后经历立托林合成酶(LS)→立托林变位酶(CYP80F1)→莨菪醛脱氢酶(HDH)形成莨菪碱,进一步羟化(莨菪碱羟化酶H6H)可生成山莨菪碱及东莨菪碱.西南大学廖志华团队在莨菪烷类生物碱的生物途径解析方面做出系列原创性贡献,先后确立了ODC、PPAR、UGT1和LS的酶学机制[86-88].2019年,廖志华团队与中国科学院昆明植物研究所黄胜雄团队利用转录组种内差异表达和种间同源基因分析[89],发现了不同植物来源的3个Ⅲ型聚酮合酶(AaPYKS、DsPYKS和AbPYKS),并验证了其能催化N-甲基吡咯啉阳离子与丙二酰辅酶A缩合形成三羰基戊二酸,随后发生自发Mannich反应得到托品烷骨架关键前体,揭示了托品烷生物碱生物合成中基本骨架形成的酶学机制[图8(b)].近期这两个团队进一步发现并鉴定了莨菪碱生物合成途径中[90],催化莨菪醛生成莨菪碱的关键酶——莨菪醛脱氢酶(hyoscyamine dehydrogenase,HDH),蛋白晶体结构及体外酶促反应研究揭示该酶在生理条件下利用NADPH将莨菪醛还原为莨菪碱[图8(c)],这些研究标志着以莨菪碱为代表的药用托品烷类化合物的生物合成途径得以完整解析,将为莨菪碱的合成生物学异源创制和工业制造提供基础. ...
A phenylpyruvic acid reductase is required for biosynthesis of tropane alkaloids
1
2018
... 来自茄科植物的莨菪烷类生物碱是神经递质抑制剂,可用于治疗神经肌肉疾病,已经被世界卫生组织列为基本药物.从天然植物获取这类药物,受到气候环境等因素的严重制约,而化学全合成路线较长、副产物较多,因此该类化合物的生物合成策略逐渐受到关注[85].莨菪烷类生物碱的生物合成途径共涉及13个酶[图8(a)]:从鸟氨酸开始,经历鸟氨酸脱羧酶(ODC)→N-甲基转移酶(PMT)→N-甲基腐胺氧化酶(MPO)→自发环化→Ⅲ型聚酮合酶(PYKS)→托品酮合成酶(CYP82M3)→托品酮还原酶(TRI)历程形成托品碱;另一方面从苯丙氨酸出发,经历氨基转移酶(ArAT4)→苯丙酮酸还原酶(PPAR)→糖基转移酶(UGT1)形成糖基化苯乳酸,随后经历立托林合成酶(LS)→立托林变位酶(CYP80F1)→莨菪醛脱氢酶(HDH)形成莨菪碱,进一步羟化(莨菪碱羟化酶H6H)可生成山莨菪碱及东莨菪碱.西南大学廖志华团队在莨菪烷类生物碱的生物途径解析方面做出系列原创性贡献,先后确立了ODC、PPAR、UGT1和LS的酶学机制[86-88].2019年,廖志华团队与中国科学院昆明植物研究所黄胜雄团队利用转录组种内差异表达和种间同源基因分析[89],发现了不同植物来源的3个Ⅲ型聚酮合酶(AaPYKS、DsPYKS和AbPYKS),并验证了其能催化N-甲基吡咯啉阳离子与丙二酰辅酶A缩合形成三羰基戊二酸,随后发生自发Mannich反应得到托品烷骨架关键前体,揭示了托品烷生物碱生物合成中基本骨架形成的酶学机制[图8(b)].近期这两个团队进一步发现并鉴定了莨菪碱生物合成途径中[90],催化莨菪醛生成莨菪碱的关键酶——莨菪醛脱氢酶(hyoscyamine dehydrogenase,HDH),蛋白晶体结构及体外酶促反应研究揭示该酶在生理条件下利用NADPH将莨菪醛还原为莨菪碱[图8(c)],这些研究标志着以莨菪碱为代表的药用托品烷类化合物的生物合成途径得以完整解析,将为莨菪碱的合成生物学异源创制和工业制造提供基础. ...
Functional genomics analysis reveals two novel genes required for littorine biosynthesis
0
2020
Engineering tropane alkaloid production based on metabolic characterization of ornithine decarboxylase in Atropa belladonna
1
2020
... 来自茄科植物的莨菪烷类生物碱是神经递质抑制剂,可用于治疗神经肌肉疾病,已经被世界卫生组织列为基本药物.从天然植物获取这类药物,受到气候环境等因素的严重制约,而化学全合成路线较长、副产物较多,因此该类化合物的生物合成策略逐渐受到关注[85].莨菪烷类生物碱的生物合成途径共涉及13个酶[图8(a)]:从鸟氨酸开始,经历鸟氨酸脱羧酶(ODC)→N-甲基转移酶(PMT)→N-甲基腐胺氧化酶(MPO)→自发环化→Ⅲ型聚酮合酶(PYKS)→托品酮合成酶(CYP82M3)→托品酮还原酶(TRI)历程形成托品碱;另一方面从苯丙氨酸出发,经历氨基转移酶(ArAT4)→苯丙酮酸还原酶(PPAR)→糖基转移酶(UGT1)形成糖基化苯乳酸,随后经历立托林合成酶(LS)→立托林变位酶(CYP80F1)→莨菪醛脱氢酶(HDH)形成莨菪碱,进一步羟化(莨菪碱羟化酶H6H)可生成山莨菪碱及东莨菪碱.西南大学廖志华团队在莨菪烷类生物碱的生物途径解析方面做出系列原创性贡献,先后确立了ODC、PPAR、UGT1和LS的酶学机制[86-88].2019年,廖志华团队与中国科学院昆明植物研究所黄胜雄团队利用转录组种内差异表达和种间同源基因分析[89],发现了不同植物来源的3个Ⅲ型聚酮合酶(AaPYKS、DsPYKS和AbPYKS),并验证了其能催化N-甲基吡咯啉阳离子与丙二酰辅酶A缩合形成三羰基戊二酸,随后发生自发Mannich反应得到托品烷骨架关键前体,揭示了托品烷生物碱生物合成中基本骨架形成的酶学机制[图8(b)].近期这两个团队进一步发现并鉴定了莨菪碱生物合成途径中[90],催化莨菪醛生成莨菪碱的关键酶——莨菪醛脱氢酶(hyoscyamine dehydrogenase,HDH),蛋白晶体结构及体外酶促反应研究揭示该酶在生理条件下利用NADPH将莨菪醛还原为莨菪碱[图8(c)],这些研究标志着以莨菪碱为代表的药用托品烷类化合物的生物合成途径得以完整解析,将为莨菪碱的合成生物学异源创制和工业制造提供基础. ...
Tropane alkaloids biosynthesis involves an unusual typeⅢ polyketide synthase and non-enzymatic condensation
1
2019
... 来自茄科植物的莨菪烷类生物碱是神经递质抑制剂,可用于治疗神经肌肉疾病,已经被世界卫生组织列为基本药物.从天然植物获取这类药物,受到气候环境等因素的严重制约,而化学全合成路线较长、副产物较多,因此该类化合物的生物合成策略逐渐受到关注[85].莨菪烷类生物碱的生物合成途径共涉及13个酶[图8(a)]:从鸟氨酸开始,经历鸟氨酸脱羧酶(ODC)→N-甲基转移酶(PMT)→N-甲基腐胺氧化酶(MPO)→自发环化→Ⅲ型聚酮合酶(PYKS)→托品酮合成酶(CYP82M3)→托品酮还原酶(TRI)历程形成托品碱;另一方面从苯丙氨酸出发,经历氨基转移酶(ArAT4)→苯丙酮酸还原酶(PPAR)→糖基转移酶(UGT1)形成糖基化苯乳酸,随后经历立托林合成酶(LS)→立托林变位酶(CYP80F1)→莨菪醛脱氢酶(HDH)形成莨菪碱,进一步羟化(莨菪碱羟化酶H6H)可生成山莨菪碱及东莨菪碱.西南大学廖志华团队在莨菪烷类生物碱的生物途径解析方面做出系列原创性贡献,先后确立了ODC、PPAR、UGT1和LS的酶学机制[86-88].2019年,廖志华团队与中国科学院昆明植物研究所黄胜雄团队利用转录组种内差异表达和种间同源基因分析[89],发现了不同植物来源的3个Ⅲ型聚酮合酶(AaPYKS、DsPYKS和AbPYKS),并验证了其能催化N-甲基吡咯啉阳离子与丙二酰辅酶A缩合形成三羰基戊二酸,随后发生自发Mannich反应得到托品烷骨架关键前体,揭示了托品烷生物碱生物合成中基本骨架形成的酶学机制[图8(b)].近期这两个团队进一步发现并鉴定了莨菪碱生物合成途径中[90],催化莨菪醛生成莨菪碱的关键酶——莨菪醛脱氢酶(hyoscyamine dehydrogenase,HDH),蛋白晶体结构及体外酶促反应研究揭示该酶在生理条件下利用NADPH将莨菪醛还原为莨菪碱[图8(c)],这些研究标志着以莨菪碱为代表的药用托品烷类化合物的生物合成途径得以完整解析,将为莨菪碱的合成生物学异源创制和工业制造提供基础. ...
Biochemical and metabolic insights into hyoscyamine dehydrogenase
1
2021
... 来自茄科植物的莨菪烷类生物碱是神经递质抑制剂,可用于治疗神经肌肉疾病,已经被世界卫生组织列为基本药物.从天然植物获取这类药物,受到气候环境等因素的严重制约,而化学全合成路线较长、副产物较多,因此该类化合物的生物合成策略逐渐受到关注[85].莨菪烷类生物碱的生物合成途径共涉及13个酶[图8(a)]:从鸟氨酸开始,经历鸟氨酸脱羧酶(ODC)→N-甲基转移酶(PMT)→N-甲基腐胺氧化酶(MPO)→自发环化→Ⅲ型聚酮合酶(PYKS)→托品酮合成酶(CYP82M3)→托品酮还原酶(TRI)历程形成托品碱;另一方面从苯丙氨酸出发,经历氨基转移酶(ArAT4)→苯丙酮酸还原酶(PPAR)→糖基转移酶(UGT1)形成糖基化苯乳酸,随后经历立托林合成酶(LS)→立托林变位酶(CYP80F1)→莨菪醛脱氢酶(HDH)形成莨菪碱,进一步羟化(莨菪碱羟化酶H6H)可生成山莨菪碱及东莨菪碱.西南大学廖志华团队在莨菪烷类生物碱的生物途径解析方面做出系列原创性贡献,先后确立了ODC、PPAR、UGT1和LS的酶学机制[86-88].2019年,廖志华团队与中国科学院昆明植物研究所黄胜雄团队利用转录组种内差异表达和种间同源基因分析[89],发现了不同植物来源的3个Ⅲ型聚酮合酶(AaPYKS、DsPYKS和AbPYKS),并验证了其能催化N-甲基吡咯啉阳离子与丙二酰辅酶A缩合形成三羰基戊二酸,随后发生自发Mannich反应得到托品烷骨架关键前体,揭示了托品烷生物碱生物合成中基本骨架形成的酶学机制[图8(b)].近期这两个团队进一步发现并鉴定了莨菪碱生物合成途径中[90],催化莨菪醛生成莨菪碱的关键酶——莨菪醛脱氢酶(hyoscyamine dehydrogenase,HDH),蛋白晶体结构及体外酶促反应研究揭示该酶在生理条件下利用NADPH将莨菪醛还原为莨菪碱[图8(c)],这些研究标志着以莨菪碱为代表的药用托品烷类化合物的生物合成途径得以完整解析,将为莨菪碱的合成生物学异源创制和工业制造提供基础. ...
