Education in green chemistry and in sustainable chemistry: perspectives towards sustainability
1
2021
... 传统的化学合成存在许多弊端,如多数反应需使用昂贵且对环境有害的过渡金属催化剂及具有毒性的有机溶剂,且往往需要严苛的反应条件,这很大程度上造成了能源浪费与环境污染[1 ] .酶(enzyme)作为一种重要的生物催化剂,具有催化效率高、专一性强且反应条件温和等优势,因此被认为是绿色且对环境友好的催化剂.然而,酶促反应也存在着诸多不足之处:①酶的热稳定性差;②酶的底物普适性较差(酶催化具有专一性,通常只能催化单一底物,对于其他结构相似的底物催化活性往往较低甚至没有);③酶对有机溶剂的耐受性差;④酶可催化的反应类型较少[2 ] .这些缺点限制了酶在大规模工业生产中的应用,因此,目前化学催化在工业化生产中仍占据着主流地位[3 ] .而定向进化技术的兴起,很大程度上改善了上述问题,大大促进了酶在化合物合成上的应用. ...
Directed evolution: creating biocatalysts for the future
1
1996
... 传统的化学合成存在许多弊端,如多数反应需使用昂贵且对环境有害的过渡金属催化剂及具有毒性的有机溶剂,且往往需要严苛的反应条件,这很大程度上造成了能源浪费与环境污染[1 ] .酶(enzyme)作为一种重要的生物催化剂,具有催化效率高、专一性强且反应条件温和等优势,因此被认为是绿色且对环境友好的催化剂.然而,酶促反应也存在着诸多不足之处:①酶的热稳定性差;②酶的底物普适性较差(酶催化具有专一性,通常只能催化单一底物,对于其他结构相似的底物催化活性往往较低甚至没有);③酶对有机溶剂的耐受性差;④酶可催化的反应类型较少[2 ] .这些缺点限制了酶在大规模工业生产中的应用,因此,目前化学催化在工业化生产中仍占据着主流地位[3 ] .而定向进化技术的兴起,很大程度上改善了上述问题,大大促进了酶在化合物合成上的应用. ...
The importance of synthetic chemistry in the pharmaceutical industry
1
2019
... 传统的化学合成存在许多弊端,如多数反应需使用昂贵且对环境有害的过渡金属催化剂及具有毒性的有机溶剂,且往往需要严苛的反应条件,这很大程度上造成了能源浪费与环境污染[1 ] .酶(enzyme)作为一种重要的生物催化剂,具有催化效率高、专一性强且反应条件温和等优势,因此被认为是绿色且对环境友好的催化剂.然而,酶促反应也存在着诸多不足之处:①酶的热稳定性差;②酶的底物普适性较差(酶催化具有专一性,通常只能催化单一底物,对于其他结构相似的底物催化活性往往较低甚至没有);③酶对有机溶剂的耐受性差;④酶可催化的反应类型较少[2 ] .这些缺点限制了酶在大规模工业生产中的应用,因此,目前化学催化在工业化生产中仍占据着主流地位[3 ] .而定向进化技术的兴起,很大程度上改善了上述问题,大大促进了酶在化合物合成上的应用. ...
Directed evolution empowered redesign of natural proteins for the sustainable production of chemicals and pharmaceuticals
1
2019
... 酶的定向进化,旨在试管中模拟自然进化过程,通过提高基因突变率和设计特殊的筛选、选择方法,快速获得拥有特定性能的酶[4 ] .因此,定向进化又被称为“代替自然选择的上帝之手”,为试管中的达尔文主义[5 ] .酶的定向进化通常包括三个步骤[6 ] :①通过对蛋白编码序列进行随机突变、定点突变或重组构建基因突变体库;②定向筛选、选择以获得具有改进表型的突变体;③以该突变体作为下一轮基因多样化的起点,进行定向进化的迭代,直到获得性能最优的突变体(图1 ). ...
Directed evolution of biocatalysts
1
1999
... 酶的定向进化,旨在试管中模拟自然进化过程,通过提高基因突变率和设计特殊的筛选、选择方法,快速获得拥有特定性能的酶[4 ] .因此,定向进化又被称为“代替自然选择的上帝之手”,为试管中的达尔文主义[5 ] .酶的定向进化通常包括三个步骤[6 ] :①通过对蛋白编码序列进行随机突变、定点突变或重组构建基因突变体库;②定向筛选、选择以获得具有改进表型的突变体;③以该突变体作为下一轮基因多样化的起点,进行定向进化的迭代,直到获得性能最优的突变体(图1 ). ...
Methods for the directed evolution of proteins
2
2015
... 酶的定向进化,旨在试管中模拟自然进化过程,通过提高基因突变率和设计特殊的筛选、选择方法,快速获得拥有特定性能的酶[4 ] .因此,定向进化又被称为“代替自然选择的上帝之手”,为试管中的达尔文主义[5 ] .酶的定向进化通常包括三个步骤[6 ] :①通过对蛋白编码序列进行随机突变、定点突变或重组构建基因突变体库;②定向筛选、选择以获得具有改进表型的突变体;③以该突变体作为下一轮基因多样化的起点,进行定向进化的迭代,直到获得性能最优的突变体(图1 ). ...
... 创建序列覆盖率高、多样性强的突变体库,能最大程度挖掘不同氨基酸序列与其对应表型之间的关系,理论上能够提高获得理想突变体的概率.然而,绝大部分筛选和选择技术通量低、准确性差,导致难以实现对目标蛋白质序列全面且深入的挖掘.因此,开发更快速、灵敏、准确的高通量筛选技术,是提高定向进化效率的关键所在.尽管将酶的活性与细胞存活率相关联能够快速评估含有超过109 突变体的突变体库[7 ] ,但该方法仅限于工程改造与抗生素耐药性或基础代谢相关的酶.另外,采用一些更精确的分析方法,如液相色谱或气相色谱法、紫外-可见分光光度法、核磁共振和质谱法等,可通过对酶催化反应的底物或产物进行定量分析,从而精确筛选酶突变体库,然而这些方法往往受限于通量较低的问题[6 ] .近年来,一些微型化、自动化和集成化的新型技术体系为一些代谢途径关键酶、优势菌株、催化元件在定向进化过程中的高通量筛选和选择提供了优良的解决方案. ...
Directed evolution: methodologies and applications
7
2021
... 在过去的40多年中,定向进化技术获得了充分的发展和应用[7 ] .一方面,结合以易错PCR(error-prone PCR)[8 ] 和同源重组(DNA recombination)[9 ] 为主的随机突变,与定点饱和突变(site-Saturation mutagenesis)[10 ] ,我们既可以对蛋白序列进行“高覆盖”采样研究,亦可针对“热点”序列进行深度挖掘,这增加了突变体库的多样性,进而提高了获得目标性状突变体的可能性[11 ] .另一方面,近年来基于基因编辑技术的体内进化策略(in vivo evolution)进一步拓宽了定向进化的应用范围[7 ] .流式分选技术(flow cytometry)[12 ] 、基质辅助激光解吸电离飞行时间质谱(MALDI-ToF,MS)[13 ] 、液滴微流控技术(droplet microfluidics)[14 ] 等高通量筛选技术的兴起以及连续定向进化(continuous evolution)技术的发展[7 ] ,使得突变体库的筛选速率提高了百倍以上.最近,综合了计算机辅助学习和设计、自动化生物合成平台和高通量筛选/选择技术的集成研究平台,更为未来针对海量酶的高效、深度进化提供了可行方案[15 -16 ] . ...
... [7 ].流式分选技术(flow cytometry)[12 ] 、基质辅助激光解吸电离飞行时间质谱(MALDI-ToF,MS)[13 ] 、液滴微流控技术(droplet microfluidics)[14 ] 等高通量筛选技术的兴起以及连续定向进化(continuous evolution)技术的发展[7 ] ,使得突变体库的筛选速率提高了百倍以上.最近,综合了计算机辅助学习和设计、自动化生物合成平台和高通量筛选/选择技术的集成研究平台,更为未来针对海量酶的高效、深度进化提供了可行方案[15 -16 ] . ...
... [7 ],使得突变体库的筛选速率提高了百倍以上.最近,综合了计算机辅助学习和设计、自动化生物合成平台和高通量筛选/选择技术的集成研究平台,更为未来针对海量酶的高效、深度进化提供了可行方案[15 -16 ] . ...
... 基因突变体库的另一大类构建方法是体内突变法,即在胞内诱导特定基因的突变,无需进行基因克隆与转化,很大程度上缩短了定向进化的实验周期.例如早期开发的基于单链DNA重组的多元自动化基因组工程技术(multiplex automated genome engineering,MAGE),是针对大肠杆菌基因组上特定基因进行体内诱变的重要策略
[25 ] .近年来,CRISPR-Cas介导的重组技术实现了在原核与真核细胞中对目的基因进行高效的片段插入、删除和替换(
图2 a),大大提高了原核与真核细胞中重组的效率
[26 -27 ] ,因此也发展了一批高效的胞内蛋白质定向进化工具.例如将CRISPR-Cas系统和MAGE整合后,基因重组效率由3%提高到98%,插入/替换效率提高至70%
[28 ] ;将CRISPR-Cas9与epPCR偶联(CasPER)可以向酵母基因组中任意基因的600 bp内引入随机突变
[29 ] .以上两种方法,皆通过带有突变点的外源核酸片段经同源重组引入突变点,因此需额外提供大量的核酸片段来覆盖整个目标基因.与其不同的是,J. Dueber 课题组和D. Schaffer课题组基于CRISPR技术,在大肠杆菌中合作开发了一种促进细胞内特定基因进化的平台——EvolvR系统
[30 ] .该系统首先由Nickase切口酶(nCas9)在目标序列切割出一个缺口,然后利用融合的高错DNA聚合酶执行切口平移,在此过程中通过改造Nickase和DNA聚合酶实现定点基因的突变(
图2 b).相对于野生型,通过该方法可使目的基因的突变率提高10
7 倍,突变范围达到350 bp.2020年该团队将EvolvR系统用于酿酒酵母中,可同时在两个目的基因的40 bp范围内引入随机突变
[31 ] .但由于突变率较低且突变窗口较小,其应用有一定局限性.
图2 基于CRISPR的体内突变方法<sup>[<xref ref-type="bibr" rid="R7">7</xref>]</sup> a—基于CRISPR-Cas9同源重组的突变;b—基于nCas9和DNA聚合酶I的突变;c—基于nCas9和碱基编辑器的突变 ...
... a—基于CRISPR-Cas9同源重组的突变;b—基于nCas9和DNA聚合酶I的突变;c—基于nCas9和碱基编辑器的突变
CRISPR-assisted <i>in vivo</i> mutagenesis<sup>[<xref ref-type="bibr" rid="R7">7</xref>]</sup> a—CRISPR-Cas9-HDR; b—Random mutagenesis induced by nCas9-E. coli DNA PolI (error-prone) hybrid proteins; c—Gene mutangenesis caused by nCas9-deaminase hybrid proteins ...
... 创建序列覆盖率高、多样性强的突变体库,能最大程度挖掘不同氨基酸序列与其对应表型之间的关系,理论上能够提高获得理想突变体的概率.然而,绝大部分筛选和选择技术通量低、准确性差,导致难以实现对目标蛋白质序列全面且深入的挖掘.因此,开发更快速、灵敏、准确的高通量筛选技术,是提高定向进化效率的关键所在.尽管将酶的活性与细胞存活率相关联能够快速评估含有超过109 突变体的突变体库[7 ] ,但该方法仅限于工程改造与抗生素耐药性或基础代谢相关的酶.另外,采用一些更精确的分析方法,如液相色谱或气相色谱法、紫外-可见分光光度法、核磁共振和质谱法等,可通过对酶催化反应的底物或产物进行定量分析,从而精确筛选酶突变体库,然而这些方法往往受限于通量较低的问题[6 ] .近年来,一些微型化、自动化和集成化的新型技术体系为一些代谢途径关键酶、优势菌株、催化元件在定向进化过程中的高通量筛选和选择提供了优良的解决方案. ...
... 早期,定向进化有效地提高了天然酶对有机溶剂的耐受性,这对于改善底物在溶剂中的溶解度以进行大规模工艺制造十分必要[7 ] .蛋白酶subtilisin E可用于水解酪蛋白(casein),但其在有机溶剂N ,N- 二甲基甲酰胺(DMF)中的稳定性极差,导致其在60% DMF溶液中的催化活性还不到其在水溶液中活性的0.5%.F. Arnold课题组[113 -114 ] 结合随机突变和定点突变,经过三轮诱变和筛选,引入10个氨基酸突变(D60N,D97G,Q103R,N218S,G131D,E156G,N181S,S182G,S188P和T255A),得到了在60% DMF溶液中的催化效率提高了256倍的subtilisin E-PC3,并且PC3的活性水平与野生型subtilisin E在水溶液中的活性水平相当.此研究展示了“定向进化”能有效提高酶在体外环境中稳定性以及催化活性. ...
A method for random mutagenesis of a defined DNA segment using a modified polymerase chain reaction
2
1989
... 在过去的40多年中,定向进化技术获得了充分的发展和应用[7 ] .一方面,结合以易错PCR(error-prone PCR)[8 ] 和同源重组(DNA recombination)[9 ] 为主的随机突变,与定点饱和突变(site-Saturation mutagenesis)[10 ] ,我们既可以对蛋白序列进行“高覆盖”采样研究,亦可针对“热点”序列进行深度挖掘,这增加了突变体库的多样性,进而提高了获得目标性状突变体的可能性[11 ] .另一方面,近年来基于基因编辑技术的体内进化策略(in vivo evolution)进一步拓宽了定向进化的应用范围[7 ] .流式分选技术(flow cytometry)[12 ] 、基质辅助激光解吸电离飞行时间质谱(MALDI-ToF,MS)[13 ] 、液滴微流控技术(droplet microfluidics)[14 ] 等高通量筛选技术的兴起以及连续定向进化(continuous evolution)技术的发展[7 ] ,使得突变体库的筛选速率提高了百倍以上.最近,综合了计算机辅助学习和设计、自动化生物合成平台和高通量筛选/选择技术的集成研究平台,更为未来针对海量酶的高效、深度进化提供了可行方案[15 -16 ] . ...
... 体外突变法主要包括可以产生随机突变的易错PCR(error-prone PCR,epPCR)[8 ] 、DNA改组(DNA shuffling)[9 ] 、可以产生非随机突变的定点饱和突变(site-saturation mutagenesis,SSM)[10 ] 、序列饱和突变(sequence saturation mutagenesis,SeSaM)[17 ] 、合成文库(synthetic library generation)[18 ] 等,这些传统体外突变技术成为单个酶定向进化的有力工具,随着分子生物学的快速发展,构建体外突变体库的新方法也不断涌现. ...
DNA shuffling by random fragmentation and reassembly: in vitro recombination for molecular evolution
2
1994
... 在过去的40多年中,定向进化技术获得了充分的发展和应用[7 ] .一方面,结合以易错PCR(error-prone PCR)[8 ] 和同源重组(DNA recombination)[9 ] 为主的随机突变,与定点饱和突变(site-Saturation mutagenesis)[10 ] ,我们既可以对蛋白序列进行“高覆盖”采样研究,亦可针对“热点”序列进行深度挖掘,这增加了突变体库的多样性,进而提高了获得目标性状突变体的可能性[11 ] .另一方面,近年来基于基因编辑技术的体内进化策略(in vivo evolution)进一步拓宽了定向进化的应用范围[7 ] .流式分选技术(flow cytometry)[12 ] 、基质辅助激光解吸电离飞行时间质谱(MALDI-ToF,MS)[13 ] 、液滴微流控技术(droplet microfluidics)[14 ] 等高通量筛选技术的兴起以及连续定向进化(continuous evolution)技术的发展[7 ] ,使得突变体库的筛选速率提高了百倍以上.最近,综合了计算机辅助学习和设计、自动化生物合成平台和高通量筛选/选择技术的集成研究平台,更为未来针对海量酶的高效、深度进化提供了可行方案[15 -16 ] . ...
... 体外突变法主要包括可以产生随机突变的易错PCR(error-prone PCR,epPCR)[8 ] 、DNA改组(DNA shuffling)[9 ] 、可以产生非随机突变的定点饱和突变(site-saturation mutagenesis,SSM)[10 ] 、序列饱和突变(sequence saturation mutagenesis,SeSaM)[17 ] 、合成文库(synthetic library generation)[18 ] 等,这些传统体外突变技术成为单个酶定向进化的有力工具,随着分子生物学的快速发展,构建体外突变体库的新方法也不断涌现. ...
Targeted random mutagenesis: the use of ambiguously synthesized oligonucleotides to mutagenize sequences immediately 5' of an ATG initiation codon
2
1983
... 在过去的40多年中,定向进化技术获得了充分的发展和应用[7 ] .一方面,结合以易错PCR(error-prone PCR)[8 ] 和同源重组(DNA recombination)[9 ] 为主的随机突变,与定点饱和突变(site-Saturation mutagenesis)[10 ] ,我们既可以对蛋白序列进行“高覆盖”采样研究,亦可针对“热点”序列进行深度挖掘,这增加了突变体库的多样性,进而提高了获得目标性状突变体的可能性[11 ] .另一方面,近年来基于基因编辑技术的体内进化策略(in vivo evolution)进一步拓宽了定向进化的应用范围[7 ] .流式分选技术(flow cytometry)[12 ] 、基质辅助激光解吸电离飞行时间质谱(MALDI-ToF,MS)[13 ] 、液滴微流控技术(droplet microfluidics)[14 ] 等高通量筛选技术的兴起以及连续定向进化(continuous evolution)技术的发展[7 ] ,使得突变体库的筛选速率提高了百倍以上.最近,综合了计算机辅助学习和设计、自动化生物合成平台和高通量筛选/选择技术的集成研究平台,更为未来针对海量酶的高效、深度进化提供了可行方案[15 -16 ] . ...
... 体外突变法主要包括可以产生随机突变的易错PCR(error-prone PCR,epPCR)[8 ] 、DNA改组(DNA shuffling)[9 ] 、可以产生非随机突变的定点饱和突变(site-saturation mutagenesis,SSM)[10 ] 、序列饱和突变(sequence saturation mutagenesis,SeSaM)[17 ] 、合成文库(synthetic library generation)[18 ] 等,这些传统体外突变技术成为单个酶定向进化的有力工具,随着分子生物学的快速发展,构建体外突变体库的新方法也不断涌现. ...
Semi-rational approaches to engineering enzyme activity: combining the benefits of directed evolution and rational design
1
2005
... 在过去的40多年中,定向进化技术获得了充分的发展和应用[7 ] .一方面,结合以易错PCR(error-prone PCR)[8 ] 和同源重组(DNA recombination)[9 ] 为主的随机突变,与定点饱和突变(site-Saturation mutagenesis)[10 ] ,我们既可以对蛋白序列进行“高覆盖”采样研究,亦可针对“热点”序列进行深度挖掘,这增加了突变体库的多样性,进而提高了获得目标性状突变体的可能性[11 ] .另一方面,近年来基于基因编辑技术的体内进化策略(in vivo evolution)进一步拓宽了定向进化的应用范围[7 ] .流式分选技术(flow cytometry)[12 ] 、基质辅助激光解吸电离飞行时间质谱(MALDI-ToF,MS)[13 ] 、液滴微流控技术(droplet microfluidics)[14 ] 等高通量筛选技术的兴起以及连续定向进化(continuous evolution)技术的发展[7 ] ,使得突变体库的筛选速率提高了百倍以上.最近,综合了计算机辅助学习和设计、自动化生物合成平台和高通量筛选/选择技术的集成研究平台,更为未来针对海量酶的高效、深度进化提供了可行方案[15 -16 ] . ...
Ultrahigh-throughput FACS-based screening for directed enzyme evolution
1
2009
... 在过去的40多年中,定向进化技术获得了充分的发展和应用[7 ] .一方面,结合以易错PCR(error-prone PCR)[8 ] 和同源重组(DNA recombination)[9 ] 为主的随机突变,与定点饱和突变(site-Saturation mutagenesis)[10 ] ,我们既可以对蛋白序列进行“高覆盖”采样研究,亦可针对“热点”序列进行深度挖掘,这增加了突变体库的多样性,进而提高了获得目标性状突变体的可能性[11 ] .另一方面,近年来基于基因编辑技术的体内进化策略(in vivo evolution)进一步拓宽了定向进化的应用范围[7 ] .流式分选技术(flow cytometry)[12 ] 、基质辅助激光解吸电离飞行时间质谱(MALDI-ToF,MS)[13 ] 、液滴微流控技术(droplet microfluidics)[14 ] 等高通量筛选技术的兴起以及连续定向进化(continuous evolution)技术的发展[7 ] ,使得突变体库的筛选速率提高了百倍以上.最近,综合了计算机辅助学习和设计、自动化生物合成平台和高通量筛选/选择技术的集成研究平台,更为未来针对海量酶的高效、深度进化提供了可行方案[15 -16 ] . ...
Profiling of microbial colonies for high-throughput engineering of multistep enzymatic reactions via optically guided matrix-assisted laser desorption/ionization mass spectrometry
2
2017
... 在过去的40多年中,定向进化技术获得了充分的发展和应用[7 ] .一方面,结合以易错PCR(error-prone PCR)[8 ] 和同源重组(DNA recombination)[9 ] 为主的随机突变,与定点饱和突变(site-Saturation mutagenesis)[10 ] ,我们既可以对蛋白序列进行“高覆盖”采样研究,亦可针对“热点”序列进行深度挖掘,这增加了突变体库的多样性,进而提高了获得目标性状突变体的可能性[11 ] .另一方面,近年来基于基因编辑技术的体内进化策略(in vivo evolution)进一步拓宽了定向进化的应用范围[7 ] .流式分选技术(flow cytometry)[12 ] 、基质辅助激光解吸电离飞行时间质谱(MALDI-ToF,MS)[13 ] 、液滴微流控技术(droplet microfluidics)[14 ] 等高通量筛选技术的兴起以及连续定向进化(continuous evolution)技术的发展[7 ] ,使得突变体库的筛选速率提高了百倍以上.最近,综合了计算机辅助学习和设计、自动化生物合成平台和高通量筛选/选择技术的集成研究平台,更为未来针对海量酶的高效、深度进化提供了可行方案[15 -16 ] . ...
... 在不需要标记的高通量筛选方法中,质谱法作为一种高灵敏度和高特异性的检测方法,在蛋白质工程中应用极广.随着检测仪器性能和检测速率的不断提高,尤其是电喷射离子化技术(electrospray ionization,ESI)[57 ] 和基质辅助激光解吸离子化技术(matrix assisted laser desorption/ionization, MALDI)[58 -59 ] 的出现,生物大分子的离子化问题得以解决,并以此为基础,涌现出了许多新型质谱离子化技术,例如,基质辅助激光解吸电离飞行时间质谱(MALDI-ToF-MS)利用激光取样,通过离子的质荷比对目标分子进行无标记定性和定量分析,因具有对样品中盐浓度耐受性高、生物分子覆盖性广泛和扫描速率高等优势,已被越来越多地应用于酶反应的表征工作[60 -61 ] .但是传统的MALDI-ToF样品制备,高度依赖像声波沉积这样的先进液体处理器以提高制备通量,有一定的应用局限性[62 ] ,在此基础上进一步发展的SAMDI(self-assembled monolayers for matrix-assisted laser desorption ionization)技术,通过将反应产物固定在连接于金表面的烷硫酯自组装单分子膜上,实现了MALDI-ToF样品的快速制备,从而提高了检测通量[63 ] .M. Mrksich课题组利用该技术,从细胞色素P411随机突变文库中以每小时数千个突变体的速度筛选出催化C(sp3 )-H键烷基化的高活性突变体[64 ] .J. Sweedler课题组和H. Zhao课题组基于MALDI-ToF技术合作开发了对微生物克隆直接采样分析的技术,通量更高,且更利于对多步酶反应进行优化和表征 [13 ] .利用该方法,该课题组表征了从核糖体前体肽合成细菌素Plantazolicin的五酶途径的底物耐受性,并以1~2.5 s/克隆的速率从定向进化突变体库中筛选出选择性高产特定单鼠李糖脂同源物的双酶途径.此外,2021年报道的声波激发与质谱耦合系统(acoustic ejection mass spectrometry,AEMS),可以在筛选过程中实现零交叉污染、高通量、高定量分析精度,并同时提供极强基质耐受性[65 ] . ...
Enabling biocatalysis by high-throughput protein engineering using droplet microfluidics coupled to mass spectrometry
1
2018
... 在过去的40多年中,定向进化技术获得了充分的发展和应用[7 ] .一方面,结合以易错PCR(error-prone PCR)[8 ] 和同源重组(DNA recombination)[9 ] 为主的随机突变,与定点饱和突变(site-Saturation mutagenesis)[10 ] ,我们既可以对蛋白序列进行“高覆盖”采样研究,亦可针对“热点”序列进行深度挖掘,这增加了突变体库的多样性,进而提高了获得目标性状突变体的可能性[11 ] .另一方面,近年来基于基因编辑技术的体内进化策略(in vivo evolution)进一步拓宽了定向进化的应用范围[7 ] .流式分选技术(flow cytometry)[12 ] 、基质辅助激光解吸电离飞行时间质谱(MALDI-ToF,MS)[13 ] 、液滴微流控技术(droplet microfluidics)[14 ] 等高通量筛选技术的兴起以及连续定向进化(continuous evolution)技术的发展[7 ] ,使得突变体库的筛选速率提高了百倍以上.最近,综合了计算机辅助学习和设计、自动化生物合成平台和高通量筛选/选择技术的集成研究平台,更为未来针对海量酶的高效、深度进化提供了可行方案[15 -16 ] . ...
Machine learning-assisted directed protein evolution with combinatorial libraries
2
2019
... 在过去的40多年中,定向进化技术获得了充分的发展和应用[7 ] .一方面,结合以易错PCR(error-prone PCR)[8 ] 和同源重组(DNA recombination)[9 ] 为主的随机突变,与定点饱和突变(site-Saturation mutagenesis)[10 ] ,我们既可以对蛋白序列进行“高覆盖”采样研究,亦可针对“热点”序列进行深度挖掘,这增加了突变体库的多样性,进而提高了获得目标性状突变体的可能性[11 ] .另一方面,近年来基于基因编辑技术的体内进化策略(in vivo evolution)进一步拓宽了定向进化的应用范围[7 ] .流式分选技术(flow cytometry)[12 ] 、基质辅助激光解吸电离飞行时间质谱(MALDI-ToF,MS)[13 ] 、液滴微流控技术(droplet microfluidics)[14 ] 等高通量筛选技术的兴起以及连续定向进化(continuous evolution)技术的发展[7 ] ,使得突变体库的筛选速率提高了百倍以上.最近,综合了计算机辅助学习和设计、自动化生物合成平台和高通量筛选/选择技术的集成研究平台,更为未来针对海量酶的高效、深度进化提供了可行方案[15 -16 ] . ...
... 传统的定向进化可以概括为对目的蛋白质反复的诱变和筛选,使用每轮中的最佳变体作为下一轮突变的起点,直至达到功能目标,这种实验思路是有效但烦琐的,而机器学习通过在学习过程中的每个进化周期内对大量蛋白质进行计算评估,实现比实验室筛选更加深入的探索[108 ] .例如J. Peng和H. Zhao课题组[111 ] 最新发明的进化环境集成神经网络(evolutionary context-integrated neural network,ECNet)深度学习算法,利用进化环境来预测蛋白质的功能适应性.该算法将来自同源序列的局部进化环境与编码大型蛋白质序列的丰富结构特征的全局进化背景相结合,实现了从序列到功能的映射.目前已有部分研究使用机器学习进行定向进化,例如F. Arnold课题组对海红藻来源能够催化碳硅键的生成的一氧化氮双加氧酶(nitric oxide dioxygenase,NOD)进行预测和改造,通过机器学习的辅助,以典型的监督学习的方式,对NOD的突变体库进行筛选和缩减,仅通过两轮进化即使野生型NOD催化硅烷和重氮乙酸乙酯经卡宾插入途径(如图6 所示)得到S -2-[二甲基(苯基)甲硅烷基]丙酸乙酯(76% ee )转变为S 构型(93% ee ),此外他们还发现VCHV-49P,51R,53L突变体可催化得到R -2-[二甲基(苯基)甲硅烷基]丙酸乙酯(79% ee )[15 ] . ...
Biosystems design by machine learning
1
2020
... 在过去的40多年中,定向进化技术获得了充分的发展和应用[7 ] .一方面,结合以易错PCR(error-prone PCR)[8 ] 和同源重组(DNA recombination)[9 ] 为主的随机突变,与定点饱和突变(site-Saturation mutagenesis)[10 ] ,我们既可以对蛋白序列进行“高覆盖”采样研究,亦可针对“热点”序列进行深度挖掘,这增加了突变体库的多样性,进而提高了获得目标性状突变体的可能性[11 ] .另一方面,近年来基于基因编辑技术的体内进化策略(in vivo evolution)进一步拓宽了定向进化的应用范围[7 ] .流式分选技术(flow cytometry)[12 ] 、基质辅助激光解吸电离飞行时间质谱(MALDI-ToF,MS)[13 ] 、液滴微流控技术(droplet microfluidics)[14 ] 等高通量筛选技术的兴起以及连续定向进化(continuous evolution)技术的发展[7 ] ,使得突变体库的筛选速率提高了百倍以上.最近,综合了计算机辅助学习和设计、自动化生物合成平台和高通量筛选/选择技术的集成研究平台,更为未来针对海量酶的高效、深度进化提供了可行方案[15 -16 ] . ...
Sequence saturation mutagenesis (SeSaM): a novel method for directed evolution
1
2004
... 体外突变法主要包括可以产生随机突变的易错PCR(error-prone PCR,epPCR)[8 ] 、DNA改组(DNA shuffling)[9 ] 、可以产生非随机突变的定点饱和突变(site-saturation mutagenesis,SSM)[10 ] 、序列饱和突变(sequence saturation mutagenesis,SeSaM)[17 ] 、合成文库(synthetic library generation)[18 ] 等,这些传统体外突变技术成为单个酶定向进化的有力工具,随着分子生物学的快速发展,构建体外突变体库的新方法也不断涌现. ...
Beating bias in the directed evolution of proteins: combining high-fidelity on-chip solid-phase gene synthesis with efficient gene assembly for combinatorial library construction
3
2018
... 体外突变法主要包括可以产生随机突变的易错PCR(error-prone PCR,epPCR)[8 ] 、DNA改组(DNA shuffling)[9 ] 、可以产生非随机突变的定点饱和突变(site-saturation mutagenesis,SSM)[10 ] 、序列饱和突变(sequence saturation mutagenesis,SeSaM)[17 ] 、合成文库(synthetic library generation)[18 ] 等,这些传统体外突变技术成为单个酶定向进化的有力工具,随着分子生物学的快速发展,构建体外突变体库的新方法也不断涌现. ...
... 此外,随着基因高通量合成技术及DNA测序技术的高速发展,传统的体外突变法存在的共有缺陷,如密码子缺乏控制、具有序列偏好性等,在一定程度上被合成文库法所改善.Twist Bioscience公司与M. Reetz课题组合作,在2018年报道了这种新型的体外突变体库构建方法,即在硅基芯片上应用大规模平行寡核苷酸合成技术,然后进行有效的基因组装,每种突变体均采用计算机模拟设计,并在合成前进行筛选,因此消除了不需要的序列偏倚、提前出现的终止密码子和多余的基序.通过实验分析,这种通过大规模基因合成构建突变体库的方法相比于利用传统的易错PCR或饱和突变等方法,具有野生型序列更少、突变体分布比例更均匀、文库多样性更丰富等突出优势[18 ] ,有利于下游筛选,目前,该方法已实现商业化应用. ...
... 除了提高通量,构建精准的突变体库也是提高定向进化效率的关键,其中最具代表性的是M. Reetz教授团队所发展的CSAT、ISM、FRISM等一系列方法,以及Twist Bioscience公司与M. Reetz教授合作,在2018年报道的依托高通量合成DNA的体外突变体库构建方法已经用于商业化[18 ] .得益于测序技术的发展,未被解析的氨基酸序列数量飞速增长,机器学习技术在预测蛋白结构、功能及活性位点等领域取得了一定成功[91 ] .然而,目前主流的两种机器学习方法仍然需要依赖庞大、多样且高质量数据点来完成学习并做出较为精确的预测[143 ] ,而蛋白质序列及折叠方式的复杂性也使得现有算法无法同时预测两个或以上的性质.AlphaFold 2将利用机器学习预测蛋白质结构的研究推向了一个新的巅峰,其已经成功预测了超过30万种蛋白质的结构,但并未能准确预测庞大的序列数据库对应的所有蛋白序列.另外,对于蛋白质功能的预测目前完全依赖基于样本的机器学习模型构建或同源分析,而现有算法虽可以通过缩小突变体库来辅助定向进化,但其精确性也较大依赖于基于对蛋白质结构及功能的理解所设定的限制条件或突变倾向[144 ] .因此,如何减少建模方法对人工设置的依赖和实现自主模型的构建、如何通过较少的样本库来达到较高的预测精确度、如何同时模拟预测多个性状都将是未来探索的重点方向. ...
通过原位易错PCR一步构建基因突变文库
1
2014
... 针对传统易错PCR过程烦琐且不能实现环状质粒的指数扩增的问题,W. Shao课题组提出原位易错PCR 法(In situ error-prone PCR,Is -epPCR),通过引入DNA连接酶建立 “变性—退火—延伸—连接”的四步循环法PCR,实现了环状质粒的PCR 指数扩增,该方法所用引物为含有与模板质粒不同筛选标记的线性双链DNA,产物转化宿主菌后,模板质粒在筛选平板上被直接剔除.原位易错PCR 法已被用于木聚糖酶和纤维素酶的定向进化中,被证实为一种快速有效的随机突变文库构建方法[19 ] . ...
One-step construction of mutagenesis libraries via in situ error-prone PCR
1
2014
... 针对传统易错PCR过程烦琐且不能实现环状质粒的指数扩增的问题,W. Shao课题组提出原位易错PCR 法(In situ error-prone PCR,Is -epPCR),通过引入DNA连接酶建立 “变性—退火—延伸—连接”的四步循环法PCR,实现了环状质粒的PCR 指数扩增,该方法所用引物为含有与模板质粒不同筛选标记的线性双链DNA,产物转化宿主菌后,模板质粒在筛选平板上被直接剔除.原位易错PCR 法已被用于木聚糖酶和纤维素酶的定向进化中,被证实为一种快速有效的随机突变文库构建方法[19 ] . ...
一种改进的基于PCR的建立饱和突变库的方法
1
2019
... 相比于易错PCR较高的随机性,饱和突变的目的性很强,操作也更为精确、简单,但是仍然存在明显的氨基酸偏好性(与易错PCR类似)、突变体库过于庞大等问题,因此在一定程度上增加了筛选成本.针对这些技术瓶颈,L. Ma课题组在2019年报道了一种改进的定点饱和突变体库的构建方法,摒弃传统的NNK或NNN密码子简并引物,采用半理性设计突变氨基酸的方法,将PCR反扩载体与T5介导的克隆方法联用,构建了柠檬烯环氧水解酶(LEH)四位点组合突变体库,突变效率高达81.25%,突变体库的偏好性大大降低,突变氨基酸的分布趋于平均,为定点饱和突变技术的应用和蛋白质工程的研究提供了一种更好的思路[20 ] .另外,2019年,M. Reetz课题组与Jia. Zhou等课题组合作,在组合活性中心饱和突变策略(combinatorial active-site saturation test, CAST)[21 ] 和迭代饱和突变技术 (iterative saturation mutagenesis, ISM)[22 ] 的基础上,进一步在有效密码子的选取方面作了改进,报道了一种聚焦理性迭代定点诱变策略(focused rational iterative site-specific mutagenesis,FRISM),以南极假丝酵母脂肪酶B(Candida antarctica lipase B, CALB)催化的酯交换反应为模型,以极小的突变株筛选工作量,获得了具有高度立体选择性的CALB突变体,目标构型的选择性都在90%以上,凸显了FRISM策略的先进性和有效性[23 ] .近年来,动力学模拟等计算技术的引入推动了以CAST策略为基础的三密码子饱和突变(triple code saturation mutagenesis,TCSM)策略的形成,即通过理性选择 3种氨基酸密码子作为饱和突变的构建单元,进一步降低了筛选工作量,单文库的筛选规模可控制在500个转化子左右[24 ] . ...
An improved PCR-based method to create saturated mutagenic library
1
2019
... 相比于易错PCR较高的随机性,饱和突变的目的性很强,操作也更为精确、简单,但是仍然存在明显的氨基酸偏好性(与易错PCR类似)、突变体库过于庞大等问题,因此在一定程度上增加了筛选成本.针对这些技术瓶颈,L. Ma课题组在2019年报道了一种改进的定点饱和突变体库的构建方法,摒弃传统的NNK或NNN密码子简并引物,采用半理性设计突变氨基酸的方法,将PCR反扩载体与T5介导的克隆方法联用,构建了柠檬烯环氧水解酶(LEH)四位点组合突变体库,突变效率高达81.25%,突变体库的偏好性大大降低,突变氨基酸的分布趋于平均,为定点饱和突变技术的应用和蛋白质工程的研究提供了一种更好的思路[20 ] .另外,2019年,M. Reetz课题组与Jia. Zhou等课题组合作,在组合活性中心饱和突变策略(combinatorial active-site saturation test, CAST)[21 ] 和迭代饱和突变技术 (iterative saturation mutagenesis, ISM)[22 ] 的基础上,进一步在有效密码子的选取方面作了改进,报道了一种聚焦理性迭代定点诱变策略(focused rational iterative site-specific mutagenesis,FRISM),以南极假丝酵母脂肪酶B(Candida antarctica lipase B, CALB)催化的酯交换反应为模型,以极小的突变株筛选工作量,获得了具有高度立体选择性的CALB突变体,目标构型的选择性都在90%以上,凸显了FRISM策略的先进性和有效性[23 ] .近年来,动力学模拟等计算技术的引入推动了以CAST策略为基础的三密码子饱和突变(triple code saturation mutagenesis,TCSM)策略的形成,即通过理性选择 3种氨基酸密码子作为饱和突变的构建单元,进一步降低了筛选工作量,单文库的筛选规模可控制在500个转化子左右[24 ] . ...
Expanding the range of substrate acceptance of enzymes: combinatorial active-site saturation test
1
2005
... 相比于易错PCR较高的随机性,饱和突变的目的性很强,操作也更为精确、简单,但是仍然存在明显的氨基酸偏好性(与易错PCR类似)、突变体库过于庞大等问题,因此在一定程度上增加了筛选成本.针对这些技术瓶颈,L. Ma课题组在2019年报道了一种改进的定点饱和突变体库的构建方法,摒弃传统的NNK或NNN密码子简并引物,采用半理性设计突变氨基酸的方法,将PCR反扩载体与T5介导的克隆方法联用,构建了柠檬烯环氧水解酶(LEH)四位点组合突变体库,突变效率高达81.25%,突变体库的偏好性大大降低,突变氨基酸的分布趋于平均,为定点饱和突变技术的应用和蛋白质工程的研究提供了一种更好的思路[20 ] .另外,2019年,M. Reetz课题组与Jia. Zhou等课题组合作,在组合活性中心饱和突变策略(combinatorial active-site saturation test, CAST)[21 ] 和迭代饱和突变技术 (iterative saturation mutagenesis, ISM)[22 ] 的基础上,进一步在有效密码子的选取方面作了改进,报道了一种聚焦理性迭代定点诱变策略(focused rational iterative site-specific mutagenesis,FRISM),以南极假丝酵母脂肪酶B(Candida antarctica lipase B, CALB)催化的酯交换反应为模型,以极小的突变株筛选工作量,获得了具有高度立体选择性的CALB突变体,目标构型的选择性都在90%以上,凸显了FRISM策略的先进性和有效性[23 ] .近年来,动力学模拟等计算技术的引入推动了以CAST策略为基础的三密码子饱和突变(triple code saturation mutagenesis,TCSM)策略的形成,即通过理性选择 3种氨基酸密码子作为饱和突变的构建单元,进一步降低了筛选工作量,单文库的筛选规模可控制在500个转化子左右[24 ] . ...
Iterative saturation mutagenesis (ISM) for rapid directed evolution of functional enzymes
1
2007
... 相比于易错PCR较高的随机性,饱和突变的目的性很强,操作也更为精确、简单,但是仍然存在明显的氨基酸偏好性(与易错PCR类似)、突变体库过于庞大等问题,因此在一定程度上增加了筛选成本.针对这些技术瓶颈,L. Ma课题组在2019年报道了一种改进的定点饱和突变体库的构建方法,摒弃传统的NNK或NNN密码子简并引物,采用半理性设计突变氨基酸的方法,将PCR反扩载体与T5介导的克隆方法联用,构建了柠檬烯环氧水解酶(LEH)四位点组合突变体库,突变效率高达81.25%,突变体库的偏好性大大降低,突变氨基酸的分布趋于平均,为定点饱和突变技术的应用和蛋白质工程的研究提供了一种更好的思路[20 ] .另外,2019年,M. Reetz课题组与Jia. Zhou等课题组合作,在组合活性中心饱和突变策略(combinatorial active-site saturation test, CAST)[21 ] 和迭代饱和突变技术 (iterative saturation mutagenesis, ISM)[22 ] 的基础上,进一步在有效密码子的选取方面作了改进,报道了一种聚焦理性迭代定点诱变策略(focused rational iterative site-specific mutagenesis,FRISM),以南极假丝酵母脂肪酶B(Candida antarctica lipase B, CALB)催化的酯交换反应为模型,以极小的突变株筛选工作量,获得了具有高度立体选择性的CALB突变体,目标构型的选择性都在90%以上,凸显了FRISM策略的先进性和有效性[23 ] .近年来,动力学模拟等计算技术的引入推动了以CAST策略为基础的三密码子饱和突变(triple code saturation mutagenesis,TCSM)策略的形成,即通过理性选择 3种氨基酸密码子作为饱和突变的构建单元,进一步降低了筛选工作量,单文库的筛选规模可控制在500个转化子左右[24 ] . ...
Stereodivergent protein engineering of a lipase to access all possible stereoisomers of chiral esters with two stereocenters
1
2019
... 相比于易错PCR较高的随机性,饱和突变的目的性很强,操作也更为精确、简单,但是仍然存在明显的氨基酸偏好性(与易错PCR类似)、突变体库过于庞大等问题,因此在一定程度上增加了筛选成本.针对这些技术瓶颈,L. Ma课题组在2019年报道了一种改进的定点饱和突变体库的构建方法,摒弃传统的NNK或NNN密码子简并引物,采用半理性设计突变氨基酸的方法,将PCR反扩载体与T5介导的克隆方法联用,构建了柠檬烯环氧水解酶(LEH)四位点组合突变体库,突变效率高达81.25%,突变体库的偏好性大大降低,突变氨基酸的分布趋于平均,为定点饱和突变技术的应用和蛋白质工程的研究提供了一种更好的思路[20 ] .另外,2019年,M. Reetz课题组与Jia. Zhou等课题组合作,在组合活性中心饱和突变策略(combinatorial active-site saturation test, CAST)[21 ] 和迭代饱和突变技术 (iterative saturation mutagenesis, ISM)[22 ] 的基础上,进一步在有效密码子的选取方面作了改进,报道了一种聚焦理性迭代定点诱变策略(focused rational iterative site-specific mutagenesis,FRISM),以南极假丝酵母脂肪酶B(Candida antarctica lipase B, CALB)催化的酯交换反应为模型,以极小的突变株筛选工作量,获得了具有高度立体选择性的CALB突变体,目标构型的选择性都在90%以上,凸显了FRISM策略的先进性和有效性[23 ] .近年来,动力学模拟等计算技术的引入推动了以CAST策略为基础的三密码子饱和突变(triple code saturation mutagenesis,TCSM)策略的形成,即通过理性选择 3种氨基酸密码子作为饱和突变的构建单元,进一步降低了筛选工作量,单文库的筛选规模可控制在500个转化子左右[24 ] . ...
Structure-guided triple-code saturation mutagenesis: efficient tuning of the stereoselectivity of an epoxide hydrolase
1
2016
... 相比于易错PCR较高的随机性,饱和突变的目的性很强,操作也更为精确、简单,但是仍然存在明显的氨基酸偏好性(与易错PCR类似)、突变体库过于庞大等问题,因此在一定程度上增加了筛选成本.针对这些技术瓶颈,L. Ma课题组在2019年报道了一种改进的定点饱和突变体库的构建方法,摒弃传统的NNK或NNN密码子简并引物,采用半理性设计突变氨基酸的方法,将PCR反扩载体与T5介导的克隆方法联用,构建了柠檬烯环氧水解酶(LEH)四位点组合突变体库,突变效率高达81.25%,突变体库的偏好性大大降低,突变氨基酸的分布趋于平均,为定点饱和突变技术的应用和蛋白质工程的研究提供了一种更好的思路[20 ] .另外,2019年,M. Reetz课题组与Jia. Zhou等课题组合作,在组合活性中心饱和突变策略(combinatorial active-site saturation test, CAST)[21 ] 和迭代饱和突变技术 (iterative saturation mutagenesis, ISM)[22 ] 的基础上,进一步在有效密码子的选取方面作了改进,报道了一种聚焦理性迭代定点诱变策略(focused rational iterative site-specific mutagenesis,FRISM),以南极假丝酵母脂肪酶B(Candida antarctica lipase B, CALB)催化的酯交换反应为模型,以极小的突变株筛选工作量,获得了具有高度立体选择性的CALB突变体,目标构型的选择性都在90%以上,凸显了FRISM策略的先进性和有效性[23 ] .近年来,动力学模拟等计算技术的引入推动了以CAST策略为基础的三密码子饱和突变(triple code saturation mutagenesis,TCSM)策略的形成,即通过理性选择 3种氨基酸密码子作为饱和突变的构建单元,进一步降低了筛选工作量,单文库的筛选规模可控制在500个转化子左右[24 ] . ...
High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides
1
2001
... 基因突变体库的另一大类构建方法是体内突变法,即在胞内诱导特定基因的突变,无需进行基因克隆与转化,很大程度上缩短了定向进化的实验周期.例如早期开发的基于单链DNA重组的多元自动化基因组工程技术(multiplex automated genome engineering,MAGE),是针对大肠杆菌基因组上特定基因进行体内诱变的重要策略[25 ] .近年来,CRISPR-Cas介导的重组技术实现了在原核与真核细胞中对目的基因进行高效的片段插入、删除和替换(图2 a),大大提高了原核与真核细胞中重组的效率[26 -27 ] ,因此也发展了一批高效的胞内蛋白质定向进化工具.例如将CRISPR-Cas系统和MAGE整合后,基因重组效率由3%提高到98%,插入/替换效率提高至70%[28 ] ;将CRISPR-Cas9与epPCR偶联(CasPER)可以向酵母基因组中任意基因的600 bp内引入随机突变[29 ] .以上两种方法,皆通过带有突变点的外源核酸片段经同源重组引入突变点,因此需额外提供大量的核酸片段来覆盖整个目标基因.与其不同的是,J. Dueber 课题组和D. Schaffer课题组基于CRISPR技术,在大肠杆菌中合作开发了一种促进细胞内特定基因进化的平台——EvolvR系统[30 ] .该系统首先由Nickase切口酶(nCas9)在目标序列切割出一个缺口,然后利用融合的高错DNA聚合酶执行切口平移,在此过程中通过改造Nickase和DNA聚合酶实现定点基因的突变(图2 b).相对于野生型,通过该方法可使目的基因的突变率提高107 倍,突变范围达到350 bp.2020年该团队将EvolvR系统用于酿酒酵母中,可同时在两个目的基因的40 bp范围内引入随机突变[31 ] .但由于突变率较低且突变窗口较小,其应用有一定局限性. ...
Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors
1
2020
... 基因突变体库的另一大类构建方法是体内突变法,即在胞内诱导特定基因的突变,无需进行基因克隆与转化,很大程度上缩短了定向进化的实验周期.例如早期开发的基于单链DNA重组的多元自动化基因组工程技术(multiplex automated genome engineering,MAGE),是针对大肠杆菌基因组上特定基因进行体内诱变的重要策略[25 ] .近年来,CRISPR-Cas介导的重组技术实现了在原核与真核细胞中对目的基因进行高效的片段插入、删除和替换(图2 a),大大提高了原核与真核细胞中重组的效率[26 -27 ] ,因此也发展了一批高效的胞内蛋白质定向进化工具.例如将CRISPR-Cas系统和MAGE整合后,基因重组效率由3%提高到98%,插入/替换效率提高至70%[28 ] ;将CRISPR-Cas9与epPCR偶联(CasPER)可以向酵母基因组中任意基因的600 bp内引入随机突变[29 ] .以上两种方法,皆通过带有突变点的外源核酸片段经同源重组引入突变点,因此需额外提供大量的核酸片段来覆盖整个目标基因.与其不同的是,J. Dueber 课题组和D. Schaffer课题组基于CRISPR技术,在大肠杆菌中合作开发了一种促进细胞内特定基因进化的平台——EvolvR系统[30 ] .该系统首先由Nickase切口酶(nCas9)在目标序列切割出一个缺口,然后利用融合的高错DNA聚合酶执行切口平移,在此过程中通过改造Nickase和DNA聚合酶实现定点基因的突变(图2 b).相对于野生型,通过该方法可使目的基因的突变率提高107 倍,突变范围达到350 bp.2020年该团队将EvolvR系统用于酿酒酵母中,可同时在两个目的基因的40 bp范围内引入随机突变[31 ] .但由于突变率较低且突变窗口较小,其应用有一定局限性. ...
Homology-integrated CRISPR-Cas (HI-CRISPR) system for one-step multigene disruption in Saccharomyces cerevisiae
1
2015
... 基因突变体库的另一大类构建方法是体内突变法,即在胞内诱导特定基因的突变,无需进行基因克隆与转化,很大程度上缩短了定向进化的实验周期.例如早期开发的基于单链DNA重组的多元自动化基因组工程技术(multiplex automated genome engineering,MAGE),是针对大肠杆菌基因组上特定基因进行体内诱变的重要策略[25 ] .近年来,CRISPR-Cas介导的重组技术实现了在原核与真核细胞中对目的基因进行高效的片段插入、删除和替换(图2 a),大大提高了原核与真核细胞中重组的效率[26 -27 ] ,因此也发展了一批高效的胞内蛋白质定向进化工具.例如将CRISPR-Cas系统和MAGE整合后,基因重组效率由3%提高到98%,插入/替换效率提高至70%[28 ] ;将CRISPR-Cas9与epPCR偶联(CasPER)可以向酵母基因组中任意基因的600 bp内引入随机突变[29 ] .以上两种方法,皆通过带有突变点的外源核酸片段经同源重组引入突变点,因此需额外提供大量的核酸片段来覆盖整个目标基因.与其不同的是,J. Dueber 课题组和D. Schaffer课题组基于CRISPR技术,在大肠杆菌中合作开发了一种促进细胞内特定基因进化的平台——EvolvR系统[30 ] .该系统首先由Nickase切口酶(nCas9)在目标序列切割出一个缺口,然后利用融合的高错DNA聚合酶执行切口平移,在此过程中通过改造Nickase和DNA聚合酶实现定点基因的突变(图2 b).相对于野生型,通过该方法可使目的基因的突变率提高107 倍,突变范围达到350 bp.2020年该团队将EvolvR系统用于酿酒酵母中,可同时在两个目的基因的40 bp范围内引入随机突变[31 ] .但由于突变率较低且突变窗口较小,其应用有一定局限性. ...
CRMAGE: CRISPR optimized MAGE recombineering
1
2016
... 基因突变体库的另一大类构建方法是体内突变法,即在胞内诱导特定基因的突变,无需进行基因克隆与转化,很大程度上缩短了定向进化的实验周期.例如早期开发的基于单链DNA重组的多元自动化基因组工程技术(multiplex automated genome engineering,MAGE),是针对大肠杆菌基因组上特定基因进行体内诱变的重要策略[25 ] .近年来,CRISPR-Cas介导的重组技术实现了在原核与真核细胞中对目的基因进行高效的片段插入、删除和替换(图2 a),大大提高了原核与真核细胞中重组的效率[26 -27 ] ,因此也发展了一批高效的胞内蛋白质定向进化工具.例如将CRISPR-Cas系统和MAGE整合后,基因重组效率由3%提高到98%,插入/替换效率提高至70%[28 ] ;将CRISPR-Cas9与epPCR偶联(CasPER)可以向酵母基因组中任意基因的600 bp内引入随机突变[29 ] .以上两种方法,皆通过带有突变点的外源核酸片段经同源重组引入突变点,因此需额外提供大量的核酸片段来覆盖整个目标基因.与其不同的是,J. Dueber 课题组和D. Schaffer课题组基于CRISPR技术,在大肠杆菌中合作开发了一种促进细胞内特定基因进化的平台——EvolvR系统[30 ] .该系统首先由Nickase切口酶(nCas9)在目标序列切割出一个缺口,然后利用融合的高错DNA聚合酶执行切口平移,在此过程中通过改造Nickase和DNA聚合酶实现定点基因的突变(图2 b).相对于野生型,通过该方法可使目的基因的突变率提高107 倍,突变范围达到350 bp.2020年该团队将EvolvR系统用于酿酒酵母中,可同时在两个目的基因的40 bp范围内引入随机突变[31 ] .但由于突变率较低且突变窗口较小,其应用有一定局限性. ...
CasPER, a method for directed evolution in genomic contexts using mutagenesis and CRISPR/Cas9
1
2018
... 基因突变体库的另一大类构建方法是体内突变法,即在胞内诱导特定基因的突变,无需进行基因克隆与转化,很大程度上缩短了定向进化的实验周期.例如早期开发的基于单链DNA重组的多元自动化基因组工程技术(multiplex automated genome engineering,MAGE),是针对大肠杆菌基因组上特定基因进行体内诱变的重要策略[25 ] .近年来,CRISPR-Cas介导的重组技术实现了在原核与真核细胞中对目的基因进行高效的片段插入、删除和替换(图2 a),大大提高了原核与真核细胞中重组的效率[26 -27 ] ,因此也发展了一批高效的胞内蛋白质定向进化工具.例如将CRISPR-Cas系统和MAGE整合后,基因重组效率由3%提高到98%,插入/替换效率提高至70%[28 ] ;将CRISPR-Cas9与epPCR偶联(CasPER)可以向酵母基因组中任意基因的600 bp内引入随机突变[29 ] .以上两种方法,皆通过带有突变点的外源核酸片段经同源重组引入突变点,因此需额外提供大量的核酸片段来覆盖整个目标基因.与其不同的是,J. Dueber 课题组和D. Schaffer课题组基于CRISPR技术,在大肠杆菌中合作开发了一种促进细胞内特定基因进化的平台——EvolvR系统[30 ] .该系统首先由Nickase切口酶(nCas9)在目标序列切割出一个缺口,然后利用融合的高错DNA聚合酶执行切口平移,在此过程中通过改造Nickase和DNA聚合酶实现定点基因的突变(图2 b).相对于野生型,通过该方法可使目的基因的突变率提高107 倍,突变范围达到350 bp.2020年该团队将EvolvR系统用于酿酒酵母中,可同时在两个目的基因的40 bp范围内引入随机突变[31 ] .但由于突变率较低且突变窗口较小,其应用有一定局限性. ...
CRISPR-guided DNA polymerases enable diversification of all nucleotides in a tunable window
1
2018
... 基因突变体库的另一大类构建方法是体内突变法,即在胞内诱导特定基因的突变,无需进行基因克隆与转化,很大程度上缩短了定向进化的实验周期.例如早期开发的基于单链DNA重组的多元自动化基因组工程技术(multiplex automated genome engineering,MAGE),是针对大肠杆菌基因组上特定基因进行体内诱变的重要策略[25 ] .近年来,CRISPR-Cas介导的重组技术实现了在原核与真核细胞中对目的基因进行高效的片段插入、删除和替换(图2 a),大大提高了原核与真核细胞中重组的效率[26 -27 ] ,因此也发展了一批高效的胞内蛋白质定向进化工具.例如将CRISPR-Cas系统和MAGE整合后,基因重组效率由3%提高到98%,插入/替换效率提高至70%[28 ] ;将CRISPR-Cas9与epPCR偶联(CasPER)可以向酵母基因组中任意基因的600 bp内引入随机突变[29 ] .以上两种方法,皆通过带有突变点的外源核酸片段经同源重组引入突变点,因此需额外提供大量的核酸片段来覆盖整个目标基因.与其不同的是,J. Dueber 课题组和D. Schaffer课题组基于CRISPR技术,在大肠杆菌中合作开发了一种促进细胞内特定基因进化的平台——EvolvR系统[30 ] .该系统首先由Nickase切口酶(nCas9)在目标序列切割出一个缺口,然后利用融合的高错DNA聚合酶执行切口平移,在此过程中通过改造Nickase和DNA聚合酶实现定点基因的突变(图2 b).相对于野生型,通过该方法可使目的基因的突变率提高107 倍,突变范围达到350 bp.2020年该团队将EvolvR系统用于酿酒酵母中,可同时在两个目的基因的40 bp范围内引入随机突变[31 ] .但由于突变率较低且突变窗口较小,其应用有一定局限性. ...
Targeted diversification in the S. cerevisiae genome with CRISPR-guided DNA polymerase I
1
2020
... 基因突变体库的另一大类构建方法是体内突变法,即在胞内诱导特定基因的突变,无需进行基因克隆与转化,很大程度上缩短了定向进化的实验周期.例如早期开发的基于单链DNA重组的多元自动化基因组工程技术(multiplex automated genome engineering,MAGE),是针对大肠杆菌基因组上特定基因进行体内诱变的重要策略[25 ] .近年来,CRISPR-Cas介导的重组技术实现了在原核与真核细胞中对目的基因进行高效的片段插入、删除和替换(图2 a),大大提高了原核与真核细胞中重组的效率[26 -27 ] ,因此也发展了一批高效的胞内蛋白质定向进化工具.例如将CRISPR-Cas系统和MAGE整合后,基因重组效率由3%提高到98%,插入/替换效率提高至70%[28 ] ;将CRISPR-Cas9与epPCR偶联(CasPER)可以向酵母基因组中任意基因的600 bp内引入随机突变[29 ] .以上两种方法,皆通过带有突变点的外源核酸片段经同源重组引入突变点,因此需额外提供大量的核酸片段来覆盖整个目标基因.与其不同的是,J. Dueber 课题组和D. Schaffer课题组基于CRISPR技术,在大肠杆菌中合作开发了一种促进细胞内特定基因进化的平台——EvolvR系统[30 ] .该系统首先由Nickase切口酶(nCas9)在目标序列切割出一个缺口,然后利用融合的高错DNA聚合酶执行切口平移,在此过程中通过改造Nickase和DNA聚合酶实现定点基因的突变(图2 b).相对于野生型,通过该方法可使目的基因的突变率提高107 倍,突变范围达到350 bp.2020年该团队将EvolvR系统用于酿酒酵母中,可同时在两个目的基因的40 bp范围内引入随机突变[31 ] .但由于突变率较低且突变窗口较小,其应用有一定局限性. ...
Scalable, continuous evolution of genes at mutation rates above genomic error thresholds
1
2018
... 此外,相较早期的胞内定向进化方法,近期开发的方法在降低脱靶率的研究上有较大突破,因此大大改善了体内诱变致使基因组不稳定的问题.例如,基于酵母细胞质线性质粒pGKL1的复制受到DNA聚合酶TP-DNAP1的严格调控,与酵母基因组的复制成正交关系的机制,C. Liu课题组开发了正交易错复制系统(orthogonal error-prone replicate system,OrthoRep)[32 ] .通过改造TP-DNAP1提高其复制错误率,在不明显提高酵母基因组突变率的前提下,使构建在质粒pGKL1上的目标基因以10-5 的突变率进行随机突变.该课题组之后利用OrthoRep,对恶性疟原虫二氢叶酸还原酶(dihydrofolate reductases,DHFR)进行连续进化,获得了高度耐乙胺嘧啶的二氢叶酸还原酶突变体.最近该课题组利用OrthoRep对海栖热袍菌的色氨酸合成酶β亚基(Tm TrpB)进行100多轮进化,提高了其通过吲哚化合物和丝氨酸底物合成色氨酸的活性,同时赋予了该酶催化混杂性[33 ] .一般地,理想化的胞内诱变工具需具有以下三个特征:低无关诱变;高突变率且随开随关;易使用.2021年X. Yi和M. Travisano等课题组合作报道了一种靶向人工DNA复制体(targeted artificial DNA replisome,TADR)的蛋白质复合物,也叫最简DNA复制体,由噬菌体蛋白CisA、细菌Rep解旋酶、T5噬菌体DNA聚合酶的出错性突变子三个蛋白质组成.该系统能够在短时间内把大量突变靶向指定的DNA,促使靶质粒的突变率提高2.3×105 倍,同时保留基因组其他部分的完整性,满足靶向性、诱变性和灵活性三个要求.此外,TADR也可以进行多种碱基的同时替换,为解决酶的定向进化中的难题提供了新的思路[34 ] . ...
Scalable continuous evolution for the generation of diverse enzyme variants encompassing promiscuous activities
1
2020
... 此外,相较早期的胞内定向进化方法,近期开发的方法在降低脱靶率的研究上有较大突破,因此大大改善了体内诱变致使基因组不稳定的问题.例如,基于酵母细胞质线性质粒pGKL1的复制受到DNA聚合酶TP-DNAP1的严格调控,与酵母基因组的复制成正交关系的机制,C. Liu课题组开发了正交易错复制系统(orthogonal error-prone replicate system,OrthoRep)[32 ] .通过改造TP-DNAP1提高其复制错误率,在不明显提高酵母基因组突变率的前提下,使构建在质粒pGKL1上的目标基因以10-5 的突变率进行随机突变.该课题组之后利用OrthoRep,对恶性疟原虫二氢叶酸还原酶(dihydrofolate reductases,DHFR)进行连续进化,获得了高度耐乙胺嘧啶的二氢叶酸还原酶突变体.最近该课题组利用OrthoRep对海栖热袍菌的色氨酸合成酶β亚基(Tm TrpB)进行100多轮进化,提高了其通过吲哚化合物和丝氨酸底物合成色氨酸的活性,同时赋予了该酶催化混杂性[33 ] .一般地,理想化的胞内诱变工具需具有以下三个特征:低无关诱变;高突变率且随开随关;易使用.2021年X. Yi和M. Travisano等课题组合作报道了一种靶向人工DNA复制体(targeted artificial DNA replisome,TADR)的蛋白质复合物,也叫最简DNA复制体,由噬菌体蛋白CisA、细菌Rep解旋酶、T5噬菌体DNA聚合酶的出错性突变子三个蛋白质组成.该系统能够在短时间内把大量突变靶向指定的DNA,促使靶质粒的突变率提高2.3×105 倍,同时保留基因组其他部分的完整性,满足靶向性、诱变性和灵活性三个要求.此外,TADR也可以进行多种碱基的同时替换,为解决酶的定向进化中的难题提供了新的思路[34 ] . ...
Plasmid hypermutation using a targeted artificial DNA replisome
1
2021
... 此外,相较早期的胞内定向进化方法,近期开发的方法在降低脱靶率的研究上有较大突破,因此大大改善了体内诱变致使基因组不稳定的问题.例如,基于酵母细胞质线性质粒pGKL1的复制受到DNA聚合酶TP-DNAP1的严格调控,与酵母基因组的复制成正交关系的机制,C. Liu课题组开发了正交易错复制系统(orthogonal error-prone replicate system,OrthoRep)[32 ] .通过改造TP-DNAP1提高其复制错误率,在不明显提高酵母基因组突变率的前提下,使构建在质粒pGKL1上的目标基因以10-5 的突变率进行随机突变.该课题组之后利用OrthoRep,对恶性疟原虫二氢叶酸还原酶(dihydrofolate reductases,DHFR)进行连续进化,获得了高度耐乙胺嘧啶的二氢叶酸还原酶突变体.最近该课题组利用OrthoRep对海栖热袍菌的色氨酸合成酶β亚基(Tm TrpB)进行100多轮进化,提高了其通过吲哚化合物和丝氨酸底物合成色氨酸的活性,同时赋予了该酶催化混杂性[33 ] .一般地,理想化的胞内诱变工具需具有以下三个特征:低无关诱变;高突变率且随开随关;易使用.2021年X. Yi和M. Travisano等课题组合作报道了一种靶向人工DNA复制体(targeted artificial DNA replisome,TADR)的蛋白质复合物,也叫最简DNA复制体,由噬菌体蛋白CisA、细菌Rep解旋酶、T5噬菌体DNA聚合酶的出错性突变子三个蛋白质组成.该系统能够在短时间内把大量突变靶向指定的DNA,促使靶质粒的突变率提高2.3×105 倍,同时保留基因组其他部分的完整性,满足靶向性、诱变性和灵活性三个要求.此外,TADR也可以进行多种碱基的同时替换,为解决酶的定向进化中的难题提供了新的思路[34 ] . ...
High-throughput antibody engineering in mammalian cells by CRISPR/Cas9-mediated homology-directed mutagenesis
1
2018
... 相较于传统的体外进化,胞内定向进化技术也为一些特殊目标蛋白的有效进化提供了解决方案.早期的定向进化,主要基于细菌和酵母,对异源表达酶的特异性、稳定性和催化活性进行改造,继而服务于能源和精细化工领域.开发能够应用于哺乳动物细胞的定向进化技术,对特殊的药用蛋白的定向进化意义重大.以抗体药物为例,在进入临床阶段前,需对其表达水平、药代动力学、溶解度和免疫原性等进行优化.此类性质的改良主要通过噬菌体和酵母展示来完成,然后改良得到的候选抗体在哺乳动物细胞中以全长IgG的形式表达,并在该环境下进行表型筛选.这个过程不但烦琐,且在微生物环境下进化得到的突变体,常会因错误的折叠修饰、细胞定位以及分子间相互作用等因素,无法在复杂的哺乳动物细胞中正常工作.另外,通过传统方法直接在哺乳动物细胞中构建基因突变体库也有弊端:一是文库多样性受限;二是每个哺乳动物细胞可能含多个抗体突变体,不利于最终的筛选.为此,S. T. Reddy 课题组开发了基于CRISPR-Cas 9的同源定向突变(homology-directed mutagenesis,HDM)技术,用于抗体的设计、改造及高通量筛选[35 ] .首先在杂交瘤细胞中,借助CRISPR-Cas9的非同源末端连接向抗体的互补决定区3(complementarity determining region 3,CDRH3)引入移码突变,关闭抗体表达.然后在Cas9的同源定向修复作用下,引入含沉默突变的单链寡核苷酸(single-stranded oligonucleotides,ssODNs),使接受了HDR的突变体重新表达出CDRH3.利用该技术,可直接在杂交瘤细胞中产生超过105 个突变体的文库,并筛选出与HEL(hen egg lysozyme)抗原有高亲和力的突变体.此外,D. F. Chen 课题组通过结合T7 RNA聚合酶与胞苷脱氨酶(cytidine deaminase),开发了针对哺乳动物细胞的TRACE(T7 polymerase-driven continuous editing)系统,能对T7启动子下游的2000 bp 基因序列,以10-6 的突变率随机引入CG→TA突变[36 ] .利用TRACE,该团队已筛选出在人恶性黑色素瘤细胞(A375)中对小分子抑制剂具有耐药性的丝裂原活化蛋白激酶激酶(mitogen-activated protein kinase kinase, MAPKK)突变体. ...
Efficient, continuous mutagenesis in human cells using a pseudo-random DNA editor
1
2020
... 相较于传统的体外进化,胞内定向进化技术也为一些特殊目标蛋白的有效进化提供了解决方案.早期的定向进化,主要基于细菌和酵母,对异源表达酶的特异性、稳定性和催化活性进行改造,继而服务于能源和精细化工领域.开发能够应用于哺乳动物细胞的定向进化技术,对特殊的药用蛋白的定向进化意义重大.以抗体药物为例,在进入临床阶段前,需对其表达水平、药代动力学、溶解度和免疫原性等进行优化.此类性质的改良主要通过噬菌体和酵母展示来完成,然后改良得到的候选抗体在哺乳动物细胞中以全长IgG的形式表达,并在该环境下进行表型筛选.这个过程不但烦琐,且在微生物环境下进化得到的突变体,常会因错误的折叠修饰、细胞定位以及分子间相互作用等因素,无法在复杂的哺乳动物细胞中正常工作.另外,通过传统方法直接在哺乳动物细胞中构建基因突变体库也有弊端:一是文库多样性受限;二是每个哺乳动物细胞可能含多个抗体突变体,不利于最终的筛选.为此,S. T. Reddy 课题组开发了基于CRISPR-Cas 9的同源定向突变(homology-directed mutagenesis,HDM)技术,用于抗体的设计、改造及高通量筛选[35 ] .首先在杂交瘤细胞中,借助CRISPR-Cas9的非同源末端连接向抗体的互补决定区3(complementarity determining region 3,CDRH3)引入移码突变,关闭抗体表达.然后在Cas9的同源定向修复作用下,引入含沉默突变的单链寡核苷酸(single-stranded oligonucleotides,ssODNs),使接受了HDR的突变体重新表达出CDRH3.利用该技术,可直接在杂交瘤细胞中产生超过105 个突变体的文库,并筛选出与HEL(hen egg lysozyme)抗原有高亲和力的突变体.此外,D. F. Chen 课题组通过结合T7 RNA聚合酶与胞苷脱氨酶(cytidine deaminase),开发了针对哺乳动物细胞的TRACE(T7 polymerase-driven continuous editing)系统,能对T7启动子下游的2000 bp 基因序列,以10-6 的突变率随机引入CG→TA突变[36 ] .利用TRACE,该团队已筛选出在人恶性黑色素瘤细胞(A375)中对小分子抑制剂具有耐药性的丝裂原活化蛋白激酶激酶(mitogen-activated protein kinase kinase, MAPKK)突变体. ...
Targeted, random mutagenesis of plant genes with dual cytosine and adenine base editors
1
2020
... 特异性胞内进化工具的开发,促使一些高等植物的内源基因工程改造得以实现.C. Gao课题组和J. Li课题组联合设计了五种饱和靶向内源基因突变碱基编辑器(saturated targeted endogenous mutagenesis editors,STEME)[37 ] ,实现了在体内对植物内源基因的突变改造.该方法将胞嘧啶脱氨酶APOBEC3A和腺嘌呤脱氨酶ecTadA-ecTadA7.10同时融合在nCas9(D10A)的N端(图2 c),并将抑制体内尿嘧啶糖基化酶活性的尿嘧啶糖酵解抑制剂UGI融合表达或自由表达于nCas9的C端,从而构建了4种形式的双碱基编辑器STEME,其可在一个sgRNA的引导下,诱导靶位点(12 bp)上C→T和A→G的随机突变,显著增加了目标基因的多样性.利用该技术,该课题组使用20条sgRNA,实现对水稻乙酰辅酶A羧化酶OsACC序列中56个氨基酸的近饱和突变(73.21%),获得了抗除草剂的水稻突变体. ...