Biosynthesis of medicinal tropane alkaloids in yeast
1
2020
... 来自斯坦福大学的Christina Smolke团队在酵母中整合多步异源表达基因,通过不同模块组装和亚细胞器定位,成功实现了莨菪碱(hyoscyamine)和东莨菪碱(scopolamine)的异源合成[91].生物合成途径如图9所示:模块Ⅰ,以天然谷氨酸为前体,通过两种不同的双酶途径,精氨酸酶(Car1)/鸟氨酸脱羧酶(spe1)或精氨酸脱羧酶(AsADC)/胍丁胺脲水解酶(speB)分别合成腐胺;模块Ⅱ,腐胺经过甲基化(AbPMT1/DsPMT1)、氨氧化(DmMPO1突变体)、自发环化合成N-甲基吡咯啉,再经聚酮合酶(AbPYKS)、托品酮合成酶(AbCYP82M3)、P450还原酶(AtATR1)和托品酮还原酶(DsTR1)催化合成托品碱;模块Ⅲ,涉及糖基化苯乳酸的合成途径,具体包括苯丙氨酸经氨基转移酶(Aro8/Aro9)、苯丙酮酸还原酶(WfPPR)、糖基转移酶(AbUGT)的催化,合成糖基化苯乳酸;模块Ⅳ,托品碱转运至液泡中,经蛋白质工程改造过的立托林(littorine)合成酶(AdLs)催化,糖基苯乳酸和托品碱转化为立托林;模块Ⅴ,立托林转运至细胞质中再经过异构化(AbCYP80F1/AtATR1)及醛还原酶(DsHDH)催化得到莨菪碱,进一步在双加氧酶(DsH6H)作用下制得东莨菪碱(图9).该工作整合了34个染色体修饰(26个基因以及8个突变基因),在不同亚细胞位置上定位了20多种酶,构建了一个完整的全细胞体系,莨菪碱和东莨菪碱的合成滴度达30 μg/L.该微生物合成平台可进一步发掘合成新的生物碱衍生物,用于新的药物开发. ...
Recent advances in the chemoenzymatic synthesis of bioactive natural products
1
2020
... 如上所述,生物合成策略在天然产物全合成中发挥越来越重要的作用,但仍面临诸多挑战性问题,如对一些来源独特的复杂天然产物,一方面,挖掘生物体内相关基因序列、解析生化反应机制以及特异性遗传修饰生物合成途径还存在较大困难;另一方面,将不同来源的天然产物生物合成基因进行重组,在异源微生物体内构建全新的代谢途径,还存在匹配和耐受性问题;此外,生物酶对底物的专一性要求非常高,天然产物分子骨架的衍生拓展性较差.在这些方面,化学合成可以发挥重要的互补优势,因而利用生物与化学交叉融合策略制备天然产物逐渐引起关注,成为天然产物及其衍生物全合成的另一个新趋势[92-93]. ...
复杂天然产物全合成: 化学合成与生物合成结合的策略
1
2018
... 如上所述,生物合成策略在天然产物全合成中发挥越来越重要的作用,但仍面临诸多挑战性问题,如对一些来源独特的复杂天然产物,一方面,挖掘生物体内相关基因序列、解析生化反应机制以及特异性遗传修饰生物合成途径还存在较大困难;另一方面,将不同来源的天然产物生物合成基因进行重组,在异源微生物体内构建全新的代谢途径,还存在匹配和耐受性问题;此外,生物酶对底物的专一性要求非常高,天然产物分子骨架的衍生拓展性较差.在这些方面,化学合成可以发挥重要的互补优势,因而利用生物与化学交叉融合策略制备天然产物逐渐引起关注,成为天然产物及其衍生物全合成的另一个新趋势[92-93]. ...
Total synthesis of complex natural products: combination of chemical synthesis and biosynthesis strategies
1
2018
... 如上所述,生物合成策略在天然产物全合成中发挥越来越重要的作用,但仍面临诸多挑战性问题,如对一些来源独特的复杂天然产物,一方面,挖掘生物体内相关基因序列、解析生化反应机制以及特异性遗传修饰生物合成途径还存在较大困难;另一方面,将不同来源的天然产物生物合成基因进行重组,在异源微生物体内构建全新的代谢途径,还存在匹配和耐受性问题;此外,生物酶对底物的专一性要求非常高,天然产物分子骨架的衍生拓展性较差.在这些方面,化学合成可以发挥重要的互补优势,因而利用生物与化学交叉融合策略制备天然产物逐渐引起关注,成为天然产物及其衍生物全合成的另一个新趋势[92-93]. ...
Expression in Escherichia coli of a chemically synthesized gene for the hormone somatostatin
1
1977
... 胰岛素分子的氨基酸序列在20世纪50年代已经解析清楚,随后科学家也尝试了很多策略用于人工合成胰岛素,但由于合成步骤太长、效率太低而无法实现大量工业生产,因此主要来源依然是通过粉碎牛或猪内脏进行提取,每生产1 kg胰岛素则需要超过8 t动物胰脏.20世纪60~70年代,重组DNA技术取得了重大突破.美国加州大学洛杉矶分校微生物学家Herb Boyer教授团队通过重组DNA技术,实现了人源胰岛素的全合成:他们采用化学从头合成的方法,先合成出编码胰岛素A链和B链两条DNA分子,将这些DNA插入细菌的基因组使它们能够合成出胰岛素A链和B链分子,再将两条肽链进行分离纯化,最后运用化学手段将两条链连接,获得人源胰岛素蛋白分子(图10)[94-96].此外,也可以通过大肠杆菌或巴斯德毕赤酵母直接合成单体或胰岛素原,进一步经过二硫键匹配和酶切合成胰岛素[97].Herb Boyer与风险投资人Robert Swanson一起创立了美国基因泰克(Genentch)生物技术公司,成功实现了人源胰岛素的工业化大量生产. ...
Chemical synthesis of genes for human insulin
0
1978
Expression in Escherichia coli of chemically synthesized genes for human insulin
1
1979
... 胰岛素分子的氨基酸序列在20世纪50年代已经解析清楚,随后科学家也尝试了很多策略用于人工合成胰岛素,但由于合成步骤太长、效率太低而无法实现大量工业生产,因此主要来源依然是通过粉碎牛或猪内脏进行提取,每生产1 kg胰岛素则需要超过8 t动物胰脏.20世纪60~70年代,重组DNA技术取得了重大突破.美国加州大学洛杉矶分校微生物学家Herb Boyer教授团队通过重组DNA技术,实现了人源胰岛素的全合成:他们采用化学从头合成的方法,先合成出编码胰岛素A链和B链两条DNA分子,将这些DNA插入细菌的基因组使它们能够合成出胰岛素A链和B链分子,再将两条肽链进行分离纯化,最后运用化学手段将两条链连接,获得人源胰岛素蛋白分子(图10)[94-96].此外,也可以通过大肠杆菌或巴斯德毕赤酵母直接合成单体或胰岛素原,进一步经过二硫键匹配和酶切合成胰岛素[97].Herb Boyer与风险投资人Robert Swanson一起创立了美国基因泰克(Genentch)生物技术公司,成功实现了人源胰岛素的工业化大量生产. ...
Making, cloning, and the expression of human insulin genes in bacteria: the path to humulin
1
2021
... 胰岛素分子的氨基酸序列在20世纪50年代已经解析清楚,随后科学家也尝试了很多策略用于人工合成胰岛素,但由于合成步骤太长、效率太低而无法实现大量工业生产,因此主要来源依然是通过粉碎牛或猪内脏进行提取,每生产1 kg胰岛素则需要超过8 t动物胰脏.20世纪60~70年代,重组DNA技术取得了重大突破.美国加州大学洛杉矶分校微生物学家Herb Boyer教授团队通过重组DNA技术,实现了人源胰岛素的全合成:他们采用化学从头合成的方法,先合成出编码胰岛素A链和B链两条DNA分子,将这些DNA插入细菌的基因组使它们能够合成出胰岛素A链和B链分子,再将两条肽链进行分离纯化,最后运用化学手段将两条链连接,获得人源胰岛素蛋白分子(图10)[94-96].此外,也可以通过大肠杆菌或巴斯德毕赤酵母直接合成单体或胰岛素原,进一步经过二硫键匹配和酶切合成胰岛素[97].Herb Boyer与风险投资人Robert Swanson一起创立了美国基因泰克(Genentch)生物技术公司,成功实现了人源胰岛素的工业化大量生产. ...
1
2015
... 青蒿素属于植物萜类天然产物,是20世纪60~70年代以屠呦呦教授为代表的我国科学家,从植物青蒿中提取分离出来的具有高效抗疟活性的有机分子[98].目前主要从黄花蒿中直接提取,或提取黄花蒿中含量较高的青蒿酸,然后经化学半合成制得.由于受气候条件和地区环境等因素影响,产量严重受限.随着越来越多关键酶基因被表征鉴定,青蒿酸的生物合成途径已获得全部解析;通过在微生物细胞中异源表达,进行生物合成青蒿酸引起科学家高度关注.在这方面最具代表性的是美国加州大学伯克利分校Jay Keasling教授实验室(图11)[99],他们从乙酰辅酶A起始,通过甲羟戊酸途径合成异戊烯基焦磷酸(isopentenyl phosphate, IPP)及其异构物二甲基烯丙基焦磷酸(dimethylallyl diphosphate, DMAPP),然后经法尼基焦磷酸合成酶(farnesyl diphosphate synthase,FPS)催化IPP聚合生成法尼基焦磷酸(farnesyl diphosphate,FPP);再采用基因工程手段,将关键编码紫穗槐-4,11-二烯合酶(amorpha-4,11-diene synthase,ADS)基因引入大肠杆菌中,实现了由FPP到紫穗槐-4,11-二烯(amorpha-4,11-diene)的催化过程;随即又在酵母中表达P450紫穗槐二烯氧化酶CYP71AV1、还原酶CPR1及细胞色素CYB5等基因,将紫穗槐二烯水解为青蒿醇(artemisinic alcohol),并进一步氧化为青蒿醛(artemisinic aldehyde);再经青蒿醛双键还原酶DBR2和醛脱氢酶ALDH1两步反应,分别将青蒿醛催化生成二氢青蒿醛(dihydroartemisinic aldehyde)以及二氢青蒿酸(dihydro-artemisinic acid,DHAA),发酵产量可以达到25 g/L.随后以青蒿酸为底物进行后续化学转化,威尔金森催化剂[RhCl(PPh3)3]催化下可以高达94∶6的非对映选择性得到二氢青蒿酸,酯化后使用钼酸锂促进的双氧水歧化反应得到烯丙基过氧化物,进一步在一锅内发生重排、氧化、成环步骤可生成最终的青蒿素,四步的化学合成总收率可以达到45%.值得注意的是,赛诺菲公司的Turconi等使用光照反应策略可将二氢青蒿酸以55%的收率转化为目标青蒿素[100],并可实现370 kg的放大量生产[图11(b)].2015年,Michael George等对光照条件下二氢青蒿酸到青蒿素这一关键步骤进行了进一步优化[101],他们发现以液态二氧化碳为溶剂可以50%的收率得到目标青蒿素,而使用四氢呋喃/水的混合溶剂体系可进一步提升收率到66%,并可省去烦琐的分离纯化步骤,这些反应条件更加符合绿色化学的发展要求,具有很好的放大应用潜力[图11(b)]. ...
1
2015
... 青蒿素属于植物萜类天然产物,是20世纪60~70年代以屠呦呦教授为代表的我国科学家,从植物青蒿中提取分离出来的具有高效抗疟活性的有机分子[98].目前主要从黄花蒿中直接提取,或提取黄花蒿中含量较高的青蒿酸,然后经化学半合成制得.由于受气候条件和地区环境等因素影响,产量严重受限.随着越来越多关键酶基因被表征鉴定,青蒿酸的生物合成途径已获得全部解析;通过在微生物细胞中异源表达,进行生物合成青蒿酸引起科学家高度关注.在这方面最具代表性的是美国加州大学伯克利分校Jay Keasling教授实验室(图11)[99],他们从乙酰辅酶A起始,通过甲羟戊酸途径合成异戊烯基焦磷酸(isopentenyl phosphate, IPP)及其异构物二甲基烯丙基焦磷酸(dimethylallyl diphosphate, DMAPP),然后经法尼基焦磷酸合成酶(farnesyl diphosphate synthase,FPS)催化IPP聚合生成法尼基焦磷酸(farnesyl diphosphate,FPP);再采用基因工程手段,将关键编码紫穗槐-4,11-二烯合酶(amorpha-4,11-diene synthase,ADS)基因引入大肠杆菌中,实现了由FPP到紫穗槐-4,11-二烯(amorpha-4,11-diene)的催化过程;随即又在酵母中表达P450紫穗槐二烯氧化酶CYP71AV1、还原酶CPR1及细胞色素CYB5等基因,将紫穗槐二烯水解为青蒿醇(artemisinic alcohol),并进一步氧化为青蒿醛(artemisinic aldehyde);再经青蒿醛双键还原酶DBR2和醛脱氢酶ALDH1两步反应,分别将青蒿醛催化生成二氢青蒿醛(dihydroartemisinic aldehyde)以及二氢青蒿酸(dihydro-artemisinic acid,DHAA),发酵产量可以达到25 g/L.随后以青蒿酸为底物进行后续化学转化,威尔金森催化剂[RhCl(PPh3)3]催化下可以高达94∶6的非对映选择性得到二氢青蒿酸,酯化后使用钼酸锂促进的双氧水歧化反应得到烯丙基过氧化物,进一步在一锅内发生重排、氧化、成环步骤可生成最终的青蒿素,四步的化学合成总收率可以达到45%.值得注意的是,赛诺菲公司的Turconi等使用光照反应策略可将二氢青蒿酸以55%的收率转化为目标青蒿素[100],并可实现370 kg的放大量生产[图11(b)].2015年,Michael George等对光照条件下二氢青蒿酸到青蒿素这一关键步骤进行了进一步优化[101],他们发现以液态二氧化碳为溶剂可以50%的收率得到目标青蒿素,而使用四氢呋喃/水的混合溶剂体系可进一步提升收率到66%,并可省去烦琐的分离纯化步骤,这些反应条件更加符合绿色化学的发展要求,具有很好的放大应用潜力[图11(b)]. ...
High-level semi-synthetic production of the potent antimalarial artemisinin
1
2013
... 青蒿素属于植物萜类天然产物,是20世纪60~70年代以屠呦呦教授为代表的我国科学家,从植物青蒿中提取分离出来的具有高效抗疟活性的有机分子[98].目前主要从黄花蒿中直接提取,或提取黄花蒿中含量较高的青蒿酸,然后经化学半合成制得.由于受气候条件和地区环境等因素影响,产量严重受限.随着越来越多关键酶基因被表征鉴定,青蒿酸的生物合成途径已获得全部解析;通过在微生物细胞中异源表达,进行生物合成青蒿酸引起科学家高度关注.在这方面最具代表性的是美国加州大学伯克利分校Jay Keasling教授实验室(图11)[99],他们从乙酰辅酶A起始,通过甲羟戊酸途径合成异戊烯基焦磷酸(isopentenyl phosphate, IPP)及其异构物二甲基烯丙基焦磷酸(dimethylallyl diphosphate, DMAPP),然后经法尼基焦磷酸合成酶(farnesyl diphosphate synthase,FPS)催化IPP聚合生成法尼基焦磷酸(farnesyl diphosphate,FPP);再采用基因工程手段,将关键编码紫穗槐-4,11-二烯合酶(amorpha-4,11-diene synthase,ADS)基因引入大肠杆菌中,实现了由FPP到紫穗槐-4,11-二烯(amorpha-4,11-diene)的催化过程;随即又在酵母中表达P450紫穗槐二烯氧化酶CYP71AV1、还原酶CPR1及细胞色素CYB5等基因,将紫穗槐二烯水解为青蒿醇(artemisinic alcohol),并进一步氧化为青蒿醛(artemisinic aldehyde);再经青蒿醛双键还原酶DBR2和醛脱氢酶ALDH1两步反应,分别将青蒿醛催化生成二氢青蒿醛(dihydroartemisinic aldehyde)以及二氢青蒿酸(dihydro-artemisinic acid,DHAA),发酵产量可以达到25 g/L.随后以青蒿酸为底物进行后续化学转化,威尔金森催化剂[RhCl(PPh3)3]催化下可以高达94∶6的非对映选择性得到二氢青蒿酸,酯化后使用钼酸锂促进的双氧水歧化反应得到烯丙基过氧化物,进一步在一锅内发生重排、氧化、成环步骤可生成最终的青蒿素,四步的化学合成总收率可以达到45%.值得注意的是,赛诺菲公司的Turconi等使用光照反应策略可将二氢青蒿酸以55%的收率转化为目标青蒿素[100],并可实现370 kg的放大量生产[图11(b)].2015年,Michael George等对光照条件下二氢青蒿酸到青蒿素这一关键步骤进行了进一步优化[101],他们发现以液态二氧化碳为溶剂可以50%的收率得到目标青蒿素,而使用四氢呋喃/水的混合溶剂体系可进一步提升收率到66%,并可省去烦琐的分离纯化步骤,这些反应条件更加符合绿色化学的发展要求,具有很好的放大应用潜力[图11(b)]. ...
Semisynthetic artemisinin, the chemical path to industrial production
1
2014
... 青蒿素属于植物萜类天然产物,是20世纪60~70年代以屠呦呦教授为代表的我国科学家,从植物青蒿中提取分离出来的具有高效抗疟活性的有机分子[98].目前主要从黄花蒿中直接提取,或提取黄花蒿中含量较高的青蒿酸,然后经化学半合成制得.由于受气候条件和地区环境等因素影响,产量严重受限.随着越来越多关键酶基因被表征鉴定,青蒿酸的生物合成途径已获得全部解析;通过在微生物细胞中异源表达,进行生物合成青蒿酸引起科学家高度关注.在这方面最具代表性的是美国加州大学伯克利分校Jay Keasling教授实验室(图11)[99],他们从乙酰辅酶A起始,通过甲羟戊酸途径合成异戊烯基焦磷酸(isopentenyl phosphate, IPP)及其异构物二甲基烯丙基焦磷酸(dimethylallyl diphosphate, DMAPP),然后经法尼基焦磷酸合成酶(farnesyl diphosphate synthase,FPS)催化IPP聚合生成法尼基焦磷酸(farnesyl diphosphate,FPP);再采用基因工程手段,将关键编码紫穗槐-4,11-二烯合酶(amorpha-4,11-diene synthase,ADS)基因引入大肠杆菌中,实现了由FPP到紫穗槐-4,11-二烯(amorpha-4,11-diene)的催化过程;随即又在酵母中表达P450紫穗槐二烯氧化酶CYP71AV1、还原酶CPR1及细胞色素CYB5等基因,将紫穗槐二烯水解为青蒿醇(artemisinic alcohol),并进一步氧化为青蒿醛(artemisinic aldehyde);再经青蒿醛双键还原酶DBR2和醛脱氢酶ALDH1两步反应,分别将青蒿醛催化生成二氢青蒿醛(dihydroartemisinic aldehyde)以及二氢青蒿酸(dihydro-artemisinic acid,DHAA),发酵产量可以达到25 g/L.随后以青蒿酸为底物进行后续化学转化,威尔金森催化剂[RhCl(PPh3)3]催化下可以高达94∶6的非对映选择性得到二氢青蒿酸,酯化后使用钼酸锂促进的双氧水歧化反应得到烯丙基过氧化物,进一步在一锅内发生重排、氧化、成环步骤可生成最终的青蒿素,四步的化学合成总收率可以达到45%.值得注意的是,赛诺菲公司的Turconi等使用光照反应策略可将二氢青蒿酸以55%的收率转化为目标青蒿素[100],并可实现370 kg的放大量生产[图11(b)].2015年,Michael George等对光照条件下二氢青蒿酸到青蒿素这一关键步骤进行了进一步优化[101],他们发现以液态二氧化碳为溶剂可以50%的收率得到目标青蒿素,而使用四氢呋喃/水的混合溶剂体系可进一步提升收率到66%,并可省去烦琐的分离纯化步骤,这些反应条件更加符合绿色化学的发展要求,具有很好的放大应用潜力[图11(b)]. ...