In vivo continuous evolution of genes and pathways in yeast
1
2016
... 更为重要的是,胞内定向进化技术的兴起也使得定向进化的对象从一个酶,拓展到了参与同一代谢途径的多个蛋白.例如H. Alper 课题组利用逆转录转座子Ty1在逆转录复制过程中的易错性,在酿酒酵母中构建了一个多基因随机突变系统.该方法将目标基因插入Ty1的长末端重复序列5′-LTR(long terminal repeat)和3′-LTR之间,在Ty1起始转录形成mRNA,随即逆转录成cDNA后重新整合到基因组的过程中,以10-5 /碱基的突变率向目标基因引入随机突变,且突变个数会随着反转座子循环而增加.该课题组利用该系统对一条全长4.5 kb,由木糖异构酶(XylA)和木酮糖激酶(XKS1)构成的代谢通路进行诱变,大大提高了酵母对木糖的代谢能力[38 ] .但是,多基因调控的复杂表型的定向进化,往往需要更大的突变框.N. Crook 课题组利用温和噬菌体实现了大片段DNA的诱导定向进化(inducible directed evolution)[39 ] .该系统由P1噬菌粒、响应阿拉伯糖信号的P1噬菌体突变质粒组成.首先,将搭载长片段基因通路的P1噬菌粒转入大肠杆菌中,在突变质粒作用下生成随机突变;然后由阿拉伯糖诱导P1噬菌体引发细胞裂解,释放携带突变的噬菌体颗粒,进行下一轮侵染.基于此,他们分别对地衣芽孢杆菌中由5个基因调控的塔格糖代谢途径以及短双歧杆菌中由10个基因调控的松三糖代谢途径进行定向进化,提高了大肠杆菌对塔格糖和松三塘的代谢能力. ...
Inducible directed evolution of complex phenotypes in bacteria
1
2022
... 更为重要的是,胞内定向进化技术的兴起也使得定向进化的对象从一个酶,拓展到了参与同一代谢途径的多个蛋白.例如H. Alper 课题组利用逆转录转座子Ty1在逆转录复制过程中的易错性,在酿酒酵母中构建了一个多基因随机突变系统.该方法将目标基因插入Ty1的长末端重复序列5′-LTR(long terminal repeat)和3′-LTR之间,在Ty1起始转录形成mRNA,随即逆转录成cDNA后重新整合到基因组的过程中,以10-5 /碱基的突变率向目标基因引入随机突变,且突变个数会随着反转座子循环而增加.该课题组利用该系统对一条全长4.5 kb,由木糖异构酶(XylA)和木酮糖激酶(XKS1)构成的代谢通路进行诱变,大大提高了酵母对木糖的代谢能力[38 ] .但是,多基因调控的复杂表型的定向进化,往往需要更大的突变框.N. Crook 课题组利用温和噬菌体实现了大片段DNA的诱导定向进化(inducible directed evolution)[39 ] .该系统由P1噬菌粒、响应阿拉伯糖信号的P1噬菌体突变质粒组成.首先,将搭载长片段基因通路的P1噬菌粒转入大肠杆菌中,在突变质粒作用下生成随机突变;然后由阿拉伯糖诱导P1噬菌体引发细胞裂解,释放携带突变的噬菌体颗粒,进行下一轮侵染.基于此,他们分别对地衣芽孢杆菌中由5个基因调控的塔格糖代谢途径以及短双歧杆菌中由10个基因调控的松三糖代谢途径进行定向进化,提高了大肠杆菌对塔格糖和松三塘的代谢能力. ...
高通量筛选系统在定向改造中的新进展
1
2021
... 平板筛选是一种简单且直接的筛选方法,通常是利用平板培养基上不同重组细胞的表型(生长、水解圈等)在视觉上产生的区分度,将含有目标突变体的重组细胞筛选出来,缺点是仅适用于突变体库的初筛且通量较低[40 ] . ...
Advances of high-throughput screening system in reengineering of biological entities
1
2021
... 平板筛选是一种简单且直接的筛选方法,通常是利用平板培养基上不同重组细胞的表型(生长、水解圈等)在视觉上产生的区分度,将含有目标突变体的重组细胞筛选出来,缺点是仅适用于突变体库的初筛且通量较低[40 ] . ...
Phage display systems and their applications
1
2006
... 相比于平板筛选法,利用展示技术进行的突变体库高通量筛选,更为有力地促进了蛋白质工程的发展.其中应用较普遍的展示技术可以分为噬菌体展示技术[41 ] 、细胞表面展示技术[42 ] 、核糖体展示技术与mRNA 展示技术 [43 ] 共三类. ...
Microbial cell-surface display
1
2003
... 相比于平板筛选法,利用展示技术进行的突变体库高通量筛选,更为有力地促进了蛋白质工程的发展.其中应用较普遍的展示技术可以分为噬菌体展示技术[41 ] 、细胞表面展示技术[42 ] 、核糖体展示技术与mRNA 展示技术 [43 ] 共三类. ...
In vitro selection of proteins with desired characteristics using mRNA-display
1
2013
... 相比于平板筛选法,利用展示技术进行的突变体库高通量筛选,更为有力地促进了蛋白质工程的发展.其中应用较普遍的展示技术可以分为噬菌体展示技术[41 ] 、细胞表面展示技术[42 ] 、核糖体展示技术与mRNA 展示技术 [43 ] 共三类. ...
Antibody-selectable filamentous fd phage vectors: affinity purification of target genes
1
1988
... 噬菌体展示技术原理为将蛋白基因插入到噬菌体外壳蛋白结构基因的适当位置,随着噬菌体的传代,融合蛋白展示在噬菌体的表面,而对应的编码基因则位于病毒颗粒内,因此大量蛋白与其DNA编码序列之间建立了直接联系,使各种靶分子(抗体、酶等)的配体通过“吸附、洗脱、扩增”的过程得以快速鉴定[44 ] .噬菌体展示技术可以应用于较大突变体库(109 )的筛选,加之具有技术成熟、不需要昂贵的试剂与设备的特点,在酶的定向进化和抗体研发领域颇受青睐.S. Vichier-Guerre课题组利用该技术,从来自水生栖热菌(Thermus aquaticus )的DNA 聚合酶 I进化出一组耐热性提高的逆转录酶[45 ] . ...
A population of thermostable reverse transcriptases evolved from Thermus aquaticus DNA polymerase I by phage display
1
2006
... 噬菌体展示技术原理为将蛋白基因插入到噬菌体外壳蛋白结构基因的适当位置,随着噬菌体的传代,融合蛋白展示在噬菌体的表面,而对应的编码基因则位于病毒颗粒内,因此大量蛋白与其DNA编码序列之间建立了直接联系,使各种靶分子(抗体、酶等)的配体通过“吸附、洗脱、扩增”的过程得以快速鉴定[44 ] .噬菌体展示技术可以应用于较大突变体库(109 )的筛选,加之具有技术成熟、不需要昂贵的试剂与设备的特点,在酶的定向进化和抗体研发领域颇受青睐.S. Vichier-Guerre课题组利用该技术,从来自水生栖热菌(Thermus aquaticus )的DNA 聚合酶 I进化出一组耐热性提高的逆转录酶[45 ] . ...
Optimizing the affinity and specificity of proteins with molecular display
1
2006
... 细胞表面展示技术是指将外源蛋白与细胞表面结构蛋白融合,并在细胞表面进行活性表达,常用的宿主有大肠杆菌、酵母、哺乳动物细胞等.和噬菌体展示技术相比,细胞表面展示技术的优势是可以展示分子量较大、结构更复杂的蛋白质,并且可以结合流式细胞术,直接分析展示蛋白的表达、稳定性以及与互作蛋白的亲和力[46 ] . ...
Directed evolution of PD-L1-targeted affibodies by mRNA display
1
2022
... 核糖体展示技术与mRNA 展示技术的本质均为利用mRNA的可复制性,建立蛋白质表型和基因型之间的物理关联,使目的蛋白得到有效鉴定和富集.两种技术均在无细胞系统中进行,可避免一些基于细胞展示系统的限制,文库容量和筛选通量得到极大提升(1012 ~1014 ).区别在于,mRNA展示系统翻译出来的蛋白质是与mRNA通过小分子接头连接在一起的,筛选更加便捷;而核糖体展示技术因要保证mRNA-核糖体-蛋白质三聚体的完整性,筛选中的不稳定因素较多.2022年,S. Millward 课题组[47 ] 首次将mRNA展示技术应用于一种特异性亲和体(programmed cell death-ligand 1,PD-L1)的定向进化,实现了靶标分子结合特异性的完全转换. ...
Directed evolution of an α1,3-fucosyltransferase using a single-cell ultrahigh-throughput screening method
2
2019
... 荧光筛选法是指针对本身无光学特性的酶反应,通过对底物进行荧光标记,根据由化学键断裂或形成导致的样本荧光信号的改变,来表征突变体的催化活性[48 ] .另外,通过合成生物学理性设计,将目标蛋白酶的反应与荧光蛋白的表达、折叠或运输过程相偶联,也是将荧光筛选技术用于工程改造不具光学特性的酶反应过程的最常用方法之一[49 ] .由于荧光筛选法操作简便、灵敏度高和灵活性强,目前已成为应用范围最广泛的高通量筛选方法,荧光标记也成为首选的高通量筛选标记. ...
... 传统的荧光筛选大多是采用小体积筛选体系在微孔板中进行,利用酶的底物或产物的荧光特性,通过荧光强度的变化鉴定酶的活性,但存在通量低、操作耗时等缺点.为解决这些问题,近年来发展出了基于荧光筛选标记的多种新型高通量筛选方法,例如荧光激活细胞分选技术(fluorescence-activated cell sorting,FACS).这是一种新兴的超高通量筛选方法,能够以105 s-1 的速率对数亿样品完成光散射能力和荧光特性的分析与分选,因其高灵敏度和高分析量等显著优势,从而成为筛选突变体库的强大工具.G. Yang课题组巧妙地利用了细胞膜表面的半乳糖透酶(LacY 基因编码)对底物及糖基化产物通透性的差异,建立了首个可以利用流式细胞仪在单细胞层面检测岩藻糖基转移酶(fucosyltransferases,FucTs)及其突变体的FACS筛选体系,筛选速度达到107 h-1 以上,一举打破了这类酶的筛选瓶颈,成功获得了目前国际报道催化效率最高的FucTs突变体[48 ] . ...
High throughput screening and selection methods for directed enzyme evolution
1
2015
... 荧光筛选法是指针对本身无光学特性的酶反应,通过对底物进行荧光标记,根据由化学键断裂或形成导致的样本荧光信号的改变,来表征突变体的催化活性[48 ] .另外,通过合成生物学理性设计,将目标蛋白酶的反应与荧光蛋白的表达、折叠或运输过程相偶联,也是将荧光筛选技术用于工程改造不具光学特性的酶反应过程的最常用方法之一[49 ] .由于荧光筛选法操作简便、灵敏度高和灵活性强,目前已成为应用范围最广泛的高通量筛选方法,荧光标记也成为首选的高通量筛选标记. ...
Ultrahigh-throughput screening in drop-based microfluidics for directed evolution
1
2010
... 在FACS筛选体系问世不久后,J. Agresti课题组提出了另一种里程碑式的、通用的超高通量筛选平台.该平台基于液滴的微流控技术,可在一次实验中对数千万个细胞进行分析,极大改变了筛选的规模和速度[50 ] .例如,D. Hilvert课题组将微流控技术与高通量荧光分选技术相结合,筛选到了能与天然Ⅰ类醛缩酶相媲美的新人工醛缩酶突变体,其酶活性较母体提高了30倍,催化速率提高了2×109 倍,而这是传统筛选技术难以实现的[51 ] .提高酶的化学选择性、区域选择性和对映选择性,对医药化工绿色工艺转型意义重大,除了提高目标酶的活性,超高通量的液滴微流控技术也能用于提高酶选择性的定向进化实验中.从大量突变体中筛选具有对映体选择性的酶往往需要进行手性色谱分析,该方法成本高且通量低.对此,G. Yang课题组开发了超高通量双通道微流控液滴筛选技术DMDS(dual-channel microfluidic droplet screening),将含突变酶的细胞和被不同荧光标记的对映体底物包裹在液滴中,当用不同波长的激发光照射时,能够对两种对映体底物的催化活性进行表征,并高效分选出阳性个体[52 ] .作者利用该系统对嗜热酯酶AFEST的对映选择性进行优化,经5轮连续进化后,获得了对S -布洛芬的对映体选择性提高700倍的突变体. ...
Emergence of a catalytic tetrad during evolution of a highly active artificial aldolase
1
2017
... 在FACS筛选体系问世不久后,J. Agresti课题组提出了另一种里程碑式的、通用的超高通量筛选平台.该平台基于液滴的微流控技术,可在一次实验中对数千万个细胞进行分析,极大改变了筛选的规模和速度[50 ] .例如,D. Hilvert课题组将微流控技术与高通量荧光分选技术相结合,筛选到了能与天然Ⅰ类醛缩酶相媲美的新人工醛缩酶突变体,其酶活性较母体提高了30倍,催化速率提高了2×109 倍,而这是传统筛选技术难以实现的[51 ] .提高酶的化学选择性、区域选择性和对映选择性,对医药化工绿色工艺转型意义重大,除了提高目标酶的活性,超高通量的液滴微流控技术也能用于提高酶选择性的定向进化实验中.从大量突变体中筛选具有对映体选择性的酶往往需要进行手性色谱分析,该方法成本高且通量低.对此,G. Yang课题组开发了超高通量双通道微流控液滴筛选技术DMDS(dual-channel microfluidic droplet screening),将含突变酶的细胞和被不同荧光标记的对映体底物包裹在液滴中,当用不同波长的激发光照射时,能够对两种对映体底物的催化活性进行表征,并高效分选出阳性个体[52 ] .作者利用该系统对嗜热酯酶AFEST的对映选择性进行优化,经5轮连续进化后,获得了对S -布洛芬的对映体选择性提高700倍的突变体. ...
Efficient molecular evolution to generate enantioselective enzymes using a dual-channel microfluidic droplet screening platform
1
2018
... 在FACS筛选体系问世不久后,J. Agresti课题组提出了另一种里程碑式的、通用的超高通量筛选平台.该平台基于液滴的微流控技术,可在一次实验中对数千万个细胞进行分析,极大改变了筛选的规模和速度[50 ] .例如,D. Hilvert课题组将微流控技术与高通量荧光分选技术相结合,筛选到了能与天然Ⅰ类醛缩酶相媲美的新人工醛缩酶突变体,其酶活性较母体提高了30倍,催化速率提高了2×109 倍,而这是传统筛选技术难以实现的[51 ] .提高酶的化学选择性、区域选择性和对映选择性,对医药化工绿色工艺转型意义重大,除了提高目标酶的活性,超高通量的液滴微流控技术也能用于提高酶选择性的定向进化实验中.从大量突变体中筛选具有对映体选择性的酶往往需要进行手性色谱分析,该方法成本高且通量低.对此,G. Yang课题组开发了超高通量双通道微流控液滴筛选技术DMDS(dual-channel microfluidic droplet screening),将含突变酶的细胞和被不同荧光标记的对映体底物包裹在液滴中,当用不同波长的激发光照射时,能够对两种对映体底物的催化活性进行表征,并高效分选出阳性个体[52 ] .作者利用该系统对嗜热酯酶AFEST的对映选择性进行优化,经5轮连续进化后,获得了对S -布洛芬的对映体选择性提高700倍的突变体. ...
Recent advances in design of fluorescence-based assays for high-throughput screening
2
2019
... 除了将荧光标记和分选或微流控技术相结合的FACS和DMDS体系,利用荧光或被荧光标记的分子所具备的物理性质而发展出的荧光偏振(fluorescence polarization,FP)技术和荧光共振能量转移(fluorescence resonance energy transfer, FRET)技术也是近年来在蛋白质工程领域应用较多的荧光筛选法.FP技术是根据体系中荧光基团与被测分子结合前后的偏振信号变化,来对分子间相互作用进行研究或定量检测目标分子,由于荧光的偏振性对于分子质量、体积、状态都非常敏感,因此FP技术具有快速、直观、灵敏等优势[53 ] .M. Tokeshi课题组通过FP技术结合免疫分析,可以实现快速筛选并定量检测到人血清中新型冠状病毒(SARS-CoV-2)抗体[54 ] .S. Mattoo课题组利用FP技术构建了稳健的、适用于追踪人类HYPE 腺苷酸转移酶活性的高通量筛选体系,对美国食品药品监督管理局(Food and Drug Administration,FDA)上市化合物、天然产物等 9680 种活性化合物进行筛选,鉴定出了第一个HYPE 腺苷酸转移酶活性调节剂[55 ] .FRET技术则是利用当供体荧光分子的发射光谱与受体荧光分子的吸收光谱重叠,且距离合适(一般小于10 nm)的两个荧光分子间会产生能量转移这一现象,来检测两个被测蛋白质之间亲和力的变化,或因其结合构象的变化引起的蛋白质-蛋白质相互作用方式的改变.同其他技术相比,FRET最大的特点是可用于研究活细胞在生理条件下的蛋白间相互作用[53 ] ,且常常需要结合杂交链式反应(hybridization chain reaction,HCR)进行荧光信号放大.例如,H. Tahara 课题组通过构建基于DNA链交换的荧光共振能量转移(DNA strand exchange fluorescence resonance energy transfer,DSE-FRET)系统,从32914种化合物中高通量筛选并鉴定出了对转录因子NF-κB特定亚型起作用的抑制剂[56 ] . ...
... [53 ],且常常需要结合杂交链式反应(hybridization chain reaction,HCR)进行荧光信号放大.例如,H. Tahara 课题组通过构建基于DNA链交换的荧光共振能量转移(DNA strand exchange fluorescence resonance energy transfer,DSE-FRET)系统,从32914种化合物中高通量筛选并鉴定出了对转录因子NF-κB特定亚型起作用的抑制剂[56 ] . ...
Facile and rapid detection of SARS-CoV-2 antibody based on a noncompetitive fluorescence polarization immunoassay in human serum samples
1
2021
... 除了将荧光标记和分选或微流控技术相结合的FACS和DMDS体系,利用荧光或被荧光标记的分子所具备的物理性质而发展出的荧光偏振(fluorescence polarization,FP)技术和荧光共振能量转移(fluorescence resonance energy transfer, FRET)技术也是近年来在蛋白质工程领域应用较多的荧光筛选法.FP技术是根据体系中荧光基团与被测分子结合前后的偏振信号变化,来对分子间相互作用进行研究或定量检测目标分子,由于荧光的偏振性对于分子质量、体积、状态都非常敏感,因此FP技术具有快速、直观、灵敏等优势[53 ] .M. Tokeshi课题组通过FP技术结合免疫分析,可以实现快速筛选并定量检测到人血清中新型冠状病毒(SARS-CoV-2)抗体[54 ] .S. Mattoo课题组利用FP技术构建了稳健的、适用于追踪人类HYPE 腺苷酸转移酶活性的高通量筛选体系,对美国食品药品监督管理局(Food and Drug Administration,FDA)上市化合物、天然产物等 9680 种活性化合物进行筛选,鉴定出了第一个HYPE 腺苷酸转移酶活性调节剂[55 ] .FRET技术则是利用当供体荧光分子的发射光谱与受体荧光分子的吸收光谱重叠,且距离合适(一般小于10 nm)的两个荧光分子间会产生能量转移这一现象,来检测两个被测蛋白质之间亲和力的变化,或因其结合构象的变化引起的蛋白质-蛋白质相互作用方式的改变.同其他技术相比,FRET最大的特点是可用于研究活细胞在生理条件下的蛋白间相互作用[53 ] ,且常常需要结合杂交链式反应(hybridization chain reaction,HCR)进行荧光信号放大.例如,H. Tahara 课题组通过构建基于DNA链交换的荧光共振能量转移(DNA strand exchange fluorescence resonance energy transfer,DSE-FRET)系统,从32914种化合物中高通量筛选并鉴定出了对转录因子NF-κB特定亚型起作用的抑制剂[56 ] . ...
A fluorescence polarization-based high-throughput screen to identify the first small-molecule modulators of the human adenylyltransferase HYPE/FICD
1
2020
... 除了将荧光标记和分选或微流控技术相结合的FACS和DMDS体系,利用荧光或被荧光标记的分子所具备的物理性质而发展出的荧光偏振(fluorescence polarization,FP)技术和荧光共振能量转移(fluorescence resonance energy transfer, FRET)技术也是近年来在蛋白质工程领域应用较多的荧光筛选法.FP技术是根据体系中荧光基团与被测分子结合前后的偏振信号变化,来对分子间相互作用进行研究或定量检测目标分子,由于荧光的偏振性对于分子质量、体积、状态都非常敏感,因此FP技术具有快速、直观、灵敏等优势[53 ] .M. Tokeshi课题组通过FP技术结合免疫分析,可以实现快速筛选并定量检测到人血清中新型冠状病毒(SARS-CoV-2)抗体[54 ] .S. Mattoo课题组利用FP技术构建了稳健的、适用于追踪人类HYPE 腺苷酸转移酶活性的高通量筛选体系,对美国食品药品监督管理局(Food and Drug Administration,FDA)上市化合物、天然产物等 9680 种活性化合物进行筛选,鉴定出了第一个HYPE 腺苷酸转移酶活性调节剂[55 ] .FRET技术则是利用当供体荧光分子的发射光谱与受体荧光分子的吸收光谱重叠,且距离合适(一般小于10 nm)的两个荧光分子间会产生能量转移这一现象,来检测两个被测蛋白质之间亲和力的变化,或因其结合构象的变化引起的蛋白质-蛋白质相互作用方式的改变.同其他技术相比,FRET最大的特点是可用于研究活细胞在生理条件下的蛋白间相互作用[53 ] ,且常常需要结合杂交链式反应(hybridization chain reaction,HCR)进行荧光信号放大.例如,H. Tahara 课题组通过构建基于DNA链交换的荧光共振能量转移(DNA strand exchange fluorescence resonance energy transfer,DSE-FRET)系统,从32914种化合物中高通量筛选并鉴定出了对转录因子NF-κB特定亚型起作用的抑制剂[56 ] . ...
Identification of a selective RelA inhibitor based on DSE-FRET screening methods
1
2020
... 除了将荧光标记和分选或微流控技术相结合的FACS和DMDS体系,利用荧光或被荧光标记的分子所具备的物理性质而发展出的荧光偏振(fluorescence polarization,FP)技术和荧光共振能量转移(fluorescence resonance energy transfer, FRET)技术也是近年来在蛋白质工程领域应用较多的荧光筛选法.FP技术是根据体系中荧光基团与被测分子结合前后的偏振信号变化,来对分子间相互作用进行研究或定量检测目标分子,由于荧光的偏振性对于分子质量、体积、状态都非常敏感,因此FP技术具有快速、直观、灵敏等优势[53 ] .M. Tokeshi课题组通过FP技术结合免疫分析,可以实现快速筛选并定量检测到人血清中新型冠状病毒(SARS-CoV-2)抗体[54 ] .S. Mattoo课题组利用FP技术构建了稳健的、适用于追踪人类HYPE 腺苷酸转移酶活性的高通量筛选体系,对美国食品药品监督管理局(Food and Drug Administration,FDA)上市化合物、天然产物等 9680 种活性化合物进行筛选,鉴定出了第一个HYPE 腺苷酸转移酶活性调节剂[55 ] .FRET技术则是利用当供体荧光分子的发射光谱与受体荧光分子的吸收光谱重叠,且距离合适(一般小于10 nm)的两个荧光分子间会产生能量转移这一现象,来检测两个被测蛋白质之间亲和力的变化,或因其结合构象的变化引起的蛋白质-蛋白质相互作用方式的改变.同其他技术相比,FRET最大的特点是可用于研究活细胞在生理条件下的蛋白间相互作用[53 ] ,且常常需要结合杂交链式反应(hybridization chain reaction,HCR)进行荧光信号放大.例如,H. Tahara 课题组通过构建基于DNA链交换的荧光共振能量转移(DNA strand exchange fluorescence resonance energy transfer,DSE-FRET)系统,从32914种化合物中高通量筛选并鉴定出了对转录因子NF-κB特定亚型起作用的抑制剂[56 ] . ...