Applying green chemistry to the photochemical route to artemisinin
1
2015
... 青蒿素属于植物萜类天然产物,是20世纪60~70年代以屠呦呦教授为代表的我国科学家,从植物青蒿中提取分离出来的具有高效抗疟活性的有机分子[98].目前主要从黄花蒿中直接提取,或提取黄花蒿中含量较高的青蒿酸,然后经化学半合成制得.由于受气候条件和地区环境等因素影响,产量严重受限.随着越来越多关键酶基因被表征鉴定,青蒿酸的生物合成途径已获得全部解析;通过在微生物细胞中异源表达,进行生物合成青蒿酸引起科学家高度关注.在这方面最具代表性的是美国加州大学伯克利分校Jay Keasling教授实验室(图11)[99],他们从乙酰辅酶A起始,通过甲羟戊酸途径合成异戊烯基焦磷酸(isopentenyl phosphate, IPP)及其异构物二甲基烯丙基焦磷酸(dimethylallyl diphosphate, DMAPP),然后经法尼基焦磷酸合成酶(farnesyl diphosphate synthase,FPS)催化IPP聚合生成法尼基焦磷酸(farnesyl diphosphate,FPP);再采用基因工程手段,将关键编码紫穗槐-4,11-二烯合酶(amorpha-4,11-diene synthase,ADS)基因引入大肠杆菌中,实现了由FPP到紫穗槐-4,11-二烯(amorpha-4,11-diene)的催化过程;随即又在酵母中表达P450紫穗槐二烯氧化酶CYP71AV1、还原酶CPR1及细胞色素CYB5等基因,将紫穗槐二烯水解为青蒿醇(artemisinic alcohol),并进一步氧化为青蒿醛(artemisinic aldehyde);再经青蒿醛双键还原酶DBR2和醛脱氢酶ALDH1两步反应,分别将青蒿醛催化生成二氢青蒿醛(dihydroartemisinic aldehyde)以及二氢青蒿酸(dihydro-artemisinic acid,DHAA),发酵产量可以达到25 g/L.随后以青蒿酸为底物进行后续化学转化,威尔金森催化剂[RhCl(PPh3)3]催化下可以高达94∶6的非对映选择性得到二氢青蒿酸,酯化后使用钼酸锂促进的双氧水歧化反应得到烯丙基过氧化物,进一步在一锅内发生重排、氧化、成环步骤可生成最终的青蒿素,四步的化学合成总收率可以达到45%.值得注意的是,赛诺菲公司的Turconi等使用光照反应策略可将二氢青蒿酸以55%的收率转化为目标青蒿素[100],并可实现370 kg的放大量生产[图11(b)].2015年,Michael George等对光照条件下二氢青蒿酸到青蒿素这一关键步骤进行了进一步优化[101],他们发现以液态二氧化碳为溶剂可以50%的收率得到目标青蒿素,而使用四氢呋喃/水的混合溶剂体系可进一步提升收率到66%,并可省去烦琐的分离纯化步骤,这些反应条件更加符合绿色化学的发展要求,具有很好的放大应用潜力[图11(b)]. ...
Chemistry and biology of the tetrahydroisoquinoline antitumor antibiotics
1
2002
... 双四氢异喹啉生物碱是一类具有重要抗肿瘤活性的天然产物[102],但化学合成路线步骤烦琐[103],而单一的生物合成方法效率偏低[104],严重制约了该类天然产物的大量获取及后续活性研究.2018年,日本东京农工大学Hideaki Oikawa与Hiroki Oguri课题组首先通过化学方法,巧妙设计并制备出醛类底物;再以磷酸泛素化Pictet-pengler环合酶SfmC为关键酶,催化形成核心的五并环骨架,该反应可在一锅内同时实现两个碳-碳键、三个碳-氮键的构建及硫酯的还原,避免了活性中间体的分离纯化,极大提高了合成效率;随后采用化学方法实现二级胺的甲基化、侧链水解切断及氧化官能团转化.该方法仅需4~5步反应,即可实现3种双四氢异喹啉生物碱(沙弗拉霉素A、Fmoc-保护的沙弗拉霉素Y3和jorunnamycin A)的全合成,是目前已报道的沙弗拉霉素最短合成路线(图12)[105].该研究充分展示了化学-生物交叉融合策略在天然产物全合成中的独特优势,也为这类生物碱的规模化制备提供了新的工艺路线. ...
Asymmetric synthesis of isoquinoline alkaloids: 2004-2015
1
2016
... 双四氢异喹啉生物碱是一类具有重要抗肿瘤活性的天然产物[102],但化学合成路线步骤烦琐[103],而单一的生物合成方法效率偏低[104],严重制约了该类天然产物的大量获取及后续活性研究.2018年,日本东京农工大学Hideaki Oikawa与Hiroki Oguri课题组首先通过化学方法,巧妙设计并制备出醛类底物;再以磷酸泛素化Pictet-pengler环合酶SfmC为关键酶,催化形成核心的五并环骨架,该反应可在一锅内同时实现两个碳-碳键、三个碳-氮键的构建及硫酯的还原,避免了活性中间体的分离纯化,极大提高了合成效率;随后采用化学方法实现二级胺的甲基化、侧链水解切断及氧化官能团转化.该方法仅需4~5步反应,即可实现3种双四氢异喹啉生物碱(沙弗拉霉素A、Fmoc-保护的沙弗拉霉素Y3和jorunnamycin A)的全合成,是目前已报道的沙弗拉霉素最短合成路线(图12)[105].该研究充分展示了化学-生物交叉融合策略在天然产物全合成中的独特优势,也为这类生物碱的规模化制备提供了新的工艺路线. ...
Recent progress in the metabolic engineering of alkaloids in plant systems
1
2013
... 双四氢异喹啉生物碱是一类具有重要抗肿瘤活性的天然产物[102],但化学合成路线步骤烦琐[103],而单一的生物合成方法效率偏低[104],严重制约了该类天然产物的大量获取及后续活性研究.2018年,日本东京农工大学Hideaki Oikawa与Hiroki Oguri课题组首先通过化学方法,巧妙设计并制备出醛类底物;再以磷酸泛素化Pictet-pengler环合酶SfmC为关键酶,催化形成核心的五并环骨架,该反应可在一锅内同时实现两个碳-碳键、三个碳-氮键的构建及硫酯的还原,避免了活性中间体的分离纯化,极大提高了合成效率;随后采用化学方法实现二级胺的甲基化、侧链水解切断及氧化官能团转化.该方法仅需4~5步反应,即可实现3种双四氢异喹啉生物碱(沙弗拉霉素A、Fmoc-保护的沙弗拉霉素Y3和jorunnamycin A)的全合成,是目前已报道的沙弗拉霉素最短合成路线(图12)[105].该研究充分展示了化学-生物交叉融合策略在天然产物全合成中的独特优势,也为这类生物碱的规模化制备提供了新的工艺路线. ...
Chemo-enzymatic total syntheses of jorunnamycin A, saframycin A, and N?Fmoc saframycin Y3
1
2018
... 双四氢异喹啉生物碱是一类具有重要抗肿瘤活性的天然产物[102],但化学合成路线步骤烦琐[103],而单一的生物合成方法效率偏低[104],严重制约了该类天然产物的大量获取及后续活性研究.2018年,日本东京农工大学Hideaki Oikawa与Hiroki Oguri课题组首先通过化学方法,巧妙设计并制备出醛类底物;再以磷酸泛素化Pictet-pengler环合酶SfmC为关键酶,催化形成核心的五并环骨架,该反应可在一锅内同时实现两个碳-碳键、三个碳-氮键的构建及硫酯的还原,避免了活性中间体的分离纯化,极大提高了合成效率;随后采用化学方法实现二级胺的甲基化、侧链水解切断及氧化官能团转化.该方法仅需4~5步反应,即可实现3种双四氢异喹啉生物碱(沙弗拉霉素A、Fmoc-保护的沙弗拉霉素Y3和jorunnamycin A)的全合成,是目前已报道的沙弗拉霉素最短合成路线(图12)[105].该研究充分展示了化学-生物交叉融合策略在天然产物全合成中的独特优势,也为这类生物碱的规模化制备提供了新的工艺路线. ...
Stereodivergent, chemoenzymatic synthesis of azaphilone natural products
1
2019
... 嗜氮酮是一类来源于真菌的并环骨架天然产物,具有抗癌、抗病毒、抗炎等多种生物活性,其中仅有的一个四取代碳手性中心对其参与的生理过程发挥重要作用,但立体多样性合成的空白严重限制了该类天然产物的药用研究.2019年,美国密歇根大学Alison Narayan小组通过序列相似性网络(sequence similarity network,SSN)策略对黄素依赖的单加氧酶进行系统分析,发现了可催化氧化去芳构化反应的一系列新酶,并从中确定了两种可实现相反对映选择性的新酶:AzaH(R选择性)和AfoD(S选择性).作者采用化学合成法得到烯酮中间体,通过酶催化的氧化去芳构化反应分别得到两种绝对构型相反的手性双环烯酮醇,再借助Knoevenagel缩合反应高选择性地实现了三并环骨架的构建,最后成功得到天然产物trichoflectin的两个对映异构体,该策略还进一步应用于嗜氮酮同系天然产物deflectin与lunatoic acid A的全合成(图13)[106-107],充分展示了化学-生物结合策略的优势互补性. ...
Chemoenzymatic total synthesis of natural products
1
2021
... 嗜氮酮是一类来源于真菌的并环骨架天然产物,具有抗癌、抗病毒、抗炎等多种生物活性,其中仅有的一个四取代碳手性中心对其参与的生理过程发挥重要作用,但立体多样性合成的空白严重限制了该类天然产物的药用研究.2019年,美国密歇根大学Alison Narayan小组通过序列相似性网络(sequence similarity network,SSN)策略对黄素依赖的单加氧酶进行系统分析,发现了可催化氧化去芳构化反应的一系列新酶,并从中确定了两种可实现相反对映选择性的新酶:AzaH(R选择性)和AfoD(S选择性).作者采用化学合成法得到烯酮中间体,通过酶催化的氧化去芳构化反应分别得到两种绝对构型相反的手性双环烯酮醇,再借助Knoevenagel缩合反应高选择性地实现了三并环骨架的构建,最后成功得到天然产物trichoflectin的两个对映异构体,该策略还进一步应用于嗜氮酮同系天然产物deflectin与lunatoic acid A的全合成(图13)[106-107],充分展示了化学-生物结合策略的优势互补性. ...
Cloning of a putative high-affinity kainate receptor expressed predominantly in hippocampal CA3 cells
1
1991
... 卡英酸(kainic acid)又称红藻酸,是一种来源于海洋藻类的天然有机分子,可作为离子型谷氨酸受体抑制剂应用于神经性疾病的治疗[108].尽管该化合物结构并不复杂,但相连的三个手性中心为其立体选择性合成带来了很大挑战,已报道的化学合成路线有70余条[109],但这些方法的实际应用仍存在巨大困难.2019年,加州大学Bradley Moore团队对产生卡英酸的两种海藻进行全基因组测序[110],鉴定出两个关键酶:N-异戊二烯基转移酶KabA与双加氧酶KabC,并在大肠杆菌中进行异源表达,尝试建立卡英酸的生物合成途径.最后为实现卡英酸的高效制备,作者发展了化学-酶交叉合成体系(图14):以谷氨酸为起始原料,与3-甲基-2-丁烯醛进行还原胺化反应,以56%收率得到卡英酸前体,再使用纯化的DsKabC酶催化后续的成环步骤,46%收率得到卡英酸;更为重要的是,作者使用大肠杆菌全细胞体系可以57%收率实现卡英酸的克级制备,避免了化学中间体及酶的分离纯化,大大提高了合成效率. ...