Electrospray ionization for mass spectrometry of large biomolecules
1
1989
... 在不需要标记的高通量筛选方法中,质谱法作为一种高灵敏度和高特异性的检测方法,在蛋白质工程中应用极广.随着检测仪器性能和检测速率的不断提高,尤其是电喷射离子化技术(electrospray ionization,ESI)[57 ] 和基质辅助激光解吸离子化技术(matrix assisted laser desorption/ionization, MALDI)[58 -59 ] 的出现,生物大分子的离子化问题得以解决,并以此为基础,涌现出了许多新型质谱离子化技术,例如,基质辅助激光解吸电离飞行时间质谱(MALDI-ToF-MS)利用激光取样,通过离子的质荷比对目标分子进行无标记定性和定量分析,因具有对样品中盐浓度耐受性高、生物分子覆盖性广泛和扫描速率高等优势,已被越来越多地应用于酶反应的表征工作[60 -61 ] .但是传统的MALDI-ToF样品制备,高度依赖像声波沉积这样的先进液体处理器以提高制备通量,有一定的应用局限性[62 ] ,在此基础上进一步发展的SAMDI(self-assembled monolayers for matrix-assisted laser desorption ionization)技术,通过将反应产物固定在连接于金表面的烷硫酯自组装单分子膜上,实现了MALDI-ToF样品的快速制备,从而提高了检测通量[63 ] .M. Mrksich课题组利用该技术,从细胞色素P411随机突变文库中以每小时数千个突变体的速度筛选出催化C(sp3 )-H键烷基化的高活性突变体[64 ] .J. Sweedler课题组和H. Zhao课题组基于MALDI-ToF技术合作开发了对微生物克隆直接采样分析的技术,通量更高,且更利于对多步酶反应进行优化和表征 [13 ] .利用该方法,该课题组表征了从核糖体前体肽合成细菌素Plantazolicin的五酶途径的底物耐受性,并以1~2.5 s/克隆的速率从定向进化突变体库中筛选出选择性高产特定单鼠李糖脂同源物的双酶途径.此外,2021年报道的声波激发与质谱耦合系统(acoustic ejection mass spectrometry,AEMS),可以在筛选过程中实现零交叉污染、高通量、高定量分析精度,并同时提供极强基质耐受性[65 ] . ...
Matrix-assisted ultraviolet laser desorption of non-volatile compounds
1
1987
... 在不需要标记的高通量筛选方法中,质谱法作为一种高灵敏度和高特异性的检测方法,在蛋白质工程中应用极广.随着检测仪器性能和检测速率的不断提高,尤其是电喷射离子化技术(electrospray ionization,ESI)[57 ] 和基质辅助激光解吸离子化技术(matrix assisted laser desorption/ionization, MALDI)[58 -59 ] 的出现,生物大分子的离子化问题得以解决,并以此为基础,涌现出了许多新型质谱离子化技术,例如,基质辅助激光解吸电离飞行时间质谱(MALDI-ToF-MS)利用激光取样,通过离子的质荷比对目标分子进行无标记定性和定量分析,因具有对样品中盐浓度耐受性高、生物分子覆盖性广泛和扫描速率高等优势,已被越来越多地应用于酶反应的表征工作[60 -61 ] .但是传统的MALDI-ToF样品制备,高度依赖像声波沉积这样的先进液体处理器以提高制备通量,有一定的应用局限性[62 ] ,在此基础上进一步发展的SAMDI(self-assembled monolayers for matrix-assisted laser desorption ionization)技术,通过将反应产物固定在连接于金表面的烷硫酯自组装单分子膜上,实现了MALDI-ToF样品的快速制备,从而提高了检测通量[63 ] .M. Mrksich课题组利用该技术,从细胞色素P411随机突变文库中以每小时数千个突变体的速度筛选出催化C(sp3 )-H键烷基化的高活性突变体[64 ] .J. Sweedler课题组和H. Zhao课题组基于MALDI-ToF技术合作开发了对微生物克隆直接采样分析的技术,通量更高,且更利于对多步酶反应进行优化和表征 [13 ] .利用该方法,该课题组表征了从核糖体前体肽合成细菌素Plantazolicin的五酶途径的底物耐受性,并以1~2.5 s/克隆的速率从定向进化突变体库中筛选出选择性高产特定单鼠李糖脂同源物的双酶途径.此外,2021年报道的声波激发与质谱耦合系统(acoustic ejection mass spectrometry,AEMS),可以在筛选过程中实现零交叉污染、高通量、高定量分析精度,并同时提供极强基质耐受性[65 ] . ...
Laser desorption ionization of proteins with molecular masses exceeding 10, 000 daltons
1
1988
... 在不需要标记的高通量筛选方法中,质谱法作为一种高灵敏度和高特异性的检测方法,在蛋白质工程中应用极广.随着检测仪器性能和检测速率的不断提高,尤其是电喷射离子化技术(electrospray ionization,ESI)[57 ] 和基质辅助激光解吸离子化技术(matrix assisted laser desorption/ionization, MALDI)[58 -59 ] 的出现,生物大分子的离子化问题得以解决,并以此为基础,涌现出了许多新型质谱离子化技术,例如,基质辅助激光解吸电离飞行时间质谱(MALDI-ToF-MS)利用激光取样,通过离子的质荷比对目标分子进行无标记定性和定量分析,因具有对样品中盐浓度耐受性高、生物分子覆盖性广泛和扫描速率高等优势,已被越来越多地应用于酶反应的表征工作[60 -61 ] .但是传统的MALDI-ToF样品制备,高度依赖像声波沉积这样的先进液体处理器以提高制备通量,有一定的应用局限性[62 ] ,在此基础上进一步发展的SAMDI(self-assembled monolayers for matrix-assisted laser desorption ionization)技术,通过将反应产物固定在连接于金表面的烷硫酯自组装单分子膜上,实现了MALDI-ToF样品的快速制备,从而提高了检测通量[63 ] .M. Mrksich课题组利用该技术,从细胞色素P411随机突变文库中以每小时数千个突变体的速度筛选出催化C(sp3 )-H键烷基化的高活性突变体[64 ] .J. Sweedler课题组和H. Zhao课题组基于MALDI-ToF技术合作开发了对微生物克隆直接采样分析的技术,通量更高,且更利于对多步酶反应进行优化和表征 [13 ] .利用该方法,该课题组表征了从核糖体前体肽合成细菌素Plantazolicin的五酶途径的底物耐受性,并以1~2.5 s/克隆的速率从定向进化突变体库中筛选出选择性高产特定单鼠李糖脂同源物的双酶途径.此外,2021年报道的声波激发与质谱耦合系统(acoustic ejection mass spectrometry,AEMS),可以在筛选过程中实现零交叉污染、高通量、高定量分析精度,并同时提供极强基质耐受性[65 ] . ...
Discovery of glycosyltransferases using carbohydrate arrays and mass spectrometry
1
2012
... 在不需要标记的高通量筛选方法中,质谱法作为一种高灵敏度和高特异性的检测方法,在蛋白质工程中应用极广.随着检测仪器性能和检测速率的不断提高,尤其是电喷射离子化技术(electrospray ionization,ESI)[57 ] 和基质辅助激光解吸离子化技术(matrix assisted laser desorption/ionization, MALDI)[58 -59 ] 的出现,生物大分子的离子化问题得以解决,并以此为基础,涌现出了许多新型质谱离子化技术,例如,基质辅助激光解吸电离飞行时间质谱(MALDI-ToF-MS)利用激光取样,通过离子的质荷比对目标分子进行无标记定性和定量分析,因具有对样品中盐浓度耐受性高、生物分子覆盖性广泛和扫描速率高等优势,已被越来越多地应用于酶反应的表征工作[60 -61 ] .但是传统的MALDI-ToF样品制备,高度依赖像声波沉积这样的先进液体处理器以提高制备通量,有一定的应用局限性[62 ] ,在此基础上进一步发展的SAMDI(self-assembled monolayers for matrix-assisted laser desorption ionization)技术,通过将反应产物固定在连接于金表面的烷硫酯自组装单分子膜上,实现了MALDI-ToF样品的快速制备,从而提高了检测通量[63 ] .M. Mrksich课题组利用该技术,从细胞色素P411随机突变文库中以每小时数千个突变体的速度筛选出催化C(sp3 )-H键烷基化的高活性突变体[64 ] .J. Sweedler课题组和H. Zhao课题组基于MALDI-ToF技术合作开发了对微生物克隆直接采样分析的技术,通量更高,且更利于对多步酶反应进行优化和表征 [13 ] .利用该方法,该课题组表征了从核糖体前体肽合成细菌素Plantazolicin的五酶途径的底物耐受性,并以1~2.5 s/克隆的速率从定向进化突变体库中筛选出选择性高产特定单鼠李糖脂同源物的双酶途径.此外,2021年报道的声波激发与质谱耦合系统(acoustic ejection mass spectrometry,AEMS),可以在筛选过程中实现零交叉污染、高通量、高定量分析精度,并同时提供极强基质耐受性[65 ] . ...
High throughput screening of enzyme activity with mass spectrometry imaging
1
2015
... 在不需要标记的高通量筛选方法中,质谱法作为一种高灵敏度和高特异性的检测方法,在蛋白质工程中应用极广.随着检测仪器性能和检测速率的不断提高,尤其是电喷射离子化技术(electrospray ionization,ESI)[57 ] 和基质辅助激光解吸离子化技术(matrix assisted laser desorption/ionization, MALDI)[58 -59 ] 的出现,生物大分子的离子化问题得以解决,并以此为基础,涌现出了许多新型质谱离子化技术,例如,基质辅助激光解吸电离飞行时间质谱(MALDI-ToF-MS)利用激光取样,通过离子的质荷比对目标分子进行无标记定性和定量分析,因具有对样品中盐浓度耐受性高、生物分子覆盖性广泛和扫描速率高等优势,已被越来越多地应用于酶反应的表征工作[60 -61 ] .但是传统的MALDI-ToF样品制备,高度依赖像声波沉积这样的先进液体处理器以提高制备通量,有一定的应用局限性[62 ] ,在此基础上进一步发展的SAMDI(self-assembled monolayers for matrix-assisted laser desorption ionization)技术,通过将反应产物固定在连接于金表面的烷硫酯自组装单分子膜上,实现了MALDI-ToF样品的快速制备,从而提高了检测通量[63 ] .M. Mrksich课题组利用该技术,从细胞色素P411随机突变文库中以每小时数千个突变体的速度筛选出催化C(sp3 )-H键烷基化的高活性突变体[64 ] .J. Sweedler课题组和H. Zhao课题组基于MALDI-ToF技术合作开发了对微生物克隆直接采样分析的技术,通量更高,且更利于对多步酶反应进行优化和表征 [13 ] .利用该方法,该课题组表征了从核糖体前体肽合成细菌素Plantazolicin的五酶途径的底物耐受性,并以1~2.5 s/克隆的速率从定向进化突变体库中筛选出选择性高产特定单鼠李糖脂同源物的双酶途径.此外,2021年报道的声波激发与质谱耦合系统(acoustic ejection mass spectrometry,AEMS),可以在筛选过程中实现零交叉污染、高通量、高定量分析精度,并同时提供极强基质耐受性[65 ] . ...
Acoustic deposition with NIMS as a high-throughput enzyme activity assay
1
2012
... 在不需要标记的高通量筛选方法中,质谱法作为一种高灵敏度和高特异性的检测方法,在蛋白质工程中应用极广.随着检测仪器性能和检测速率的不断提高,尤其是电喷射离子化技术(electrospray ionization,ESI)[57 ] 和基质辅助激光解吸离子化技术(matrix assisted laser desorption/ionization, MALDI)[58 -59 ] 的出现,生物大分子的离子化问题得以解决,并以此为基础,涌现出了许多新型质谱离子化技术,例如,基质辅助激光解吸电离飞行时间质谱(MALDI-ToF-MS)利用激光取样,通过离子的质荷比对目标分子进行无标记定性和定量分析,因具有对样品中盐浓度耐受性高、生物分子覆盖性广泛和扫描速率高等优势,已被越来越多地应用于酶反应的表征工作[60 -61 ] .但是传统的MALDI-ToF样品制备,高度依赖像声波沉积这样的先进液体处理器以提高制备通量,有一定的应用局限性[62 ] ,在此基础上进一步发展的SAMDI(self-assembled monolayers for matrix-assisted laser desorption ionization)技术,通过将反应产物固定在连接于金表面的烷硫酯自组装单分子膜上,实现了MALDI-ToF样品的快速制备,从而提高了检测通量[63 ] .M. Mrksich课题组利用该技术,从细胞色素P411随机突变文库中以每小时数千个突变体的速度筛选出催化C(sp3 )-H键烷基化的高活性突变体[64 ] .J. Sweedler课题组和H. Zhao课题组基于MALDI-ToF技术合作开发了对微生物克隆直接采样分析的技术,通量更高,且更利于对多步酶反应进行优化和表征 [13 ] .利用该方法,该课题组表征了从核糖体前体肽合成细菌素Plantazolicin的五酶途径的底物耐受性,并以1~2.5 s/克隆的速率从定向进化突变体库中筛选出选择性高产特定单鼠李糖脂同源物的双酶途径.此外,2021年报道的声波激发与质谱耦合系统(acoustic ejection mass spectrometry,AEMS),可以在筛选过程中实现零交叉污染、高通量、高定量分析精度,并同时提供极强基质耐受性[65 ] . ...
Mass spectrometry of self-assembled monolayers: a new tool for molecular surface science
1
2008
... 在不需要标记的高通量筛选方法中,质谱法作为一种高灵敏度和高特异性的检测方法,在蛋白质工程中应用极广.随着检测仪器性能和检测速率的不断提高,尤其是电喷射离子化技术(electrospray ionization,ESI)[57 ] 和基质辅助激光解吸离子化技术(matrix assisted laser desorption/ionization, MALDI)[58 -59 ] 的出现,生物大分子的离子化问题得以解决,并以此为基础,涌现出了许多新型质谱离子化技术,例如,基质辅助激光解吸电离飞行时间质谱(MALDI-ToF-MS)利用激光取样,通过离子的质荷比对目标分子进行无标记定性和定量分析,因具有对样品中盐浓度耐受性高、生物分子覆盖性广泛和扫描速率高等优势,已被越来越多地应用于酶反应的表征工作[60 -61 ] .但是传统的MALDI-ToF样品制备,高度依赖像声波沉积这样的先进液体处理器以提高制备通量,有一定的应用局限性[62 ] ,在此基础上进一步发展的SAMDI(self-assembled monolayers for matrix-assisted laser desorption ionization)技术,通过将反应产物固定在连接于金表面的烷硫酯自组装单分子膜上,实现了MALDI-ToF样品的快速制备,从而提高了检测通量[63 ] .M. Mrksich课题组利用该技术,从细胞色素P411随机突变文库中以每小时数千个突变体的速度筛选出催化C(sp3 )-H键烷基化的高活性突变体[64 ] .J. Sweedler课题组和H. Zhao课题组基于MALDI-ToF技术合作开发了对微生物克隆直接采样分析的技术,通量更高,且更利于对多步酶反应进行优化和表征 [13 ] .利用该方法,该课题组表征了从核糖体前体肽合成细菌素Plantazolicin的五酶途径的底物耐受性,并以1~2.5 s/克隆的速率从定向进化突变体库中筛选出选择性高产特定单鼠李糖脂同源物的双酶途径.此外,2021年报道的声波激发与质谱耦合系统(acoustic ejection mass spectrometry,AEMS),可以在筛选过程中实现零交叉污染、高通量、高定量分析精度,并同时提供极强基质耐受性[65 ] . ...
High throughput screening with SAMDI mass spectrometry for directed evolution
1
2020
... 在不需要标记的高通量筛选方法中,质谱法作为一种高灵敏度和高特异性的检测方法,在蛋白质工程中应用极广.随着检测仪器性能和检测速率的不断提高,尤其是电喷射离子化技术(electrospray ionization,ESI)[57 ] 和基质辅助激光解吸离子化技术(matrix assisted laser desorption/ionization, MALDI)[58 -59 ] 的出现,生物大分子的离子化问题得以解决,并以此为基础,涌现出了许多新型质谱离子化技术,例如,基质辅助激光解吸电离飞行时间质谱(MALDI-ToF-MS)利用激光取样,通过离子的质荷比对目标分子进行无标记定性和定量分析,因具有对样品中盐浓度耐受性高、生物分子覆盖性广泛和扫描速率高等优势,已被越来越多地应用于酶反应的表征工作[60 -61 ] .但是传统的MALDI-ToF样品制备,高度依赖像声波沉积这样的先进液体处理器以提高制备通量,有一定的应用局限性[62 ] ,在此基础上进一步发展的SAMDI(self-assembled monolayers for matrix-assisted laser desorption ionization)技术,通过将反应产物固定在连接于金表面的烷硫酯自组装单分子膜上,实现了MALDI-ToF样品的快速制备,从而提高了检测通量[63 ] .M. Mrksich课题组利用该技术,从细胞色素P411随机突变文库中以每小时数千个突变体的速度筛选出催化C(sp3 )-H键烷基化的高活性突变体[64 ] .J. Sweedler课题组和H. Zhao课题组基于MALDI-ToF技术合作开发了对微生物克隆直接采样分析的技术,通量更高,且更利于对多步酶反应进行优化和表征 [13 ] .利用该方法,该课题组表征了从核糖体前体肽合成细菌素Plantazolicin的五酶途径的底物耐受性,并以1~2.5 s/克隆的速率从定向进化突变体库中筛选出选择性高产特定单鼠李糖脂同源物的双酶途径.此外,2021年报道的声波激发与质谱耦合系统(acoustic ejection mass spectrometry,AEMS),可以在筛选过程中实现零交叉污染、高通量、高定量分析精度,并同时提供极强基质耐受性[65 ] . ...
Fluid dynamics of the open port interface for high-speed nanoliter volume sampling mass spectrometry
1
2021
... 在不需要标记的高通量筛选方法中,质谱法作为一种高灵敏度和高特异性的检测方法,在蛋白质工程中应用极广.随着检测仪器性能和检测速率的不断提高,尤其是电喷射离子化技术(electrospray ionization,ESI)[57 ] 和基质辅助激光解吸离子化技术(matrix assisted laser desorption/ionization, MALDI)[58 -59 ] 的出现,生物大分子的离子化问题得以解决,并以此为基础,涌现出了许多新型质谱离子化技术,例如,基质辅助激光解吸电离飞行时间质谱(MALDI-ToF-MS)利用激光取样,通过离子的质荷比对目标分子进行无标记定性和定量分析,因具有对样品中盐浓度耐受性高、生物分子覆盖性广泛和扫描速率高等优势,已被越来越多地应用于酶反应的表征工作[60 -61 ] .但是传统的MALDI-ToF样品制备,高度依赖像声波沉积这样的先进液体处理器以提高制备通量,有一定的应用局限性[62 ] ,在此基础上进一步发展的SAMDI(self-assembled monolayers for matrix-assisted laser desorption ionization)技术,通过将反应产物固定在连接于金表面的烷硫酯自组装单分子膜上,实现了MALDI-ToF样品的快速制备,从而提高了检测通量[63 ] .M. Mrksich课题组利用该技术,从细胞色素P411随机突变文库中以每小时数千个突变体的速度筛选出催化C(sp3 )-H键烷基化的高活性突变体[64 ] .J. Sweedler课题组和H. Zhao课题组基于MALDI-ToF技术合作开发了对微生物克隆直接采样分析的技术,通量更高,且更利于对多步酶反应进行优化和表征 [13 ] .利用该方法,该课题组表征了从核糖体前体肽合成细菌素Plantazolicin的五酶途径的底物耐受性,并以1~2.5 s/克隆的速率从定向进化突变体库中筛选出选择性高产特定单鼠李糖脂同源物的双酶途径.此外,2021年报道的声波激发与质谱耦合系统(acoustic ejection mass spectrometry,AEMS),可以在筛选过程中实现零交叉污染、高通量、高定量分析精度,并同时提供极强基质耐受性[65 ] . ...
IR-thermographic screening of thermoneutral or endothermic transformations: the ring-closing olefin metathesis reaction
1
2000
... 基于反应热力学变化的红外检测法是一种有发展前景的高通量筛选方法,原理是所有物质都能发射红外线,且一系列先进的红外检测器可以灵敏地检测酶促反应过程中的热量变化.M. Reetz课题组[66 ] 采用ThermaCAM SC1000红外成像仪来检测脂酶催化乙烯基乙酸酯手性拆分反应中的热量变化,R 和S 底物被成对地放在孔板中进行单独检测,R 底物对应的是放热反应,成像中表现为“热点”,相反S 底物表现为“冷点”.通过对热点的分辨他们检出了R 底物特异性酶,即首次报道了热成像检测辅助手性酶筛选的实例.红外检测法的优点是不需要生色基团或荧光基团的引入,缺点是该方法容易受到样品不均一、底物催化副反应等的影响[67 ] . ...
生物酶活力的原位红外光谱测定
2
2015
... 基于反应热力学变化的红外检测法是一种有发展前景的高通量筛选方法,原理是所有物质都能发射红外线,且一系列先进的红外检测器可以灵敏地检测酶促反应过程中的热量变化.M. Reetz课题组[66 ] 采用ThermaCAM SC1000红外成像仪来检测脂酶催化乙烯基乙酸酯手性拆分反应中的热量变化,R 和S 底物被成对地放在孔板中进行单独检测,R 底物对应的是放热反应,成像中表现为“热点”,相反S 底物表现为“冷点”.通过对热点的分辨他们检出了R 底物特异性酶,即首次报道了热成像检测辅助手性酶筛选的实例.红外检测法的优点是不需要生色基团或荧光基团的引入,缺点是该方法容易受到样品不均一、底物催化副反应等的影响[67 ] . ...
... 以上提到的自动化平台,都是基于一种胞内进化方法而设计的,在实现更高通量的胞内连续进化方面取得了重要突破.但是,这些技术平台受限于改造其胞内进化技术所适用的特定目标蛋白.目前,建设基于自动化的生物合成平台已在全世界兴起,目标是通过集成式的设施,实现常规合成生物学实验操作的自动化完成,如基因克隆、蛋白表征、代谢表征等;并结合人工智能设备,实现生物技术研发过程中“设计—构建—测试—学习”四大模块的自动化迭代.例如美国伊利诺伊大学的H. Zhao课题组[86 ] 基于全自动生物合成平台iBioFAB,实现了大肠杆菌和酿酒酵母的自动转化、培养和性状表征筛选等操作,并结合机器学习开发了BioAutomata平台,通过结合不同T7启动子突变体与核糖体结合位点合成24组有不同表达水平的表达系统,对番茄红素合成路径中的三个关键基因的表达水平进行动态调控,同时结合基于贝叶斯算法(Bayesian algorithm)的计算机学习模型,对产生的数据进行学习反馈,进一步优化动态调控策略,最终获得高产番茄红素的蛋白表达体系[67 ] .同时,我国天津工业生物技术研究所的Y. Ma课题组[87 ] 开发了针对谷氨酸棒状杆菌(Corynebacterium glutamicum )代谢工程改造的多元自动化基因组编辑方法MACBETH(multiplex automated Corynebacterium glutamicum base editing method),其整合基于基因组编辑技术的基因敲除技术、目标菌株筛选和表型验证的全自动流程设备,以100%的编辑效率,构建了94个调控因子单独失活的C.glutamicum 菌株变异库存,有望提高谷氨酸的生成量.综合来看,自动化合成生物平台有望将传统的劳动密集型酶定向进化研究转化为高效便捷的工业化流程,为定向进化的发展开辟无数的可能性. ...