Total Syntheses of (-)‐α‐kainic acid
1
2012
... 卡英酸(kainic acid)又称红藻酸,是一种来源于海洋藻类的天然有机分子,可作为离子型谷氨酸受体抑制剂应用于神经性疾病的治疗[108].尽管该化合物结构并不复杂,但相连的三个手性中心为其立体选择性合成带来了很大挑战,已报道的化学合成路线有70余条[109],但这些方法的实际应用仍存在巨大困难.2019年,加州大学Bradley Moore团队对产生卡英酸的两种海藻进行全基因组测序[110],鉴定出两个关键酶:N-异戊二烯基转移酶KabA与双加氧酶KabC,并在大肠杆菌中进行异源表达,尝试建立卡英酸的生物合成途径.最后为实现卡英酸的高效制备,作者发展了化学-酶交叉合成体系(图14):以谷氨酸为起始原料,与3-甲基-2-丁烯醛进行还原胺化反应,以56%收率得到卡英酸前体,再使用纯化的DsKabC酶催化后续的成环步骤,46%收率得到卡英酸;更为重要的是,作者使用大肠杆菌全细胞体系可以57%收率实现卡英酸的克级制备,避免了化学中间体及酶的分离纯化,大大提高了合成效率. ...
Scalable biosynthesis of the seaweed neurochemical, kainic acid
1
2019
... 卡英酸(kainic acid)又称红藻酸,是一种来源于海洋藻类的天然有机分子,可作为离子型谷氨酸受体抑制剂应用于神经性疾病的治疗[108].尽管该化合物结构并不复杂,但相连的三个手性中心为其立体选择性合成带来了很大挑战,已报道的化学合成路线有70余条[109],但这些方法的实际应用仍存在巨大困难.2019年,加州大学Bradley Moore团队对产生卡英酸的两种海藻进行全基因组测序[110],鉴定出两个关键酶:N-异戊二烯基转移酶KabA与双加氧酶KabC,并在大肠杆菌中进行异源表达,尝试建立卡英酸的生物合成途径.最后为实现卡英酸的高效制备,作者发展了化学-酶交叉合成体系(图14):以谷氨酸为起始原料,与3-甲基-2-丁烯醛进行还原胺化反应,以56%收率得到卡英酸前体,再使用纯化的DsKabC酶催化后续的成环步骤,46%收率得到卡英酸;更为重要的是,作者使用大肠杆菌全细胞体系可以57%收率实现卡英酸的克级制备,避免了化学中间体及酶的分离纯化,大大提高了合成效率. ...
Podophyllotoxin: history, recent advances and future prospects
1
2021
... 作为植物来源的木质素类天然代谢产物,鬼臼毒素(podophyllotoxin)及衍生物因其所特有的抗肿瘤、抗病毒等生物活性而备受关注[111].鬼臼毒素的传统来源为植物提取,受限于植物生长缓慢,同时过度开采带来环境破坏等不利因素,不能满足人类需要.相比之下,近年来利用化学-酶法全合成鬼臼毒素取得了显著进展.2019年,Chang Weichen等报道了一种α-酮戊二酸依赖型双加氧酶(2-ODD-PH)[112],可催化脱氧鬼臼毒素(deoxypodophyllotoxin)到亚太因(yatein)的碳-碳成键反应.基于此发现,Michael Fuchs和Hans Renata先后发展了两种化学-酶法合成鬼臼毒素新策略.Michael Fuchs课题组提供的路线[图15(a)],首先利用金属锌促进胡椒醛与烯丙基溴化物反应制得高烯丙醇中间体,并在铑催化剂作用下合成消旋的羟化亚太因(rac-hydroxyyatein),再用2-ODD-PH酶催化环化,通过动力学拆分以39%收率及95% ee对映选择性得到鬼臼毒素的差向异构体,最后经化学法反转C7位点上的羟基手性获得鬼臼毒素[113].同时Hans Renata课题组发展了手性底物转化的合成路线[图15(b)],第一步采用化学缩合反应制得亚太因,第二步借助2-ODD-PH酶催化亚太因进行环合反应,获得鬼臼毒素前体,第三步通过化学法在C7位点进行羟基化反应,从而合成终产物鬼臼毒素,其分离收率达到58%[114].与化学全合成方法相比,上述两条化学-生物融合策略在收率及立体控制上均有明显提升,有望实现鬼臼毒素规模化全合成. ...
Reaction mechanism of a nonheme iron enzyme catalyzed oxidative cyclization via C-C bond formation
1
2019
... 作为植物来源的木质素类天然代谢产物,鬼臼毒素(podophyllotoxin)及衍生物因其所特有的抗肿瘤、抗病毒等生物活性而备受关注[111].鬼臼毒素的传统来源为植物提取,受限于植物生长缓慢,同时过度开采带来环境破坏等不利因素,不能满足人类需要.相比之下,近年来利用化学-酶法全合成鬼臼毒素取得了显著进展.2019年,Chang Weichen等报道了一种α-酮戊二酸依赖型双加氧酶(2-ODD-PH)[112],可催化脱氧鬼臼毒素(deoxypodophyllotoxin)到亚太因(yatein)的碳-碳成键反应.基于此发现,Michael Fuchs和Hans Renata先后发展了两种化学-酶法合成鬼臼毒素新策略.Michael Fuchs课题组提供的路线[图15(a)],首先利用金属锌促进胡椒醛与烯丙基溴化物反应制得高烯丙醇中间体,并在铑催化剂作用下合成消旋的羟化亚太因(rac-hydroxyyatein),再用2-ODD-PH酶催化环化,通过动力学拆分以39%收率及95% ee对映选择性得到鬼臼毒素的差向异构体,最后经化学法反转C7位点上的羟基手性获得鬼臼毒素[113].同时Hans Renata课题组发展了手性底物转化的合成路线[图15(b)],第一步采用化学缩合反应制得亚太因,第二步借助2-ODD-PH酶催化亚太因进行环合反应,获得鬼臼毒素前体,第三步通过化学法在C7位点进行羟基化反应,从而合成终产物鬼臼毒素,其分离收率达到58%[114].与化学全合成方法相比,上述两条化学-生物融合策略在收率及立体控制上均有明显提升,有望实现鬼臼毒素规模化全合成. ...
Chemoenzymatic total synthesis of deoxy-, epi-, and podophyllotoxin and a biocatalytic kinetic resolution of dibenzylbutyrolactones
1
2019
... 作为植物来源的木质素类天然代谢产物,鬼臼毒素(podophyllotoxin)及衍生物因其所特有的抗肿瘤、抗病毒等生物活性而备受关注[111].鬼臼毒素的传统来源为植物提取,受限于植物生长缓慢,同时过度开采带来环境破坏等不利因素,不能满足人类需要.相比之下,近年来利用化学-酶法全合成鬼臼毒素取得了显著进展.2019年,Chang Weichen等报道了一种α-酮戊二酸依赖型双加氧酶(2-ODD-PH)[112],可催化脱氧鬼臼毒素(deoxypodophyllotoxin)到亚太因(yatein)的碳-碳成键反应.基于此发现,Michael Fuchs和Hans Renata先后发展了两种化学-酶法合成鬼臼毒素新策略.Michael Fuchs课题组提供的路线[图15(a)],首先利用金属锌促进胡椒醛与烯丙基溴化物反应制得高烯丙醇中间体,并在铑催化剂作用下合成消旋的羟化亚太因(rac-hydroxyyatein),再用2-ODD-PH酶催化环化,通过动力学拆分以39%收率及95% ee对映选择性得到鬼臼毒素的差向异构体,最后经化学法反转C7位点上的羟基手性获得鬼臼毒素[113].同时Hans Renata课题组发展了手性底物转化的合成路线[图15(b)],第一步采用化学缩合反应制得亚太因,第二步借助2-ODD-PH酶催化亚太因进行环合反应,获得鬼臼毒素前体,第三步通过化学法在C7位点进行羟基化反应,从而合成终产物鬼臼毒素,其分离收率达到58%[114].与化学全合成方法相比,上述两条化学-生物融合策略在收率及立体控制上均有明显提升,有望实现鬼臼毒素规模化全合成. ...
Asymmetric chemoenzymatic synthesis of ((-))-podophyllotoxin and related aryltetralin lignans
1
2019
... 作为植物来源的木质素类天然代谢产物,鬼臼毒素(podophyllotoxin)及衍生物因其所特有的抗肿瘤、抗病毒等生物活性而备受关注[111].鬼臼毒素的传统来源为植物提取,受限于植物生长缓慢,同时过度开采带来环境破坏等不利因素,不能满足人类需要.相比之下,近年来利用化学-酶法全合成鬼臼毒素取得了显著进展.2019年,Chang Weichen等报道了一种α-酮戊二酸依赖型双加氧酶(2-ODD-PH)[112],可催化脱氧鬼臼毒素(deoxypodophyllotoxin)到亚太因(yatein)的碳-碳成键反应.基于此发现,Michael Fuchs和Hans Renata先后发展了两种化学-酶法合成鬼臼毒素新策略.Michael Fuchs课题组提供的路线[图15(a)],首先利用金属锌促进胡椒醛与烯丙基溴化物反应制得高烯丙醇中间体,并在铑催化剂作用下合成消旋的羟化亚太因(rac-hydroxyyatein),再用2-ODD-PH酶催化环化,通过动力学拆分以39%收率及95% ee对映选择性得到鬼臼毒素的差向异构体,最后经化学法反转C7位点上的羟基手性获得鬼臼毒素[113].同时Hans Renata课题组发展了手性底物转化的合成路线[图15(b)],第一步采用化学缩合反应制得亚太因,第二步借助2-ODD-PH酶催化亚太因进行环合反应,获得鬼臼毒素前体,第三步通过化学法在C7位点进行羟基化反应,从而合成终产物鬼臼毒素,其分离收率达到58%[114].与化学全合成方法相比,上述两条化学-生物融合策略在收率及立体控制上均有明显提升,有望实现鬼臼毒素规模化全合成. ...
Producing aglycons of ginsenosides in bakers' yeast
1
2014
... 药物开发离不开种类多样、功能丰富的天然产物.近两个世纪来,得益于化学分离手段的进步,人类逐渐摆脱了利用动植物/微生物混合提取物的原始用药方式,并迈向了使用高纯度单体天然产物的阶段;更为重要的是,随着有机合成方法和技术的不断提高,天然产物的化学全合成得到快速全面发展,化学家甚至为众多药物活性天然产物分子开发出了多条全合成工艺路线.然而,对很多结构复杂的活性天然产物分子,由于合成步骤多、工艺路线长、选择性控制难、总收率低等问题,严重制约了其实际生产和广泛应用.随着分子生物学、系统生物学及合成生物学的快速发展,尤其是定向进化及基因组测序技术的不断进步,成千上万的微生物及动植物基因组信息不断被挖掘,人类已探明生物体内众多天然产物的全基因合成途径与酶学机制,这为构建细胞工厂从头异源合成天然产物提供了便利条件,同时也为合成生物学变革天然产物全合成提供契机.聚酮类、生物碱类、萜类、甾体类、大环内酰胺类、核苷类等复杂天然产物的生物合成已取得较大进展;值得一提的是,我国科学家在牛胰岛素、酵母丙氨酸转移核糖核酸、青蒿素及人参皂苷等多个重要天然产物的生物合成方面也取得多项突破性进展[115],形成了良好的研究基础.尽管如此,天然产物生物全合成仍存在诸多挑战性问题亟待解决,如类天然产物新分子的生物合成一直是该领域的重要课题[116-120];含有多个手性中心的立体复杂天然产物的生物合成还存在效率较低、机制不明、途径优化困难等问题;此外结合金属催化、光催化、电催化及自由基化学的最新进展,如何实现生物合成与惰性键活化及多级串联反应等新型策略的有机融合,实现天然产物的多样性全合成也是具有潜力的发展方向. ...
合成生物学在含氟化合物生产中的应用
1
2020
... 药物开发离不开种类多样、功能丰富的天然产物.近两个世纪来,得益于化学分离手段的进步,人类逐渐摆脱了利用动植物/微生物混合提取物的原始用药方式,并迈向了使用高纯度单体天然产物的阶段;更为重要的是,随着有机合成方法和技术的不断提高,天然产物的化学全合成得到快速全面发展,化学家甚至为众多药物活性天然产物分子开发出了多条全合成工艺路线.然而,对很多结构复杂的活性天然产物分子,由于合成步骤多、工艺路线长、选择性控制难、总收率低等问题,严重制约了其实际生产和广泛应用.随着分子生物学、系统生物学及合成生物学的快速发展,尤其是定向进化及基因组测序技术的不断进步,成千上万的微生物及动植物基因组信息不断被挖掘,人类已探明生物体内众多天然产物的全基因合成途径与酶学机制,这为构建细胞工厂从头异源合成天然产物提供了便利条件,同时也为合成生物学变革天然产物全合成提供契机.聚酮类、生物碱类、萜类、甾体类、大环内酰胺类、核苷类等复杂天然产物的生物合成已取得较大进展;值得一提的是,我国科学家在牛胰岛素、酵母丙氨酸转移核糖核酸、青蒿素及人参皂苷等多个重要天然产物的生物合成方面也取得多项突破性进展[115],形成了良好的研究基础.尽管如此,天然产物生物全合成仍存在诸多挑战性问题亟待解决,如类天然产物新分子的生物合成一直是该领域的重要课题[116-120];含有多个手性中心的立体复杂天然产物的生物合成还存在效率较低、机制不明、途径优化困难等问题;此外结合金属催化、光催化、电催化及自由基化学的最新进展,如何实现生物合成与惰性键活化及多级串联反应等新型策略的有机融合,实现天然产物的多样性全合成也是具有潜力的发展方向. ...
Applications of synthetic biology in the production of fluorinated compounds
1
2020
... 药物开发离不开种类多样、功能丰富的天然产物.近两个世纪来,得益于化学分离手段的进步,人类逐渐摆脱了利用动植物/微生物混合提取物的原始用药方式,并迈向了使用高纯度单体天然产物的阶段;更为重要的是,随着有机合成方法和技术的不断提高,天然产物的化学全合成得到快速全面发展,化学家甚至为众多药物活性天然产物分子开发出了多条全合成工艺路线.然而,对很多结构复杂的活性天然产物分子,由于合成步骤多、工艺路线长、选择性控制难、总收率低等问题,严重制约了其实际生产和广泛应用.随着分子生物学、系统生物学及合成生物学的快速发展,尤其是定向进化及基因组测序技术的不断进步,成千上万的微生物及动植物基因组信息不断被挖掘,人类已探明生物体内众多天然产物的全基因合成途径与酶学机制,这为构建细胞工厂从头异源合成天然产物提供了便利条件,同时也为合成生物学变革天然产物全合成提供契机.聚酮类、生物碱类、萜类、甾体类、大环内酰胺类、核苷类等复杂天然产物的生物合成已取得较大进展;值得一提的是,我国科学家在牛胰岛素、酵母丙氨酸转移核糖核酸、青蒿素及人参皂苷等多个重要天然产物的生物合成方面也取得多项突破性进展[115],形成了良好的研究基础.尽管如此,天然产物生物全合成仍存在诸多挑战性问题亟待解决,如类天然产物新分子的生物合成一直是该领域的重要课题[116-120];含有多个手性中心的立体复杂天然产物的生物合成还存在效率较低、机制不明、途径优化困难等问题;此外结合金属催化、光催化、电催化及自由基化学的最新进展,如何实现生物合成与惰性键活化及多级串联反应等新型策略的有机融合,实现天然产物的多样性全合成也是具有潜力的发展方向. ...
Biochemistry: biosynthesis of an organofluorine molecule
0
2002
Synthetic biology approaches to fluorinated polyketides
0
2015
Asymmetric construction of stereogenic carbon centers featuring a trifluoromethyl group from prochiral trifluoromethylated substrates
0
2011
Update 1 of: asymmetric fluorination, trifluoromethylation, and perfluoroalkylation reactions
1
2008
... 药物开发离不开种类多样、功能丰富的天然产物.近两个世纪来,得益于化学分离手段的进步,人类逐渐摆脱了利用动植物/微生物混合提取物的原始用药方式,并迈向了使用高纯度单体天然产物的阶段;更为重要的是,随着有机合成方法和技术的不断提高,天然产物的化学全合成得到快速全面发展,化学家甚至为众多药物活性天然产物分子开发出了多条全合成工艺路线.然而,对很多结构复杂的活性天然产物分子,由于合成步骤多、工艺路线长、选择性控制难、总收率低等问题,严重制约了其实际生产和广泛应用.随着分子生物学、系统生物学及合成生物学的快速发展,尤其是定向进化及基因组测序技术的不断进步,成千上万的微生物及动植物基因组信息不断被挖掘,人类已探明生物体内众多天然产物的全基因合成途径与酶学机制,这为构建细胞工厂从头异源合成天然产物提供了便利条件,同时也为合成生物学变革天然产物全合成提供契机.聚酮类、生物碱类、萜类、甾体类、大环内酰胺类、核苷类等复杂天然产物的生物合成已取得较大进展;值得一提的是,我国科学家在牛胰岛素、酵母丙氨酸转移核糖核酸、青蒿素及人参皂苷等多个重要天然产物的生物合成方面也取得多项突破性进展[115],形成了良好的研究基础.尽管如此,天然产物生物全合成仍存在诸多挑战性问题亟待解决,如类天然产物新分子的生物合成一直是该领域的重要课题[116-120];含有多个手性中心的立体复杂天然产物的生物合成还存在效率较低、机制不明、途径优化困难等问题;此外结合金属催化、光催化、电催化及自由基化学的最新进展,如何实现生物合成与惰性键活化及多级串联反应等新型策略的有机融合,实现天然产物的多样性全合成也是具有潜力的发展方向. ...
Engineering Escherichia coli for the synthesis of taxadiene, a key intermediate in the biosynthesis of taxol
1
2001
... 总之,对天然产物的全合成,无论化学方法还是生物学方法,都各有其特点,如何将两者优势互补,相互促进,从而实现更多天然产物高效全合成,是未来重要发展方向;例如广谱抗癌药物紫杉醇的微生物异源生物合成,国内外多个团队已经取得了良好进展,可以获得三环紫杉二烯(taxadiene)及三环紫杉二烯醇(taxadienols)[121-127],在大肠杆菌中的产量比之前报道提高了万倍以上,相信不久的将来借助化学和生物互补的途径定能实现其高效全合成.同时,如何借助当今蓬勃发展的人工智能技术,实现生物元件(如酶元件、启动子元件等)的挖掘、设计与改造,以及人工生物合成途径拼装等过程的智能化、自动化、高效化,助力天然产物异源再造,将成为本领域未来发展的另一新趋势[128-130].展望生物合成策略在天然产物及其类似物合成中的应用,更大的进步将会不断涌现,并为人类健康与社会发展贡献更大力量. ...
Microbial transformation of cephalomannine by Luteibacter sp
0
2007
Isoprenoid pathway optimization for taxol precursor overproduction in Escherichia coli
0
2010
Production of the anticancer drug taxol in Taxus Baccata suspension cultures: a review
0
2011
Paclitaxel: biosynthesis, production and future prospects
0
2014
Paclitaxel biosynthesis: adenylation and thiolation domains of an NRPS TycA PheAT module produce various arylisoserine CoA thioesters
0
2017
Fungal cell wall and methyl-β-cyclodextrin synergistically enhance paclitaxel biosynthesis and secretion in Corylus avellana cell suspension culture
1
2020
... 总之,对天然产物的全合成,无论化学方法还是生物学方法,都各有其特点,如何将两者优势互补,相互促进,从而实现更多天然产物高效全合成,是未来重要发展方向;例如广谱抗癌药物紫杉醇的微生物异源生物合成,国内外多个团队已经取得了良好进展,可以获得三环紫杉二烯(taxadiene)及三环紫杉二烯醇(taxadienols)[121-127],在大肠杆菌中的产量比之前报道提高了万倍以上,相信不久的将来借助化学和生物互补的途径定能实现其高效全合成.同时,如何借助当今蓬勃发展的人工智能技术,实现生物元件(如酶元件、启动子元件等)的挖掘、设计与改造,以及人工生物合成途径拼装等过程的智能化、自动化、高效化,助力天然产物异源再造,将成为本领域未来发展的另一新趋势[128-130].展望生物合成策略在天然产物及其类似物合成中的应用,更大的进步将会不断涌现,并为人类健康与社会发展贡献更大力量. ...