In situ infrared spectroscopic determination of enzyme activity
2
2015
... 基于反应热力学变化的红外检测法是一种有发展前景的高通量筛选方法,原理是所有物质都能发射红外线,且一系列先进的红外检测器可以灵敏地检测酶促反应过程中的热量变化.M. Reetz课题组[66 ] 采用ThermaCAM SC1000红外成像仪来检测脂酶催化乙烯基乙酸酯手性拆分反应中的热量变化,R 和S 底物被成对地放在孔板中进行单独检测,R 底物对应的是放热反应,成像中表现为“热点”,相反S 底物表现为“冷点”.通过对热点的分辨他们检出了R 底物特异性酶,即首次报道了热成像检测辅助手性酶筛选的实例.红外检测法的优点是不需要生色基团或荧光基团的引入,缺点是该方法容易受到样品不均一、底物催化副反应等的影响[67 ] . ...
... 以上提到的自动化平台,都是基于一种胞内进化方法而设计的,在实现更高通量的胞内连续进化方面取得了重要突破.但是,这些技术平台受限于改造其胞内进化技术所适用的特定目标蛋白.目前,建设基于自动化的生物合成平台已在全世界兴起,目标是通过集成式的设施,实现常规合成生物学实验操作的自动化完成,如基因克隆、蛋白表征、代谢表征等;并结合人工智能设备,实现生物技术研发过程中“设计—构建—测试—学习”四大模块的自动化迭代.例如美国伊利诺伊大学的H. Zhao课题组[86 ] 基于全自动生物合成平台iBioFAB,实现了大肠杆菌和酿酒酵母的自动转化、培养和性状表征筛选等操作,并结合机器学习开发了BioAutomata平台,通过结合不同T7启动子突变体与核糖体结合位点合成24组有不同表达水平的表达系统,对番茄红素合成路径中的三个关键基因的表达水平进行动态调控,同时结合基于贝叶斯算法(Bayesian algorithm)的计算机学习模型,对产生的数据进行学习反馈,进一步优化动态调控策略,最终获得高产番茄红素的蛋白表达体系[67 ] .同时,我国天津工业生物技术研究所的Y. Ma课题组[87 ] 开发了针对谷氨酸棒状杆菌(Corynebacterium glutamicum )代谢工程改造的多元自动化基因组编辑方法MACBETH(multiplex automated Corynebacterium glutamicum base editing method),其整合基于基因组编辑技术的基因敲除技术、目标菌株筛选和表型验证的全自动流程设备,以100%的编辑效率,构建了94个调控因子单独失活的C.glutamicum 菌株变异库存,有望提高谷氨酸的生成量.综合来看,自动化合成生物平台有望将传统的劳动密集型酶定向进化研究转化为高效便捷的工业化流程,为定向进化的发展开辟无数的可能性. ...
High-throughput screening technology in industrial biotechnology
2
2020
... 除了质谱法和红外检测法,另一大类不需要标记的筛选策略也已在工业生物技术中得到应用,即基于拉曼光谱、傅里叶变换红外光谱(Fourier transform infrared spectroscopy, FTIR)等特殊光谱的高通量筛选方法[68 ] ,这类方法主要优势在于对细胞无侵害性,且常与微流控技术联用.其中傅里叶变换红外光谱(FTIR)可利用特征吸收谱带强度的改变对混合物及化合物进行定量分析,还可根据特征吸收谱带的频率推断分子中存在某一基团或键,由特征吸收谱带频率的变化推测邻近的基团或化学键,进而确定分子的化学结构.Z. Wang课题组利用FTIR对109株金黄色葡萄球菌进行筛选,最终获得了2株能合成脂质和碳水化合物的菌株[69 ] .虽然目前还没有酶定向进化的例子,但是FTIR能实现同时定量分析细胞内和细胞外的分子含量[70 ] ,未来可用于分泌型小分子的定向进化,大大提高其筛选通量、效率以及准确性. ...
... 另外,面对数据资源的快速增长,将自动化技术应用于“设计—构建—测试—学习”的循环以建立一个全自动生物合成平台,能够有效提高酶的进化效率.目前,已有部分自动化生物合成平台或方法被建立并投入使用,例如伊利诺伊大学的iBioFAB(illinois biological foundry for advanced biomanufacturing)[142 ] 、中国科学院天津工业生物技术研究所的MACBETH(multiplex automated Corynebacterium glutamicum base editing method)[68 ] 等,这些方法通过计算机与自动化设备实现了某些宿主或定向进化中某些步骤的自动化,然而这尚不能实现定向进化过程的完全自动.在未来,随着突变与筛选过程中的实验步骤自动化技术以及人工智能技术的不断精进,智能制造的理念将不断引入合成生物学,定向进化也终将走向全自动. ...
High-throughput biochemical fingerprinting of oleaginous Aurantiochytrium sp. strains by fourier transform infrared spectroscopy (FT-IR) for lipid and carbohydrate productions
1
2019
... 除了质谱法和红外检测法,另一大类不需要标记的筛选策略也已在工业生物技术中得到应用,即基于拉曼光谱、傅里叶变换红外光谱(Fourier transform infrared spectroscopy, FTIR)等特殊光谱的高通量筛选方法[68 ] ,这类方法主要优势在于对细胞无侵害性,且常与微流控技术联用.其中傅里叶变换红外光谱(FTIR)可利用特征吸收谱带强度的改变对混合物及化合物进行定量分析,还可根据特征吸收谱带的频率推断分子中存在某一基团或键,由特征吸收谱带频率的变化推测邻近的基团或化学键,进而确定分子的化学结构.Z. Wang课题组利用FTIR对109株金黄色葡萄球菌进行筛选,最终获得了2株能合成脂质和碳水化合物的菌株[69 ] .虽然目前还没有酶定向进化的例子,但是FTIR能实现同时定量分析细胞内和细胞外的分子含量[70 ] ,未来可用于分泌型小分子的定向进化,大大提高其筛选通量、效率以及准确性. ...
FTIR spectroscopy as a unified method for simultaneous analysis of intra-and extracellular metabolites in high-throughput screening of microbial bioprocesses
1
2017
... 除了质谱法和红外检测法,另一大类不需要标记的筛选策略也已在工业生物技术中得到应用,即基于拉曼光谱、傅里叶变换红外光谱(Fourier transform infrared spectroscopy, FTIR)等特殊光谱的高通量筛选方法[68 ] ,这类方法主要优势在于对细胞无侵害性,且常与微流控技术联用.其中傅里叶变换红外光谱(FTIR)可利用特征吸收谱带强度的改变对混合物及化合物进行定量分析,还可根据特征吸收谱带的频率推断分子中存在某一基团或键,由特征吸收谱带频率的变化推测邻近的基团或化学键,进而确定分子的化学结构.Z. Wang课题组利用FTIR对109株金黄色葡萄球菌进行筛选,最终获得了2株能合成脂质和碳水化合物的菌株[69 ] .虽然目前还没有酶定向进化的例子,但是FTIR能实现同时定量分析细胞内和细胞外的分子含量[70 ] ,未来可用于分泌型小分子的定向进化,大大提高其筛选通量、效率以及准确性. ...
A system for the continuous directed evolution of biomolecules
5
2011
... 连续进化旨在无人为干预的情况下完成基因突变、蛋白表达、表型选择与筛选的迭代实验.连续定向进化通过缩短每轮的进化时间来增加迭代次数,从而提高获得目标性状突变体的概率.由D. Liu课题组开发的噬菌体辅助的连续进化系统(phage-assisted continuous evolution,PACE)是近年来最经典的案例.PACE将目标蛋白编码序列引入筛选噬菌体DNA载体中(selection phage,SP),并通过敲除pⅢ蛋白编码区而阻断噬菌体侵染力,同时将含有pⅢ的辅助质粒(accessory plasmid,AP)和提高大肠杆菌基因突变率的突变质粒(mutator plasmid,MP)引入大肠杆菌内.通过设计特定的基因回路,将pⅢ的表达与目标蛋白的活性相偶联,再通过类似“潟湖”的液路控制系统使得含有目标活性突变体的噬菌体迭代富集,从而实现进化与筛选自动循环.由于从噬菌体侵染到再组装的周期较为短暂,因此PACE系统可以在24 h内完成30轮以上的蛋白质进化,达到其他定向进化手段难以企及的迭代次数(图3 )[71 ] . ...
... [
71 ]
Progress of phage-assisted continuous evolution (PACE)<sup>[<xref ref-type="bibr" rid="R71">71</xref>]</sup> Fig. 3 ![]()
自2011年以来,D. Liu课题组利用PACE已成功完成了对多种蛋白的改造,如基于蛋白与DNA序列互作原理,成功对T7聚合酶[71 -72 ] 、TALEN[73 ] 、spCas9[74 ] 、胞嘧啶碱基编辑器(CBEs)[75 ] 、腺嘌呤碱基编辑器(ABEs)[76 ] 进行工程改造,改变了其识别序列的范围或提高了其识别特异性.同时,基于蛋白与蛋白相互作用原理,利用PACE系统,增强了对苏云金芽孢杆菌δ-内毒素与毛滴虫的钙黏蛋白样受体结合的亲和力[77 ] ,并实现了多种抗体和麦芽糖结合蛋白的可溶性高表达[78 ] (表1 ). ...
... [
71 ]
Fig. 3 ![]()
自2011年以来,D. Liu课题组利用PACE已成功完成了对多种蛋白的改造,如基于蛋白与DNA序列互作原理,成功对T7聚合酶[71 -72 ] 、TALEN[73 ] 、spCas9[74 ] 、胞嘧啶碱基编辑器(CBEs)[75 ] 、腺嘌呤碱基编辑器(ABEs)[76 ] 进行工程改造,改变了其识别序列的范围或提高了其识别特异性.同时,基于蛋白与蛋白相互作用原理,利用PACE系统,增强了对苏云金芽孢杆菌δ-内毒素与毛滴虫的钙黏蛋白样受体结合的亲和力[77 ] ,并实现了多种抗体和麦芽糖结合蛋白的可溶性高表达[78 ] (表1 ). ...
... 自2011年以来,D. Liu课题组利用PACE已成功完成了对多种蛋白的改造,如基于蛋白与DNA序列互作原理,成功对T7聚合酶[71 -72 ] 、TALEN[73 ] 、spCas9[74 ] 、胞嘧啶碱基编辑器(CBEs)[75 ] 、腺嘌呤碱基编辑器(ABEs)[76 ] 进行工程改造,改变了其识别序列的范围或提高了其识别特异性.同时,基于蛋白与蛋白相互作用原理,利用PACE系统,增强了对苏云金芽孢杆菌δ-内毒素与毛滴虫的钙黏蛋白样受体结合的亲和力[77 ] ,并实现了多种抗体和麦芽糖结合蛋白的可溶性高表达[78 ] (表1 ). ...
... Genetic circuit design
T7聚合酶 拓宽可识别的启动子范围[71 ] T7聚合酶 增强识别人工启动子的特异性[72 ] TALEN DNA序列识别特异性[73 ] spCas9 拓宽可识别的PAM序列[74 ] 胞嘧啶碱基编 辑器(CBEs) 拓宽可编辑的基因序列范围(例如GC丰富的序列)[75 ] 腺嘌呤碱基编 辑器(ABEs) 提高与Cas结构域的兼容性和编辑活性[76 ] 苏云金芽孢杆菌δ-内毒素 增强与毛滴虫的钙黏蛋白样受体结合亲和力[77 ] 抗体、麦芽糖 结合蛋白 增强目标蛋白 ...
Negative selection and stringency modulation in phage-assisted continuous evolution
2
2014
... 自2011年以来,D. Liu课题组利用PACE已成功完成了对多种蛋白的改造,如基于蛋白与DNA序列互作原理,成功对T7聚合酶[71 -72 ] 、TALEN[73 ] 、spCas9[74 ] 、胞嘧啶碱基编辑器(CBEs)[75 ] 、腺嘌呤碱基编辑器(ABEs)[76 ] 进行工程改造,改变了其识别序列的范围或提高了其识别特异性.同时,基于蛋白与蛋白相互作用原理,利用PACE系统,增强了对苏云金芽孢杆菌δ-内毒素与毛滴虫的钙黏蛋白样受体结合的亲和力[77 ] ,并实现了多种抗体和麦芽糖结合蛋白的可溶性高表达[78 ] (表1 ). ...
... Genetic circuit design
T7聚合酶 拓宽可识别的启动子范围[71 ] T7聚合酶 增强识别人工启动子的特异性[72 ] TALEN DNA序列识别特异性[73 ] spCas9 拓宽可识别的PAM序列[74 ] 胞嘧啶碱基编 辑器(CBEs) 拓宽可编辑的基因序列范围(例如GC丰富的序列)[75 ] 腺嘌呤碱基编 辑器(ABEs) 提高与Cas结构域的兼容性和编辑活性[76 ] 苏云金芽孢杆菌δ-内毒素 增强与毛滴虫的钙黏蛋白样受体结合亲和力[77 ] 抗体、麦芽糖 结合蛋白 增强目标蛋白 ...
Continuous directed evolution of DNA-binding proteins to improve TALEN specificity
2
2015
... 自2011年以来,D. Liu课题组利用PACE已成功完成了对多种蛋白的改造,如基于蛋白与DNA序列互作原理,成功对T7聚合酶[71 -72 ] 、TALEN[73 ] 、spCas9[74 ] 、胞嘧啶碱基编辑器(CBEs)[75 ] 、腺嘌呤碱基编辑器(ABEs)[76 ] 进行工程改造,改变了其识别序列的范围或提高了其识别特异性.同时,基于蛋白与蛋白相互作用原理,利用PACE系统,增强了对苏云金芽孢杆菌δ-内毒素与毛滴虫的钙黏蛋白样受体结合的亲和力[77 ] ,并实现了多种抗体和麦芽糖结合蛋白的可溶性高表达[78 ] (表1 ). ...
... Genetic circuit design
T7聚合酶 拓宽可识别的启动子范围[71 ] T7聚合酶 增强识别人工启动子的特异性[72 ] TALEN DNA序列识别特异性[73 ] spCas9 拓宽可识别的PAM序列[74 ] 胞嘧啶碱基编 辑器(CBEs) 拓宽可编辑的基因序列范围(例如GC丰富的序列)[75 ] 腺嘌呤碱基编 辑器(ABEs) 提高与Cas结构域的兼容性和编辑活性[76 ] 苏云金芽孢杆菌δ-内毒素 增强与毛滴虫的钙黏蛋白样受体结合亲和力[77 ] 抗体、麦芽糖 结合蛋白 增强目标蛋白 ...
Evolved Cas9 variants with broad PAM compatibility and high DNA specificity
2
2018
... 自2011年以来,D. Liu课题组利用PACE已成功完成了对多种蛋白的改造,如基于蛋白与DNA序列互作原理,成功对T7聚合酶[71 -72 ] 、TALEN[73 ] 、spCas9[74 ] 、胞嘧啶碱基编辑器(CBEs)[75 ] 、腺嘌呤碱基编辑器(ABEs)[76 ] 进行工程改造,改变了其识别序列的范围或提高了其识别特异性.同时,基于蛋白与蛋白相互作用原理,利用PACE系统,增强了对苏云金芽孢杆菌δ-内毒素与毛滴虫的钙黏蛋白样受体结合的亲和力[77 ] ,并实现了多种抗体和麦芽糖结合蛋白的可溶性高表达[78 ] (表1 ). ...
... Genetic circuit design
T7聚合酶 拓宽可识别的启动子范围[71 ] T7聚合酶 增强识别人工启动子的特异性[72 ] TALEN DNA序列识别特异性[73 ] spCas9 拓宽可识别的PAM序列[74 ] 胞嘧啶碱基编 辑器(CBEs) 拓宽可编辑的基因序列范围(例如GC丰富的序列)[75 ] 腺嘌呤碱基编 辑器(ABEs) 提高与Cas结构域的兼容性和编辑活性[76 ] 苏云金芽孢杆菌δ-内毒素 增强与毛滴虫的钙黏蛋白样受体结合亲和力[77 ] 抗体、麦芽糖 结合蛋白 增强目标蛋白 ...
Continuous evolution of base editors with expanded target compatibility and improved activity
2
2019
... 自2011年以来,D. Liu课题组利用PACE已成功完成了对多种蛋白的改造,如基于蛋白与DNA序列互作原理,成功对T7聚合酶[71 -72 ] 、TALEN[73 ] 、spCas9[74 ] 、胞嘧啶碱基编辑器(CBEs)[75 ] 、腺嘌呤碱基编辑器(ABEs)[76 ] 进行工程改造,改变了其识别序列的范围或提高了其识别特异性.同时,基于蛋白与蛋白相互作用原理,利用PACE系统,增强了对苏云金芽孢杆菌δ-内毒素与毛滴虫的钙黏蛋白样受体结合的亲和力[77 ] ,并实现了多种抗体和麦芽糖结合蛋白的可溶性高表达[78 ] (表1 ). ...
... Genetic circuit design
T7聚合酶 拓宽可识别的启动子范围[71 ] T7聚合酶 增强识别人工启动子的特异性[72 ] TALEN DNA序列识别特异性[73 ] spCas9 拓宽可识别的PAM序列[74 ] 胞嘧啶碱基编 辑器(CBEs) 拓宽可编辑的基因序列范围(例如GC丰富的序列)[75 ] 腺嘌呤碱基编 辑器(ABEs) 提高与Cas结构域的兼容性和编辑活性[76 ] 苏云金芽孢杆菌δ-内毒素 增强与毛滴虫的钙黏蛋白样受体结合亲和力[77 ] 抗体、麦芽糖 结合蛋白 增强目标蛋白 ...
Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity
2
2020
... 自2011年以来,D. Liu课题组利用PACE已成功完成了对多种蛋白的改造,如基于蛋白与DNA序列互作原理,成功对T7聚合酶[71 -72 ] 、TALEN[73 ] 、spCas9[74 ] 、胞嘧啶碱基编辑器(CBEs)[75 ] 、腺嘌呤碱基编辑器(ABEs)[76 ] 进行工程改造,改变了其识别序列的范围或提高了其识别特异性.同时,基于蛋白与蛋白相互作用原理,利用PACE系统,增强了对苏云金芽孢杆菌δ-内毒素与毛滴虫的钙黏蛋白样受体结合的亲和力[77 ] ,并实现了多种抗体和麦芽糖结合蛋白的可溶性高表达[78 ] (表1 ). ...
... Genetic circuit design
T7聚合酶 拓宽可识别的启动子范围[71 ] T7聚合酶 增强识别人工启动子的特异性[72 ] TALEN DNA序列识别特异性[73 ] spCas9 拓宽可识别的PAM序列[74 ] 胞嘧啶碱基编 辑器(CBEs) 拓宽可编辑的基因序列范围(例如GC丰富的序列)[75 ] 腺嘌呤碱基编 辑器(ABEs) 提高与Cas结构域的兼容性和编辑活性[76 ] 苏云金芽孢杆菌δ-内毒素 增强与毛滴虫的钙黏蛋白样受体结合亲和力[77 ] 抗体、麦芽糖 结合蛋白 增强目标蛋白 ...
Continuous evolution of Bacillus thuringiensis toxins overcomes insect resistance
2
2016
... 自2011年以来,D. Liu课题组利用PACE已成功完成了对多种蛋白的改造,如基于蛋白与DNA序列互作原理,成功对T7聚合酶[71 -72 ] 、TALEN[73 ] 、spCas9[74 ] 、胞嘧啶碱基编辑器(CBEs)[75 ] 、腺嘌呤碱基编辑器(ABEs)[76 ] 进行工程改造,改变了其识别序列的范围或提高了其识别特异性.同时,基于蛋白与蛋白相互作用原理,利用PACE系统,增强了对苏云金芽孢杆菌δ-内毒素与毛滴虫的钙黏蛋白样受体结合的亲和力[77 ] ,并实现了多种抗体和麦芽糖结合蛋白的可溶性高表达[78 ] (表1 ). ...
... Genetic circuit design
T7聚合酶 拓宽可识别的启动子范围[71 ] T7聚合酶 增强识别人工启动子的特异性[72 ] TALEN DNA序列识别特异性[73 ] spCas9 拓宽可识别的PAM序列[74 ] 胞嘧啶碱基编 辑器(CBEs) 拓宽可编辑的基因序列范围(例如GC丰富的序列)[75 ] 腺嘌呤碱基编 辑器(ABEs) 提高与Cas结构域的兼容性和编辑活性[76 ] 苏云金芽孢杆菌δ-内毒素 增强与毛滴虫的钙黏蛋白样受体结合亲和力[77 ] 抗体、麦芽糖 结合蛋白 增强目标蛋白 ...
Continuous directed evolution of proteins with improved soluble expression
2
2018
... 自2011年以来,D. Liu课题组利用PACE已成功完成了对多种蛋白的改造,如基于蛋白与DNA序列互作原理,成功对T7聚合酶[71 -72 ] 、TALEN[73 ] 、spCas9[74 ] 、胞嘧啶碱基编辑器(CBEs)[75 ] 、腺嘌呤碱基编辑器(ABEs)[76 ] 进行工程改造,改变了其识别序列的范围或提高了其识别特异性.同时,基于蛋白与蛋白相互作用原理,利用PACE系统,增强了对苏云金芽孢杆菌δ-内毒素与毛滴虫的钙黏蛋白样受体结合的亲和力[77 ] ,并实现了多种抗体和麦芽糖结合蛋白的可溶性高表达[78 ] (表1 ). ...
... 可溶性表达[78 ] ...
A system for the continuous directed evolution of proteases rapidly reveals drug-resistance mutations
2
2014
蛋白水解酶 提高水解酶催化活性及底物特异性[79 -80 ] 肉毒神 经毒素 使肉毒神经毒素有可编程的特异性[81 ] 氨酰-tRNA 合成酶 生产高活性和选择性的正交氨基酰-tRNA合成酶[82 ] 近期,PACE的应用延伸至改造有催化活性的蛋白酶.在lysozyme+linker+T3/T7 RNAP复合体中,T3/T7 RNAP的活性与linker是否能被蛋白酶水解相关联,通过人工设计linker序列,可实现针对蛋白酶特异性及活性的筛选[79 -80 ] .利用该系统,D. Liu课题组对三种肉毒神经毒素(BoNT)的轻链蛋白酶进行改造,成功获得优先切割囊泡相关膜蛋白VAMP4和Ykt6的BoNT/X变体、选择性切割非天然底物VAMP7的BoNT/F突变体、能选择性切割磷酸酶和张力蛋白同系物(PTEN)但不识别神经元中的任何天然BoNT蛋白酶底物的特异性BoNT/E突变体[81 ] .另外,通过将T7 RNAP的全长表达与氨基酰-tRNA合成酶相偶联,进化获得了可以在蛋白质序列中安装非标准氨基酸的正交氨基酰-tRNA合成酶[82 ] . ...
... 近期,PACE的应用延伸至改造有催化活性的蛋白酶.在lysozyme+linker+T3/T7 RNAP复合体中,T3/T7 RNAP的活性与linker是否能被蛋白酶水解相关联,通过人工设计linker序列,可实现针对蛋白酶特异性及活性的筛选[79 -80 ] .利用该系统,D. Liu课题组对三种肉毒神经毒素(BoNT)的轻链蛋白酶进行改造,成功获得优先切割囊泡相关膜蛋白VAMP4和Ykt6的BoNT/X变体、选择性切割非天然底物VAMP7的BoNT/F突变体、能选择性切割磷酸酶和张力蛋白同系物(PTEN)但不识别神经元中的任何天然BoNT蛋白酶底物的特异性BoNT/E突变体[81 ] .另外,通过将T7 RNAP的全长表达与氨基酰-tRNA合成酶相偶联,进化获得了可以在蛋白质序列中安装非标准氨基酸的正交氨基酰-tRNA合成酶[82 ] . ...
Phage-assisted continuous evolution of proteases with altered substrate specificity
2
2017
... 可溶性表达
[78 ] 蛋白水解酶 提高水解酶催化活性及底物特异性[79 -80 ] 肉毒神 经毒素 使肉毒神经毒素有可编程的特异性[81 ] 氨酰-tRNA 合成酶 生产高活性和选择性的正交氨基酰-tRNA合成酶[82 ] 近期,PACE的应用延伸至改造有催化活性的蛋白酶.在lysozyme+linker+T3/T7 RNAP复合体中,T3/T7 RNAP的活性与linker是否能被蛋白酶水解相关联,通过人工设计linker序列,可实现针对蛋白酶特异性及活性的筛选[79 -80 ] .利用该系统,D. Liu课题组对三种肉毒神经毒素(BoNT)的轻链蛋白酶进行改造,成功获得优先切割囊泡相关膜蛋白VAMP4和Ykt6的BoNT/X变体、选择性切割非天然底物VAMP7的BoNT/F突变体、能选择性切割磷酸酶和张力蛋白同系物(PTEN)但不识别神经元中的任何天然BoNT蛋白酶底物的特异性BoNT/E突变体[81 ] .另外,通过将T7 RNAP的全长表达与氨基酰-tRNA合成酶相偶联,进化获得了可以在蛋白质序列中安装非标准氨基酸的正交氨基酰-tRNA合成酶[82 ] . ...
... 近期,PACE的应用延伸至改造有催化活性的蛋白酶.在lysozyme+linker+T3/T7 RNAP复合体中,T3/T7 RNAP的活性与linker是否能被蛋白酶水解相关联,通过人工设计linker序列,可实现针对蛋白酶特异性及活性的筛选[79 -80 ] .利用该系统,D. Liu课题组对三种肉毒神经毒素(BoNT)的轻链蛋白酶进行改造,成功获得优先切割囊泡相关膜蛋白VAMP4和Ykt6的BoNT/X变体、选择性切割非天然底物VAMP7的BoNT/F突变体、能选择性切割磷酸酶和张力蛋白同系物(PTEN)但不识别神经元中的任何天然BoNT蛋白酶底物的特异性BoNT/E突变体[81 ] .另外,通过将T7 RNAP的全长表达与氨基酰-tRNA合成酶相偶联,进化获得了可以在蛋白质序列中安装非标准氨基酸的正交氨基酰-tRNA合成酶[82 ] . ...
Phage-assisted evolution of botulinum neurotoxin proteases with reprogrammed specificity
2
2021
... 可溶性表达
[78 ] 蛋白水解酶 提高水解酶催化活性及底物特异性[79 -80 ] 肉毒神 经毒素 使肉毒神经毒素有可编程的特异性[81 ] 氨酰-tRNA 合成酶 生产高活性和选择性的正交氨基酰-tRNA合成酶[82 ] 近期,PACE的应用延伸至改造有催化活性的蛋白酶.在lysozyme+linker+T3/T7 RNAP复合体中,T3/T7 RNAP的活性与linker是否能被蛋白酶水解相关联,通过人工设计linker序列,可实现针对蛋白酶特异性及活性的筛选[79 -80 ] .利用该系统,D. Liu课题组对三种肉毒神经毒素(BoNT)的轻链蛋白酶进行改造,成功获得优先切割囊泡相关膜蛋白VAMP4和Ykt6的BoNT/X变体、选择性切割非天然底物VAMP7的BoNT/F突变体、能选择性切割磷酸酶和张力蛋白同系物(PTEN)但不识别神经元中的任何天然BoNT蛋白酶底物的特异性BoNT/E突变体[81 ] .另外,通过将T7 RNAP的全长表达与氨基酰-tRNA合成酶相偶联,进化获得了可以在蛋白质序列中安装非标准氨基酸的正交氨基酰-tRNA合成酶[82 ] . ...
... 近期,PACE的应用延伸至改造有催化活性的蛋白酶.在lysozyme+linker+T3/T7 RNAP复合体中,T3/T7 RNAP的活性与linker是否能被蛋白酶水解相关联,通过人工设计linker序列,可实现针对蛋白酶特异性及活性的筛选[79 -80 ] .利用该系统,D. Liu课题组对三种肉毒神经毒素(BoNT)的轻链蛋白酶进行改造,成功获得优先切割囊泡相关膜蛋白VAMP4和Ykt6的BoNT/X变体、选择性切割非天然底物VAMP7的BoNT/F突变体、能选择性切割磷酸酶和张力蛋白同系物(PTEN)但不识别神经元中的任何天然BoNT蛋白酶底物的特异性BoNT/E突变体[81 ] .另外,通过将T7 RNAP的全长表达与氨基酰-tRNA合成酶相偶联,进化获得了可以在蛋白质序列中安装非标准氨基酸的正交氨基酰-tRNA合成酶[82 ] . ...
Continuous directed evolution of aminoacyl-tRNA synthetases
2
2017
... 可溶性表达
[78 ] 蛋白水解酶 提高水解酶催化活性及底物特异性[79 -80 ] 肉毒神 经毒素 使肉毒神经毒素有可编程的特异性[81 ] 氨酰-tRNA 合成酶 生产高活性和选择性的正交氨基酰-tRNA合成酶[82 ] 近期,PACE的应用延伸至改造有催化活性的蛋白酶.在lysozyme+linker+T3/T7 RNAP复合体中,T3/T7 RNAP的活性与linker是否能被蛋白酶水解相关联,通过人工设计linker序列,可实现针对蛋白酶特异性及活性的筛选[79 -80 ] .利用该系统,D. Liu课题组对三种肉毒神经毒素(BoNT)的轻链蛋白酶进行改造,成功获得优先切割囊泡相关膜蛋白VAMP4和Ykt6的BoNT/X变体、选择性切割非天然底物VAMP7的BoNT/F突变体、能选择性切割磷酸酶和张力蛋白同系物(PTEN)但不识别神经元中的任何天然BoNT蛋白酶底物的特异性BoNT/E突变体[81 ] .另外,通过将T7 RNAP的全长表达与氨基酰-tRNA合成酶相偶联,进化获得了可以在蛋白质序列中安装非标准氨基酸的正交氨基酰-tRNA合成酶[82 ] . ...
... 近期,PACE的应用延伸至改造有催化活性的蛋白酶.在lysozyme+linker+T3/T7 RNAP复合体中,T3/T7 RNAP的活性与linker是否能被蛋白酶水解相关联,通过人工设计linker序列,可实现针对蛋白酶特异性及活性的筛选[79 -80 ] .利用该系统,D. Liu课题组对三种肉毒神经毒素(BoNT)的轻链蛋白酶进行改造,成功获得优先切割囊泡相关膜蛋白VAMP4和Ykt6的BoNT/X变体、选择性切割非天然底物VAMP7的BoNT/F突变体、能选择性切割磷酸酶和张力蛋白同系物(PTEN)但不识别神经元中的任何天然BoNT蛋白酶底物的特异性BoNT/E突变体[81 ] .另外,通过将T7 RNAP的全长表达与氨基酰-tRNA合成酶相偶联,进化获得了可以在蛋白质序列中安装非标准氨基酸的正交氨基酰-tRNA合成酶[82 ] . ...
Precise, automated control of conditions for high-throughput growth of yeast and bacteria with eVOLVER
1
2018
... 胞内定向进化技术虽然能更便捷地准备较大的基因突变体库,但其表型的准确筛选需要依赖于能够精确控制菌株生长条件的智能化设备.对此,C. Bashor课题组和A. Khalil课题组合作开发了一种用于细菌或酵母培养的自动化连续进化设备:eVOLVER[83 ] .eVOLVER采用可设计的开源软件和模块化的微生物培养与测试装置,通过搭建微生物反应器中各项参数与电脑程序之间的自动识别与反馈机制,实现了对生物反应器的精准调控.eVOLVER理论上可满足任何类型的菌株生长,可以同时监控数百种不同的培养条件,如温度、培养密度、培养基组成等,实现对任意时间尺度数据的实时测量和收集.借助该设备,研究人员对酵母突变体库施以不同的温度调控,筛选到了对温度变化敏感的酵母菌株;同时,通过改变酵母培养基的组分,追踪到了含半乳糖报告基因的酵母菌株.最近研究人员还将其与体内进化技术OrthoRep整合,开发了一个自动连续进化平台(automated continuous evolution,ACE)[84 ] ,用于酵母胞内蛋白的定向进化.在该系统中,首先通过OrthoRep实现特定基因的多轮突变,然后依靠eVOLVER进行严格的条件筛选和实时的反馈调整,对恶性疟原虫二氢叶酸还原酶进行迭代进化,成功提高了其对乙胺嘧啶的耐药性,比仅靠OrthoRep进化节约了200多小时. ...
Automated continuous evolution of proteins in vivo
1
2020
... 胞内定向进化技术虽然能更便捷地准备较大的基因突变体库,但其表型的准确筛选需要依赖于能够精确控制菌株生长条件的智能化设备.对此,C. Bashor课题组和A. Khalil课题组合作开发了一种用于细菌或酵母培养的自动化连续进化设备:eVOLVER[83 ] .eVOLVER采用可设计的开源软件和模块化的微生物培养与测试装置,通过搭建微生物反应器中各项参数与电脑程序之间的自动识别与反馈机制,实现了对生物反应器的精准调控.eVOLVER理论上可满足任何类型的菌株生长,可以同时监控数百种不同的培养条件,如温度、培养密度、培养基组成等,实现对任意时间尺度数据的实时测量和收集.借助该设备,研究人员对酵母突变体库施以不同的温度调控,筛选到了对温度变化敏感的酵母菌株;同时,通过改变酵母培养基的组分,追踪到了含半乳糖报告基因的酵母菌株.最近研究人员还将其与体内进化技术OrthoRep整合,开发了一个自动连续进化平台(automated continuous evolution,ACE)[84 ] ,用于酵母胞内蛋白的定向进化.在该系统中,首先通过OrthoRep实现特定基因的多轮突变,然后依靠eVOLVER进行严格的条件筛选和实时的反馈调整,对恶性疟原虫二氢叶酸还原酶进行迭代进化,成功提高了其对乙胺嘧啶的耐药性,比仅靠OrthoRep进化节约了200多小时. ...
Systematic molecular evolution enables robust biomolecule discovery
1
2022
... PACE系统近年来取得诸多成功,为了进一步增加其通量和精准性,K. M. Esvelt课题组开发了一个机器人辅助的高通量PACE进化平台(phage- and robotics-assisted near-continuous evolution,PRANCE)[85 ] .PRANCE利用96孔板,创造了96个平行的“潟湖”反应器,并与计算机辅助的生长条件调控模块进行结合,能实时测量每个分子的活性,进而调整每个PACE反应器的实验条件,从而达到精准进化.相较于普通的PACE系统而言,PRANCE能达到更大的进化通量,对近百个目标蛋白进行平行优化,实现了进化的可重复性和精准调控. ...
Automated multiplex genome-scale engineering in yeast
1
2017
... 以上提到的自动化平台,都是基于一种胞内进化方法而设计的,在实现更高通量的胞内连续进化方面取得了重要突破.但是,这些技术平台受限于改造其胞内进化技术所适用的特定目标蛋白.目前,建设基于自动化的生物合成平台已在全世界兴起,目标是通过集成式的设施,实现常规合成生物学实验操作的自动化完成,如基因克隆、蛋白表征、代谢表征等;并结合人工智能设备,实现生物技术研发过程中“设计—构建—测试—学习”四大模块的自动化迭代.例如美国伊利诺伊大学的H. Zhao课题组[86 ] 基于全自动生物合成平台iBioFAB,实现了大肠杆菌和酿酒酵母的自动转化、培养和性状表征筛选等操作,并结合机器学习开发了BioAutomata平台,通过结合不同T7启动子突变体与核糖体结合位点合成24组有不同表达水平的表达系统,对番茄红素合成路径中的三个关键基因的表达水平进行动态调控,同时结合基于贝叶斯算法(Bayesian algorithm)的计算机学习模型,对产生的数据进行学习反馈,进一步优化动态调控策略,最终获得高产番茄红素的蛋白表达体系[67 ] .同时,我国天津工业生物技术研究所的Y. Ma课题组[87 ] 开发了针对谷氨酸棒状杆菌(Corynebacterium glutamicum )代谢工程改造的多元自动化基因组编辑方法MACBETH(multiplex automated Corynebacterium glutamicum base editing method),其整合基于基因组编辑技术的基因敲除技术、目标菌株筛选和表型验证的全自动流程设备,以100%的编辑效率,构建了94个调控因子单独失活的C.glutamicum 菌株变异库存,有望提高谷氨酸的生成量.综合来看,自动化合成生物平台有望将传统的劳动密集型酶定向进化研究转化为高效便捷的工业化流程,为定向进化的发展开辟无数的可能性. ...
MACBETH: Multiplex automated Corynebacterium glutamicum base editing method
1
2018
... 以上提到的自动化平台,都是基于一种胞内进化方法而设计的,在实现更高通量的胞内连续进化方面取得了重要突破.但是,这些技术平台受限于改造其胞内进化技术所适用的特定目标蛋白.目前,建设基于自动化的生物合成平台已在全世界兴起,目标是通过集成式的设施,实现常规合成生物学实验操作的自动化完成,如基因克隆、蛋白表征、代谢表征等;并结合人工智能设备,实现生物技术研发过程中“设计—构建—测试—学习”四大模块的自动化迭代.例如美国伊利诺伊大学的H. Zhao课题组[86 ] 基于全自动生物合成平台iBioFAB,实现了大肠杆菌和酿酒酵母的自动转化、培养和性状表征筛选等操作,并结合机器学习开发了BioAutomata平台,通过结合不同T7启动子突变体与核糖体结合位点合成24组有不同表达水平的表达系统,对番茄红素合成路径中的三个关键基因的表达水平进行动态调控,同时结合基于贝叶斯算法(Bayesian algorithm)的计算机学习模型,对产生的数据进行学习反馈,进一步优化动态调控策略,最终获得高产番茄红素的蛋白表达体系[67 ] .同时,我国天津工业生物技术研究所的Y. Ma课题组[87 ] 开发了针对谷氨酸棒状杆菌(Corynebacterium glutamicum )代谢工程改造的多元自动化基因组编辑方法MACBETH(multiplex automated Corynebacterium glutamicum base editing method),其整合基于基因组编辑技术的基因敲除技术、目标菌株筛选和表型验证的全自动流程设备,以100%的编辑效率,构建了94个调控因子单独失活的C.glutamicum 菌株变异库存,有望提高谷氨酸的生成量.综合来看,自动化合成生物平台有望将传统的劳动密集型酶定向进化研究转化为高效便捷的工业化流程,为定向进化的发展开辟无数的可能性. ...
Hot spots-making directed evolution easier
2
2022
... 在传统的定向进化实验中,由于无法确定与功能相关的结构域,基因多样化的目标通常是整段基因.这种实验方法虽然能够获得更多的突变样本,但极大程度地增加了实验负担.一段仅由30个碱基对组成的基因片段的突变体,便能产生超过1013 种不同的氨基酸序列,因此以整段基因为突变目标的方法很难覆盖理论中的突变文库.现如今,通过生物信息学、结构生物学和分子动力学等方法预测蛋白质功能域,并以功能域中特定位点或特定区域为突变目标进行突变改造的思路越发普遍.该方法可以通过创建“小”而“精”的突变文库来大大减轻实验负担并降低筛选难度,而这种计算机辅助的定向进化通常依赖于对蛋白质三维结构的分析,以寻找其可能与功能相关的区域[88 ] . ...
... 然而,仅仅获得蛋白质的三维结构并不足以完成对其活性中心或突变热点的寻找,即使可以通过人工与已知同工酶或同源蛋白的序列的比对来推测其关键位点和保守序列[98 -100 ] ,但人工筛选的低效和可能存在的疏漏仍表明计算机辅助是更优的选择.常见的寻找酶活性中心的方法是通过序列比对预测(ClustalW[101 ] )和酶-底物对接模拟(AutoDock[102 ] 、Rosetta[93 ] )等[88 ] .其中,Rosetta是一个广泛使用且功能强大的软件,该软件自开发至今不过二十余年,却已经衍生出超过80种方法,其平台现在由来自约100所大学和实验室的开发人员和科学家贡献的代码组成(图5 ),功能包括但不限于蛋白质结构预测、蛋白质-蛋白质对接、配体对接、蛋白质设计等[93 ] .S. Khare课题组[103 ] 使用Rosetta对蛋白质框架进行设计,通过计算半胱氨酸侧链的氧化还原稳定性,确定了蛋白质支架内交联的最佳位置,随后成功改造出热稳定性和化学稳定性显著提高的环丙烷酶. ...
MGnify: the microbiome analysis resource in 2020
1
2020
... 截至2022年,NCBI Reference Sequence Database(RefSeq)数据库共有超过2.24亿种特异性的蛋白序列被记录,且随着DNA测序技术的高速发展,该数量每24个月增加约1倍.虽然X射线衍射、核磁共振和冷冻电镜技术的发展使超过十万种蛋白质的三维结构得到解析[89 -90 ] ,但其也只占总蛋白质的小部分,且即使使用高分辨冷冻电镜技术,有时也难以获得分辨率足够高的照片来精准解析目标蛋白结构.再者,确定目标蛋白的结构常常需要数月的努力,这使得以结构生物学为手段获得蛋白质结构信息的效率较低.近年来随着人工智能的高速发展,借助同源建模与机器学习对蛋白质结构进行预测的准确性日益增加[91 ] ,不同的算法及软件被开发以用于蛋白质结构的计算.包括Modeller[92 ] 、Rosetta[93 ] 、AlphaFold 2[94 ] 等在内的多种方法已被广泛用于蛋白质结构的预测.其中,目前最广为人知的计算方法当属AlphaFold 2(图4 ),这种算法通过生物信息学和物理方法结合的方法进行预测,它的运算过程可以分为两部分:一部分与传统的同源建模类似,通过多序列比对(multiple sequence alignments,MSA)寻找目的序列的同源序列,由已知的结构预测新的结构;另一部分则通过每一个氨基酸的物理和空间几何约束的神经网络构架相互作用进行预测.AlphaFold 2预测的平均分辨率能高达0.96Å,置信度95%的区间为0.85~1.16Å[94 ] .迄今为止,利用AlphaFold 2已成功预测源自不同生物的超过30万个蛋白质结构,其算法的开源也使得许多研究可以借助其功能完成蛋白质催化中心、进化和功能关系等方面的预测[95 -96 ] ,例如G. Carman课题组利用AlphaFold 2预测了突变的酿酒酵母磷脂酸磷酸酶的结构,通过其结构预测了其催化关键位点,并推测了其催化活性机理[97 ] . ...
Protein data bank: The single global archive for 3D macromolecular structure data
1
2018
... 截至2022年,NCBI Reference Sequence Database(RefSeq)数据库共有超过2.24亿种特异性的蛋白序列被记录,且随着DNA测序技术的高速发展,该数量每24个月增加约1倍.虽然X射线衍射、核磁共振和冷冻电镜技术的发展使超过十万种蛋白质的三维结构得到解析[89 -90 ] ,但其也只占总蛋白质的小部分,且即使使用高分辨冷冻电镜技术,有时也难以获得分辨率足够高的照片来精准解析目标蛋白结构.再者,确定目标蛋白的结构常常需要数月的努力,这使得以结构生物学为手段获得蛋白质结构信息的效率较低.近年来随着人工智能的高速发展,借助同源建模与机器学习对蛋白质结构进行预测的准确性日益增加[91 ] ,不同的算法及软件被开发以用于蛋白质结构的计算.包括Modeller[92 ] 、Rosetta[93 ] 、AlphaFold 2[94 ] 等在内的多种方法已被广泛用于蛋白质结构的预测.其中,目前最广为人知的计算方法当属AlphaFold 2(图4 ),这种算法通过生物信息学和物理方法结合的方法进行预测,它的运算过程可以分为两部分:一部分与传统的同源建模类似,通过多序列比对(multiple sequence alignments,MSA)寻找目的序列的同源序列,由已知的结构预测新的结构;另一部分则通过每一个氨基酸的物理和空间几何约束的神经网络构架相互作用进行预测.AlphaFold 2预测的平均分辨率能高达0.96Å,置信度95%的区间为0.85~1.16Å[94 ] .迄今为止,利用AlphaFold 2已成功预测源自不同生物的超过30万个蛋白质结构,其算法的开源也使得许多研究可以借助其功能完成蛋白质催化中心、进化和功能关系等方面的预测[95 -96 ] ,例如G. Carman课题组利用AlphaFold 2预测了突变的酿酒酵母磷脂酸磷酸酶的结构,通过其结构预测了其催化关键位点,并推测了其催化活性机理[97 ] . ...
Deep learning techniques have significantly impacted protein structure prediction and protein design
2
2021
... 截至2022年,NCBI Reference Sequence Database(RefSeq)数据库共有超过2.24亿种特异性的蛋白序列被记录,且随着DNA测序技术的高速发展,该数量每24个月增加约1倍.虽然X射线衍射、核磁共振和冷冻电镜技术的发展使超过十万种蛋白质的三维结构得到解析[89 -90 ] ,但其也只占总蛋白质的小部分,且即使使用高分辨冷冻电镜技术,有时也难以获得分辨率足够高的照片来精准解析目标蛋白结构.再者,确定目标蛋白的结构常常需要数月的努力,这使得以结构生物学为手段获得蛋白质结构信息的效率较低.近年来随着人工智能的高速发展,借助同源建模与机器学习对蛋白质结构进行预测的准确性日益增加[91 ] ,不同的算法及软件被开发以用于蛋白质结构的计算.包括Modeller[92 ] 、Rosetta[93 ] 、AlphaFold 2[94 ] 等在内的多种方法已被广泛用于蛋白质结构的预测.其中,目前最广为人知的计算方法当属AlphaFold 2(图4 ),这种算法通过生物信息学和物理方法结合的方法进行预测,它的运算过程可以分为两部分:一部分与传统的同源建模类似,通过多序列比对(multiple sequence alignments,MSA)寻找目的序列的同源序列,由已知的结构预测新的结构;另一部分则通过每一个氨基酸的物理和空间几何约束的神经网络构架相互作用进行预测.AlphaFold 2预测的平均分辨率能高达0.96Å,置信度95%的区间为0.85~1.16Å[94 ] .迄今为止,利用AlphaFold 2已成功预测源自不同生物的超过30万个蛋白质结构,其算法的开源也使得许多研究可以借助其功能完成蛋白质催化中心、进化和功能关系等方面的预测[95 -96 ] ,例如G. Carman课题组利用AlphaFold 2预测了突变的酿酒酵母磷脂酸磷酸酶的结构,通过其结构预测了其催化关键位点,并推测了其催化活性机理[97 ] . ...