机器学习助力酶定向进化
1
2020
... 总之,对天然产物的全合成,无论化学方法还是生物学方法,都各有其特点,如何将两者优势互补,相互促进,从而实现更多天然产物高效全合成,是未来重要发展方向;例如广谱抗癌药物紫杉醇的微生物异源生物合成,国内外多个团队已经取得了良好进展,可以获得三环紫杉二烯(taxadiene)及三环紫杉二烯醇(taxadienols)[121-127],在大肠杆菌中的产量比之前报道提高了万倍以上,相信不久的将来借助化学和生物互补的途径定能实现其高效全合成.同时,如何借助当今蓬勃发展的人工智能技术,实现生物元件(如酶元件、启动子元件等)的挖掘、设计与改造,以及人工生物合成途径拼装等过程的智能化、自动化、高效化,助力天然产物异源再造,将成为本领域未来发展的另一新趋势[128-130].展望生物合成策略在天然产物及其类似物合成中的应用,更大的进步将会不断涌现,并为人类健康与社会发展贡献更大力量. ...
Machine learning-assisted enzyme directed evolution
1
2020
... 总之,对天然产物的全合成,无论化学方法还是生物学方法,都各有其特点,如何将两者优势互补,相互促进,从而实现更多天然产物高效全合成,是未来重要发展方向;例如广谱抗癌药物紫杉醇的微生物异源生物合成,国内外多个团队已经取得了良好进展,可以获得三环紫杉二烯(taxadiene)及三环紫杉二烯醇(taxadienols)[121-127],在大肠杆菌中的产量比之前报道提高了万倍以上,相信不久的将来借助化学和生物互补的途径定能实现其高效全合成.同时,如何借助当今蓬勃发展的人工智能技术,实现生物元件(如酶元件、启动子元件等)的挖掘、设计与改造,以及人工生物合成途径拼装等过程的智能化、自动化、高效化,助力天然产物异源再造,将成为本领域未来发展的另一新趋势[128-130].展望生物合成策略在天然产物及其类似物合成中的应用,更大的进步将会不断涌现,并为人类健康与社会发展贡献更大力量. ...
Can machine learning revolutionize directed evolution of selective enzymes?
0
2019
Introduction to machine learning
1
2014
... 总之,对天然产物的全合成,无论化学方法还是生物学方法,都各有其特点,如何将两者优势互补,相互促进,从而实现更多天然产物高效全合成,是未来重要发展方向;例如广谱抗癌药物紫杉醇的微生物异源生物合成,国内外多个团队已经取得了良好进展,可以获得三环紫杉二烯(taxadiene)及三环紫杉二烯醇(taxadienols)[121-127],在大肠杆菌中的产量比之前报道提高了万倍以上,相信不久的将来借助化学和生物互补的途径定能实现其高效全合成.同时,如何借助当今蓬勃发展的人工智能技术,实现生物元件(如酶元件、启动子元件等)的挖掘、设计与改造,以及人工生物合成途径拼装等过程的智能化、自动化、高效化,助力天然产物异源再造,将成为本领域未来发展的另一新趋势[128-130].展望生物合成策略在天然产物及其类似物合成中的应用,更大的进步将会不断涌现,并为人类健康与社会发展贡献更大力量. ...





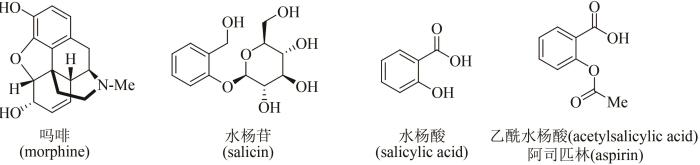
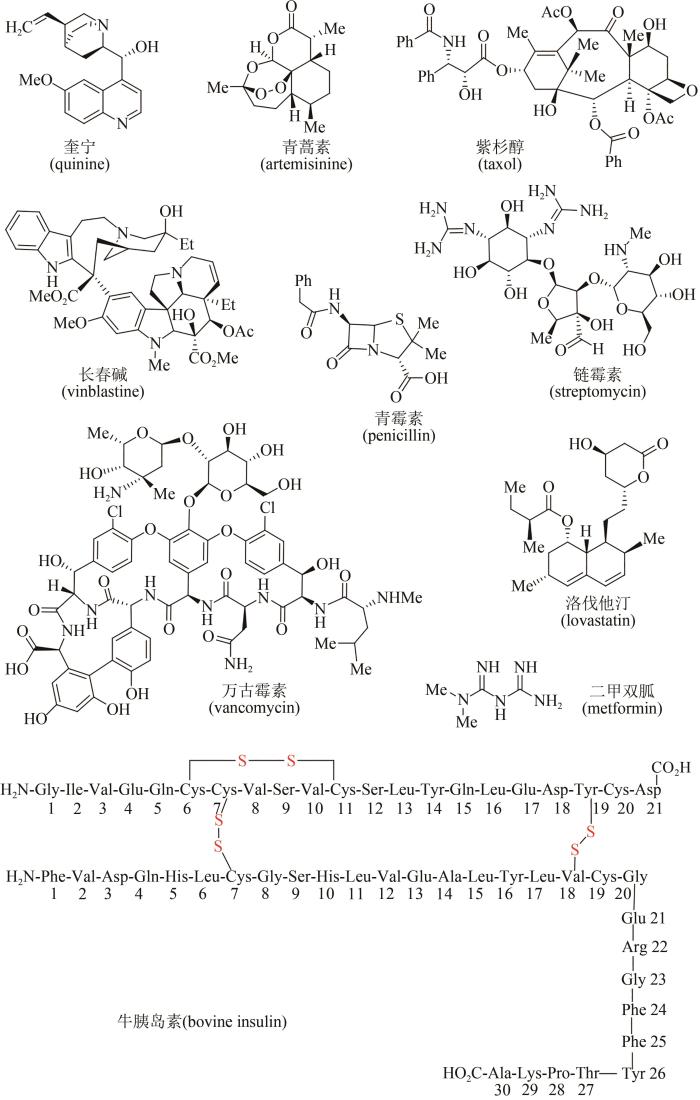
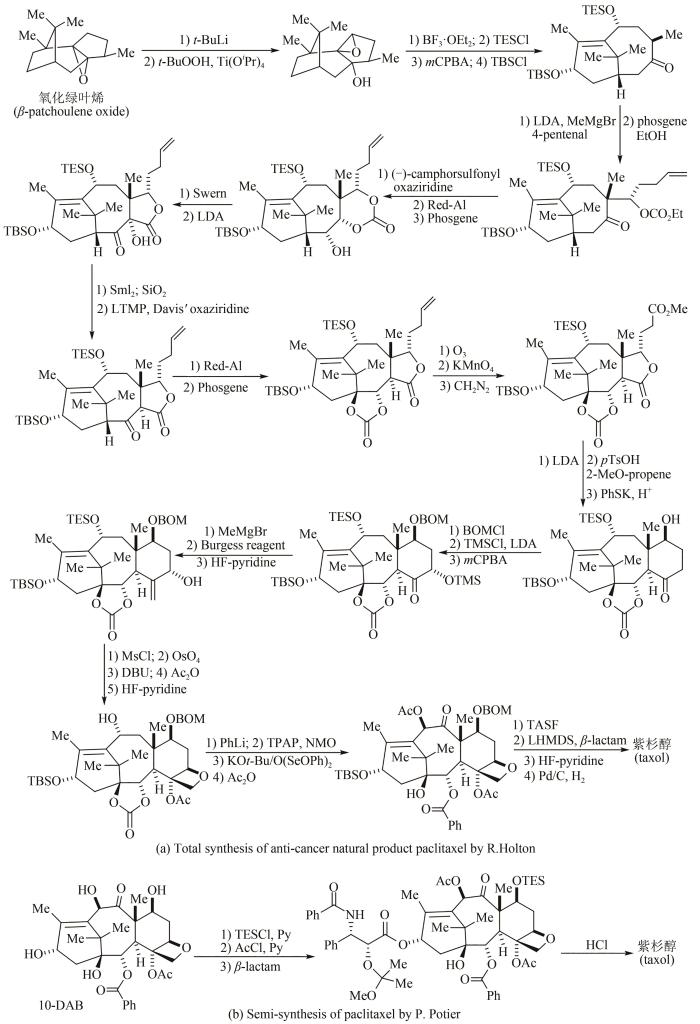
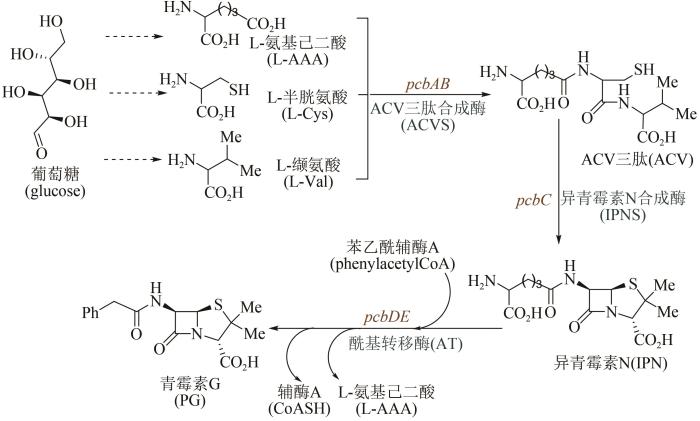
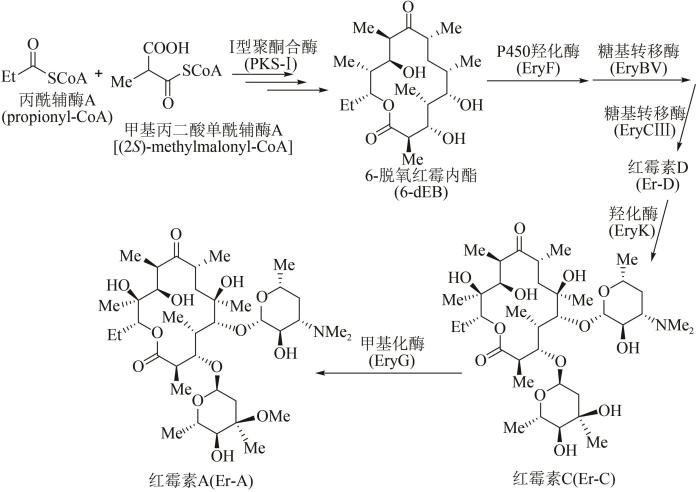
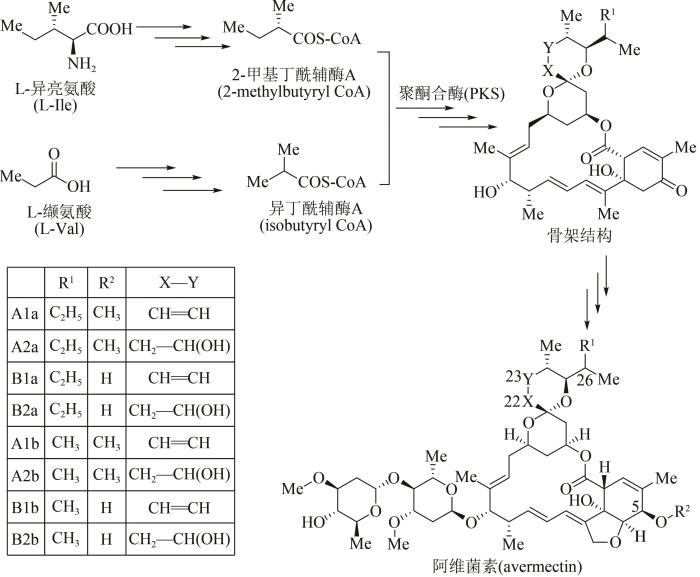
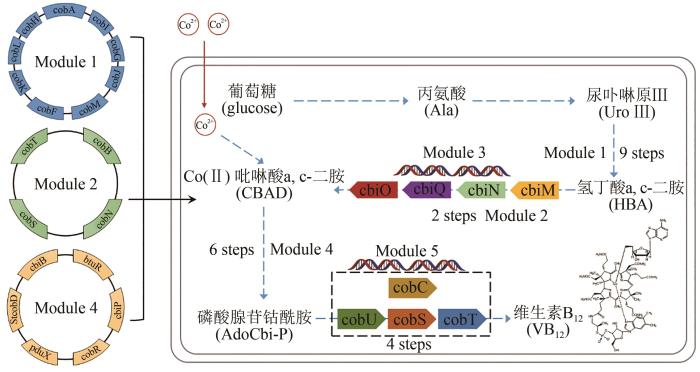
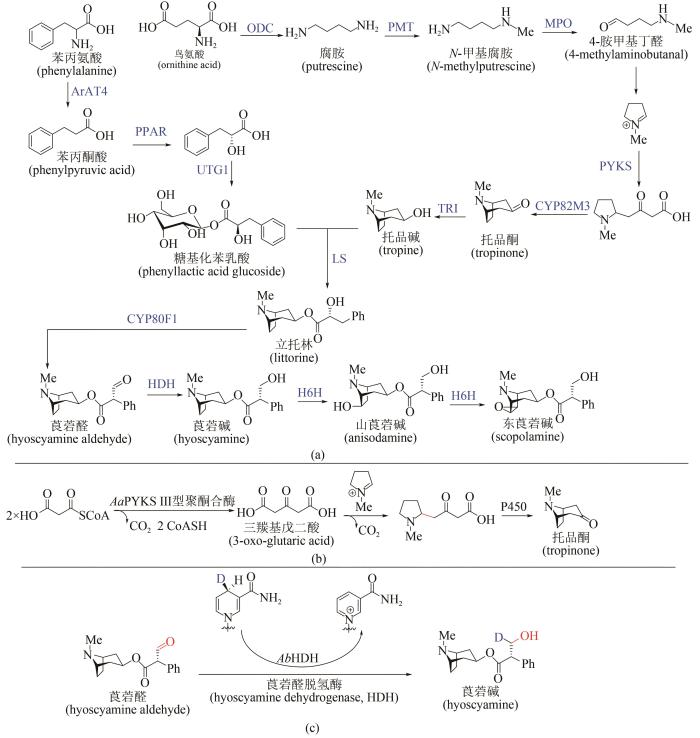
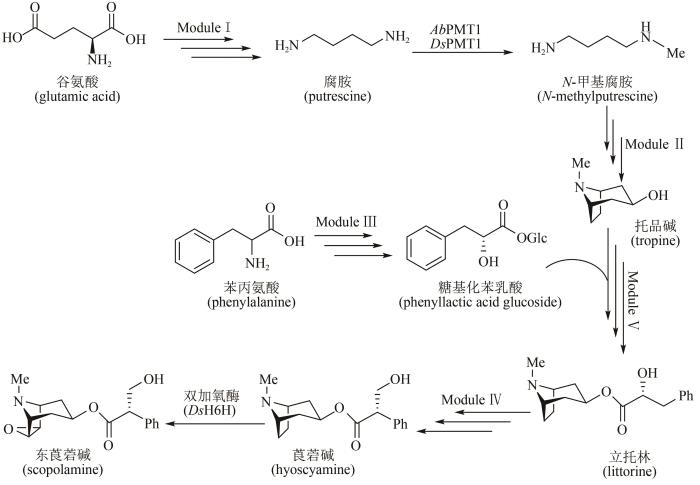
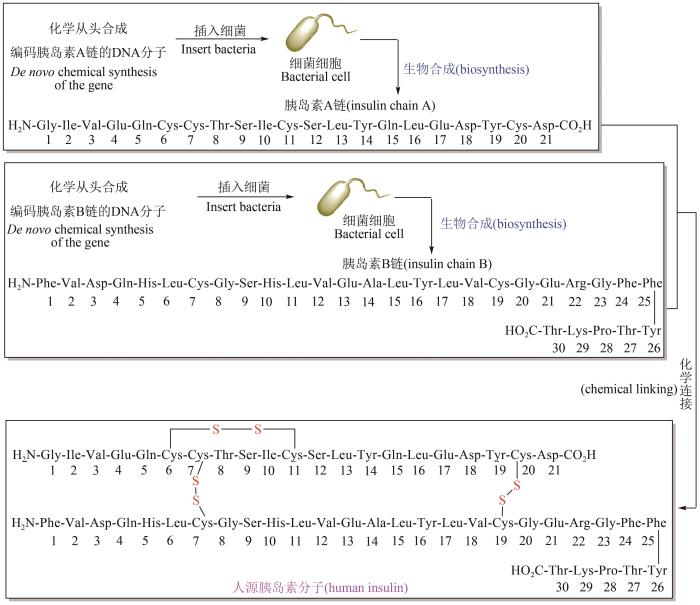
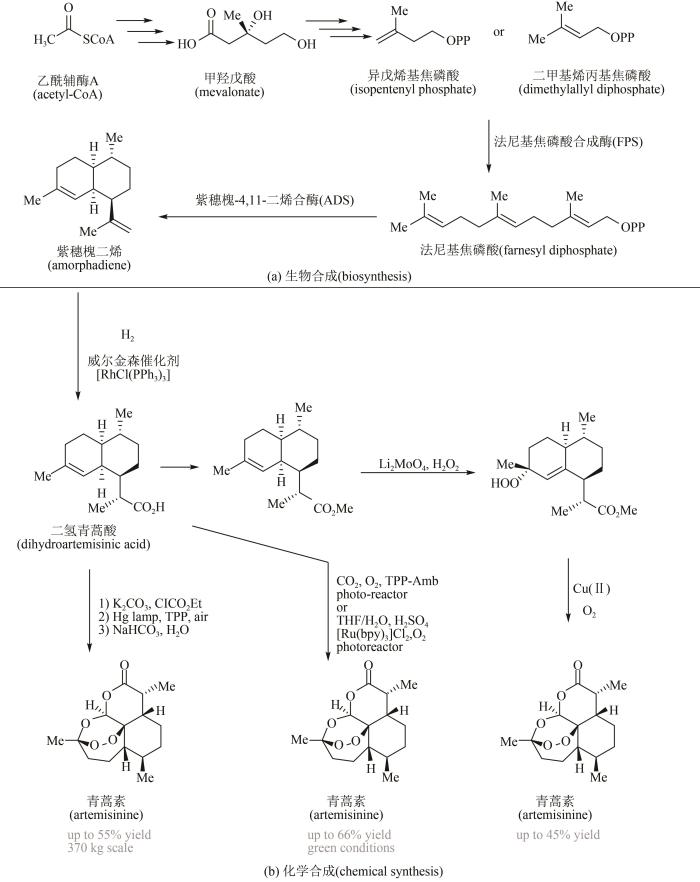
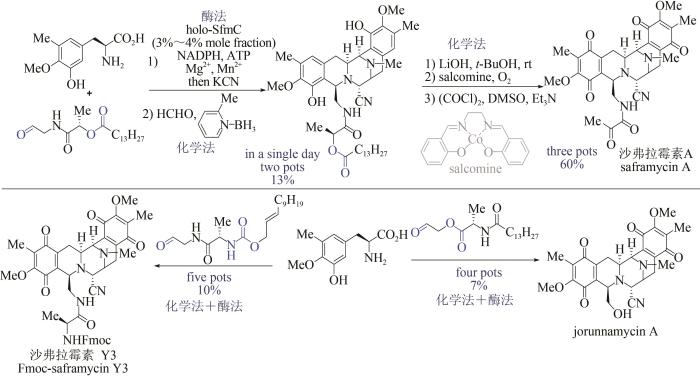
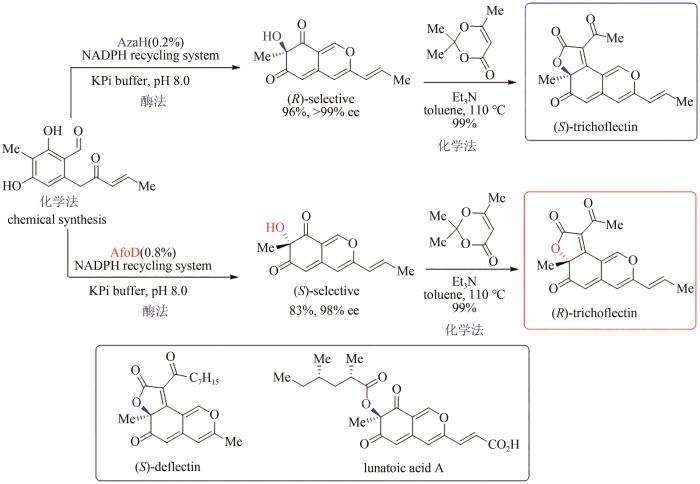
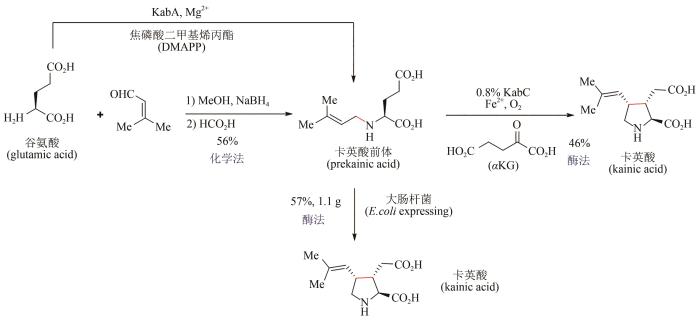
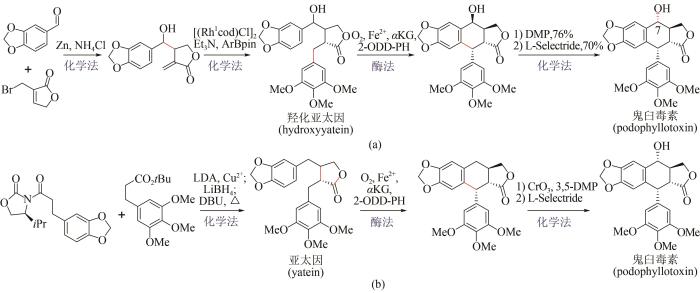


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}