... 除了提高通量,构建精准的突变体库也是提高定向进化效率的关键,其中最具代表性的是M. Reetz教授团队所发展的CSAT、ISM、FRISM等一系列方法,以及Twist Bioscience公司与M. Reetz教授合作,在2018年报道的依托高通量合成DNA的体外突变体库构建方法已经用于商业化[18 ] .得益于测序技术的发展,未被解析的氨基酸序列数量飞速增长,机器学习技术在预测蛋白结构、功能及活性位点等领域取得了一定成功[91 ] .然而,目前主流的两种机器学习方法仍然需要依赖庞大、多样且高质量数据点来完成学习并做出较为精确的预测[143 ] ,而蛋白质序列及折叠方式的复杂性也使得现有算法无法同时预测两个或以上的性质.AlphaFold 2将利用机器学习预测蛋白质结构的研究推向了一个新的巅峰,其已经成功预测了超过30万种蛋白质的结构,但并未能准确预测庞大的序列数据库对应的所有蛋白序列.另外,对于蛋白质功能的预测目前完全依赖基于样本的机器学习模型构建或同源分析,而现有算法虽可以通过缩小突变体库来辅助定向进化,但其精确性也较大依赖于基于对蛋白质结构及功能的理解所设定的限制条件或突变倾向[144 ] .因此,如何减少建模方法对人工设置的依赖和实现自主模型的构建、如何通过较少的样本库来达到较高的预测精确度、如何同时模拟预测多个性状都将是未来探索的重点方向. ...
Comparative protein structure modeling using MODELLER
1
2016
... 截至2022年,NCBI Reference Sequence Database(RefSeq)数据库共有超过2.24亿种特异性的蛋白序列被记录,且随着DNA测序技术的高速发展,该数量每24个月增加约1倍.虽然X射线衍射、核磁共振和冷冻电镜技术的发展使超过十万种蛋白质的三维结构得到解析[89 -90 ] ,但其也只占总蛋白质的小部分,且即使使用高分辨冷冻电镜技术,有时也难以获得分辨率足够高的照片来精准解析目标蛋白结构.再者,确定目标蛋白的结构常常需要数月的努力,这使得以结构生物学为手段获得蛋白质结构信息的效率较低.近年来随着人工智能的高速发展,借助同源建模与机器学习对蛋白质结构进行预测的准确性日益增加[91 ] ,不同的算法及软件被开发以用于蛋白质结构的计算.包括Modeller[92 ] 、Rosetta[93 ] 、AlphaFold 2[94 ] 等在内的多种方法已被广泛用于蛋白质结构的预测.其中,目前最广为人知的计算方法当属AlphaFold 2(图4 ),这种算法通过生物信息学和物理方法结合的方法进行预测,它的运算过程可以分为两部分:一部分与传统的同源建模类似,通过多序列比对(multiple sequence alignments,MSA)寻找目的序列的同源序列,由已知的结构预测新的结构;另一部分则通过每一个氨基酸的物理和空间几何约束的神经网络构架相互作用进行预测.AlphaFold 2预测的平均分辨率能高达0.96Å,置信度95%的区间为0.85~1.16Å[94 ] .迄今为止,利用AlphaFold 2已成功预测源自不同生物的超过30万个蛋白质结构,其算法的开源也使得许多研究可以借助其功能完成蛋白质催化中心、进化和功能关系等方面的预测[95 -96 ] ,例如G. Carman课题组利用AlphaFold 2预测了突变的酿酒酵母磷脂酸磷酸酶的结构,通过其结构预测了其催化关键位点,并推测了其催化活性机理[97 ] . ...
Macromolecular modeling and design in Rosetta: recent methods and frameworks
5
2020
... 截至2022年,NCBI Reference Sequence Database(RefSeq)数据库共有超过2.24亿种特异性的蛋白序列被记录,且随着DNA测序技术的高速发展,该数量每24个月增加约1倍.虽然X射线衍射、核磁共振和冷冻电镜技术的发展使超过十万种蛋白质的三维结构得到解析[89 -90 ] ,但其也只占总蛋白质的小部分,且即使使用高分辨冷冻电镜技术,有时也难以获得分辨率足够高的照片来精准解析目标蛋白结构.再者,确定目标蛋白的结构常常需要数月的努力,这使得以结构生物学为手段获得蛋白质结构信息的效率较低.近年来随着人工智能的高速发展,借助同源建模与机器学习对蛋白质结构进行预测的准确性日益增加[91 ] ,不同的算法及软件被开发以用于蛋白质结构的计算.包括Modeller[92 ] 、Rosetta[93 ] 、AlphaFold 2[94 ] 等在内的多种方法已被广泛用于蛋白质结构的预测.其中,目前最广为人知的计算方法当属AlphaFold 2(图4 ),这种算法通过生物信息学和物理方法结合的方法进行预测,它的运算过程可以分为两部分:一部分与传统的同源建模类似,通过多序列比对(multiple sequence alignments,MSA)寻找目的序列的同源序列,由已知的结构预测新的结构;另一部分则通过每一个氨基酸的物理和空间几何约束的神经网络构架相互作用进行预测.AlphaFold 2预测的平均分辨率能高达0.96Å,置信度95%的区间为0.85~1.16Å[94 ] .迄今为止,利用AlphaFold 2已成功预测源自不同生物的超过30万个蛋白质结构,其算法的开源也使得许多研究可以借助其功能完成蛋白质催化中心、进化和功能关系等方面的预测[95 -96 ] ,例如G. Carman课题组利用AlphaFold 2预测了突变的酿酒酵母磷脂酸磷酸酶的结构,通过其结构预测了其催化关键位点,并推测了其催化活性机理[97 ] . ...
... 然而,仅仅获得蛋白质的三维结构并不足以完成对其活性中心或突变热点的寻找,即使可以通过人工与已知同工酶或同源蛋白的序列的比对来推测其关键位点和保守序列[98 -100 ] ,但人工筛选的低效和可能存在的疏漏仍表明计算机辅助是更优的选择.常见的寻找酶活性中心的方法是通过序列比对预测(ClustalW[101 ] )和酶-底物对接模拟(AutoDock[102 ] 、Rosetta[93 ] )等[88 ] .其中,Rosetta是一个广泛使用且功能强大的软件,该软件自开发至今不过二十余年,却已经衍生出超过80种方法,其平台现在由来自约100所大学和实验室的开发人员和科学家贡献的代码组成(图5 ),功能包括但不限于蛋白质结构预测、蛋白质-蛋白质对接、配体对接、蛋白质设计等[93 ] .S. Khare课题组[103 ] 使用Rosetta对蛋白质框架进行设计,通过计算半胱氨酸侧链的氧化还原稳定性,确定了蛋白质支架内交联的最佳位置,随后成功改造出热稳定性和化学稳定性显著提高的环丙烷酶. ...
... [93 ].S. Khare课题组[103 ] 使用Rosetta对蛋白质框架进行设计,通过计算半胱氨酸侧链的氧化还原稳定性,确定了蛋白质支架内交联的最佳位置,随后成功改造出热稳定性和化学稳定性显著提高的环丙烷酶. ...
... [
93 ]
Some popular tasks that can be addressed in Rosetta (blue) and major systems that can be modeled (red)<sup>[<xref ref-type="bibr" rid="R93">93</xref>]</sup> Fig. 5 ![]()
另外,随着计算机技术的迅速发展,机器学习技术在近几十年中迅速崛起.不同于早期的生物信息学算法和软件,机器学习基于大量具有不同特征的数据点的训练找到一种通用的模型,并套用该模型解决问题[104 ] .机器学习的两种主要类型为无监督学习和监督学习.监督学习即为算法提供一个或几个目标属性作为标签,使用标记的数据集,根据标签为数据进行分类分析;而无监督学习则不提供标签,由计算机在学习过程中对样本进行分类,在学习过程中通常将高尺度的数据压缩或转换为低尺度的数据,以数学方法对特定信息进行分析[105 ] .此外,这两种类型结合的方法被称为半监督学习. ...
... [
93 ]
Fig. 5 ![]()
另外,随着计算机技术的迅速发展,机器学习技术在近几十年中迅速崛起.不同于早期的生物信息学算法和软件,机器学习基于大量具有不同特征的数据点的训练找到一种通用的模型,并套用该模型解决问题[104 ] .机器学习的两种主要类型为无监督学习和监督学习.监督学习即为算法提供一个或几个目标属性作为标签,使用标记的数据集,根据标签为数据进行分类分析;而无监督学习则不提供标签,由计算机在学习过程中对样本进行分类,在学习过程中通常将高尺度的数据压缩或转换为低尺度的数据,以数学方法对特定信息进行分析[105 ] .此外,这两种类型结合的方法被称为半监督学习. ...
Highly accurate protein structure prediction with AlphaFold
3
2021
... 截至2022年,NCBI Reference Sequence Database(RefSeq)数据库共有超过2.24亿种特异性的蛋白序列被记录,且随着DNA测序技术的高速发展,该数量每24个月增加约1倍.虽然X射线衍射、核磁共振和冷冻电镜技术的发展使超过十万种蛋白质的三维结构得到解析[89 -90 ] ,但其也只占总蛋白质的小部分,且即使使用高分辨冷冻电镜技术,有时也难以获得分辨率足够高的照片来精准解析目标蛋白结构.再者,确定目标蛋白的结构常常需要数月的努力,这使得以结构生物学为手段获得蛋白质结构信息的效率较低.近年来随着人工智能的高速发展,借助同源建模与机器学习对蛋白质结构进行预测的准确性日益增加[91 ] ,不同的算法及软件被开发以用于蛋白质结构的计算.包括Modeller[92 ] 、Rosetta[93 ] 、AlphaFold 2[94 ] 等在内的多种方法已被广泛用于蛋白质结构的预测.其中,目前最广为人知的计算方法当属AlphaFold 2(图4 ),这种算法通过生物信息学和物理方法结合的方法进行预测,它的运算过程可以分为两部分:一部分与传统的同源建模类似,通过多序列比对(multiple sequence alignments,MSA)寻找目的序列的同源序列,由已知的结构预测新的结构;另一部分则通过每一个氨基酸的物理和空间几何约束的神经网络构架相互作用进行预测.AlphaFold 2预测的平均分辨率能高达0.96Å,置信度95%的区间为0.85~1.16Å[94 ] .迄今为止,利用AlphaFold 2已成功预测源自不同生物的超过30万个蛋白质结构,其算法的开源也使得许多研究可以借助其功能完成蛋白质催化中心、进化和功能关系等方面的预测[95 -96 ] ,例如G. Carman课题组利用AlphaFold 2预测了突变的酿酒酵母磷脂酸磷酸酶的结构,通过其结构预测了其催化关键位点,并推测了其催化活性机理[97 ] . ...
... [94 ].迄今为止,利用AlphaFold 2已成功预测源自不同生物的超过30万个蛋白质结构,其算法的开源也使得许多研究可以借助其功能完成蛋白质催化中心、进化和功能关系等方面的预测[95 -96 ] ,例如G. Carman课题组利用AlphaFold 2预测了突变的酿酒酵母磷脂酸磷酸酶的结构,通过其结构预测了其催化关键位点,并推测了其催化活性机理[97 ] . ...
... [
94 ]
Fig. 4 ![]()
然而,仅仅获得蛋白质的三维结构并不足以完成对其活性中心或突变热点的寻找,即使可以通过人工与已知同工酶或同源蛋白的序列的比对来推测其关键位点和保守序列[98 -100 ] ,但人工筛选的低效和可能存在的疏漏仍表明计算机辅助是更优的选择.常见的寻找酶活性中心的方法是通过序列比对预测(ClustalW[101 ] )和酶-底物对接模拟(AutoDock[102 ] 、Rosetta[93 ] )等[88 ] .其中,Rosetta是一个广泛使用且功能强大的软件,该软件自开发至今不过二十余年,却已经衍生出超过80种方法,其平台现在由来自约100所大学和实验室的开发人员和科学家贡献的代码组成(图5 ),功能包括但不限于蛋白质结构预测、蛋白质-蛋白质对接、配体对接、蛋白质设计等[93 ] .S. Khare课题组[103 ] 使用Rosetta对蛋白质框架进行设计,通过计算半胱氨酸侧链的氧化还原稳定性,确定了蛋白质支架内交联的最佳位置,随后成功改造出热稳定性和化学稳定性显著提高的环丙烷酶. ...
Molecular modeling the proteins from the exo-xis region of Lambda and Shigatoxigenic bacteriophages
1
2021
... 截至2022年,NCBI Reference Sequence Database(RefSeq)数据库共有超过2.24亿种特异性的蛋白序列被记录,且随着DNA测序技术的高速发展,该数量每24个月增加约1倍.虽然X射线衍射、核磁共振和冷冻电镜技术的发展使超过十万种蛋白质的三维结构得到解析[89 -90 ] ,但其也只占总蛋白质的小部分,且即使使用高分辨冷冻电镜技术,有时也难以获得分辨率足够高的照片来精准解析目标蛋白结构.再者,确定目标蛋白的结构常常需要数月的努力,这使得以结构生物学为手段获得蛋白质结构信息的效率较低.近年来随着人工智能的高速发展,借助同源建模与机器学习对蛋白质结构进行预测的准确性日益增加[91 ] ,不同的算法及软件被开发以用于蛋白质结构的计算.包括Modeller[92 ] 、Rosetta[93 ] 、AlphaFold 2[94 ] 等在内的多种方法已被广泛用于蛋白质结构的预测.其中,目前最广为人知的计算方法当属AlphaFold 2(图4 ),这种算法通过生物信息学和物理方法结合的方法进行预测,它的运算过程可以分为两部分:一部分与传统的同源建模类似,通过多序列比对(multiple sequence alignments,MSA)寻找目的序列的同源序列,由已知的结构预测新的结构;另一部分则通过每一个氨基酸的物理和空间几何约束的神经网络构架相互作用进行预测.AlphaFold 2预测的平均分辨率能高达0.96Å,置信度95%的区间为0.85~1.16Å[94 ] .迄今为止,利用AlphaFold 2已成功预测源自不同生物的超过30万个蛋白质结构,其算法的开源也使得许多研究可以借助其功能完成蛋白质催化中心、进化和功能关系等方面的预测[95 -96 ] ,例如G. Carman课题组利用AlphaFold 2预测了突变的酿酒酵母磷脂酸磷酸酶的结构,通过其结构预测了其催化关键位点,并推测了其催化活性机理[97 ] . ...
AlphaFold-predicted structures of KCTD proteins unravel previously undetected relationships among the members of the family
1
2021
... 截至2022年,NCBI Reference Sequence Database(RefSeq)数据库共有超过2.24亿种特异性的蛋白序列被记录,且随着DNA测序技术的高速发展,该数量每24个月增加约1倍.虽然X射线衍射、核磁共振和冷冻电镜技术的发展使超过十万种蛋白质的三维结构得到解析[89 -90 ] ,但其也只占总蛋白质的小部分,且即使使用高分辨冷冻电镜技术,有时也难以获得分辨率足够高的照片来精准解析目标蛋白结构.再者,确定目标蛋白的结构常常需要数月的努力,这使得以结构生物学为手段获得蛋白质结构信息的效率较低.近年来随着人工智能的高速发展,借助同源建模与机器学习对蛋白质结构进行预测的准确性日益增加[91 ] ,不同的算法及软件被开发以用于蛋白质结构的计算.包括Modeller[92 ] 、Rosetta[93 ] 、AlphaFold 2[94 ] 等在内的多种方法已被广泛用于蛋白质结构的预测.其中,目前最广为人知的计算方法当属AlphaFold 2(图4 ),这种算法通过生物信息学和物理方法结合的方法进行预测,它的运算过程可以分为两部分:一部分与传统的同源建模类似,通过多序列比对(multiple sequence alignments,MSA)寻找目的序列的同源序列,由已知的结构预测新的结构;另一部分则通过每一个氨基酸的物理和空间几何约束的神经网络构架相互作用进行预测.AlphaFold 2预测的平均分辨率能高达0.96Å,置信度95%的区间为0.85~1.16Å[94 ] .迄今为止,利用AlphaFold 2已成功预测源自不同生物的超过30万个蛋白质结构,其算法的开源也使得许多研究可以借助其功能完成蛋白质催化中心、进化和功能关系等方面的预测[95 -96 ] ,例如G. Carman课题组利用AlphaFold 2预测了突变的酿酒酵母磷脂酸磷酸酶的结构,通过其结构预测了其催化关键位点,并推测了其催化活性机理[97 ] . ...
Mutant phosphatidate phosphatase Pah1-W637A exhibits altered phosphorylation, membrane association, and enzyme function in yeast
1
2022
... 截至2022年,NCBI Reference Sequence Database(RefSeq)数据库共有超过2.24亿种特异性的蛋白序列被记录,且随着DNA测序技术的高速发展,该数量每24个月增加约1倍.虽然X射线衍射、核磁共振和冷冻电镜技术的发展使超过十万种蛋白质的三维结构得到解析[89 -90 ] ,但其也只占总蛋白质的小部分,且即使使用高分辨冷冻电镜技术,有时也难以获得分辨率足够高的照片来精准解析目标蛋白结构.再者,确定目标蛋白的结构常常需要数月的努力,这使得以结构生物学为手段获得蛋白质结构信息的效率较低.近年来随着人工智能的高速发展,借助同源建模与机器学习对蛋白质结构进行预测的准确性日益增加[91 ] ,不同的算法及软件被开发以用于蛋白质结构的计算.包括Modeller[92 ] 、Rosetta[93 ] 、AlphaFold 2[94 ] 等在内的多种方法已被广泛用于蛋白质结构的预测.其中,目前最广为人知的计算方法当属AlphaFold 2(图4 ),这种算法通过生物信息学和物理方法结合的方法进行预测,它的运算过程可以分为两部分:一部分与传统的同源建模类似,通过多序列比对(multiple sequence alignments,MSA)寻找目的序列的同源序列,由已知的结构预测新的结构;另一部分则通过每一个氨基酸的物理和空间几何约束的神经网络构架相互作用进行预测.AlphaFold 2预测的平均分辨率能高达0.96Å,置信度95%的区间为0.85~1.16Å[94 ] .迄今为止,利用AlphaFold 2已成功预测源自不同生物的超过30万个蛋白质结构,其算法的开源也使得许多研究可以借助其功能完成蛋白质催化中心、进化和功能关系等方面的预测[95 -96 ] ,例如G. Carman课题组利用AlphaFold 2预测了突变的酿酒酵母磷脂酸磷酸酶的结构,通过其结构预测了其催化关键位点,并推测了其催化活性机理[97 ] . ...
Small design from big alignment: engineering proteins with multiple sequence alignment as the starting point
1
2020
... 然而,仅仅获得蛋白质的三维结构并不足以完成对其活性中心或突变热点的寻找,即使可以通过人工与已知同工酶或同源蛋白的序列的比对来推测其关键位点和保守序列[98 -100 ] ,但人工筛选的低效和可能存在的疏漏仍表明计算机辅助是更优的选择.常见的寻找酶活性中心的方法是通过序列比对预测(ClustalW[101 ] )和酶-底物对接模拟(AutoDock[102 ] 、Rosetta[93 ] )等[88 ] .其中,Rosetta是一个广泛使用且功能强大的软件,该软件自开发至今不过二十余年,却已经衍生出超过80种方法,其平台现在由来自约100所大学和实验室的开发人员和科学家贡献的代码组成(图5 ),功能包括但不限于蛋白质结构预测、蛋白质-蛋白质对接、配体对接、蛋白质设计等[93 ] .S. Khare课题组[103 ] 使用Rosetta对蛋白质框架进行设计,通过计算半胱氨酸侧链的氧化还原稳定性,确定了蛋白质支架内交联的最佳位置,随后成功改造出热稳定性和化学稳定性显著提高的环丙烷酶. ...
Protein sequence selection method that enables full consensus design of artificial L-threonine 3-dehydrogenases with unique enzymatic properties
1
2020
... 再则便是对于选择性的优化,在化学合成的众多选择性中(如化学选择性、区域选择性、对映选择性、立体选择性),最为重要的便是立体选择性与对映选择性,即手性分子与不对称合成[117 ] .1957年,沙利度胺在欧洲上市,不计其数的孕妇使用该药物缓解妊娠反应,因此又名“反应停”.但随后人们发现,沙利度胺可致婴儿畸形.而究其原因则在于沙利度胺为手性分子,存在R 与S 两种构型:其中R 构型具有镇静作用,S 构型则会引发畸形.该事件后,人们意识到手性分子在化学纯的基础上还存在着更高的纯度级别,即光学纯.随后FDA规定药品若是手性分子,则需分别对其两种对映异构体进行药理毒理学研究.因此,工业界的需求推动了合成方法学对于不对称合成的研究.直至目前,不对称合成与催化仍是有机化学领域最前沿、最具挑战性的研究方向之一.相较于化学催化方法使用昂贵的手性配体以控制产物的对映选择性,酶由于其活性中心存在立体构象,只有与活性中心相契合的底物才可与酶相结合.因此,通过定向进化提高酶活性中心与底物的契合度,可简便高效地获得高对映选择性产物.如手性胺是许多手性药物的重要合成中间体,手性胺的制备也是有机合成领域极具挑战性的课题[118 ] .在对西格列汀(sitagliptin)合成工艺优化过程中[119 ] ,Merck和Codexis公司合作,选择了一种天然的转氨酶催化甲基酮和小分子环酮的R -特异性转氨化作为起点,使用计算模型和体外协同进化方法生成了一种活性较弱的转氨酶变体.之后再经过11轮随机和定点饱和突变,得到了一个含有27个突变位点的转氨酶,与野生型相比,其最终催化活性提高了27000倍,并且可以在200 g/L的规模下生产>99.95% ee 的西格列汀(图7 ).通过该策略,可直接将酮转化为所需的手性胺,避免了高压反应体系和过渡金属催化剂铑(Rh)的使用[99 ] ,也是“绿色化学”在现代药物合成中应用的典范案例.与化学催化方法相比,进化后的转氨酶用于“西格列汀”的合成在产率上提升了10%~13%,立体选择性几乎完美(99.95% ee ),日产量增长了53%,工业废料减少了19%,避免了重金属的使用,缩短了反应步骤,无需高压条件以及高压设备,大幅降低了工业生产成本. ...
Consensus sequence design as a general strategy to create hyperstable, biologically active proteins
1
2019
... 然而,仅仅获得蛋白质的三维结构并不足以完成对其活性中心或突变热点的寻找,即使可以通过人工与已知同工酶或同源蛋白的序列的比对来推测其关键位点和保守序列[98 -100 ] ,但人工筛选的低效和可能存在的疏漏仍表明计算机辅助是更优的选择.常见的寻找酶活性中心的方法是通过序列比对预测(ClustalW[101 ] )和酶-底物对接模拟(AutoDock[102 ] 、Rosetta[93 ] )等[88 ] .其中,Rosetta是一个广泛使用且功能强大的软件,该软件自开发至今不过二十余年,却已经衍生出超过80种方法,其平台现在由来自约100所大学和实验室的开发人员和科学家贡献的代码组成(图5 ),功能包括但不限于蛋白质结构预测、蛋白质-蛋白质对接、配体对接、蛋白质设计等[93 ] .S. Khare课题组[103 ] 使用Rosetta对蛋白质框架进行设计,通过计算半胱氨酸侧链的氧化还原稳定性,确定了蛋白质支架内交联的最佳位置,随后成功改造出热稳定性和化学稳定性显著提高的环丙烷酶. ...
Sequence alignment and homology search with BLAST and ClustalW
1
2016
... 然而,仅仅获得蛋白质的三维结构并不足以完成对其活性中心或突变热点的寻找,即使可以通过人工与已知同工酶或同源蛋白的序列的比对来推测其关键位点和保守序列[98 -100 ] ,但人工筛选的低效和可能存在的疏漏仍表明计算机辅助是更优的选择.常见的寻找酶活性中心的方法是通过序列比对预测(ClustalW[101 ] )和酶-底物对接模拟(AutoDock[102 ] 、Rosetta[93 ] )等[88 ] .其中,Rosetta是一个广泛使用且功能强大的软件,该软件自开发至今不过二十余年,却已经衍生出超过80种方法,其平台现在由来自约100所大学和实验室的开发人员和科学家贡献的代码组成(图5 ),功能包括但不限于蛋白质结构预测、蛋白质-蛋白质对接、配体对接、蛋白质设计等[93 ] .S. Khare课题组[103 ] 使用Rosetta对蛋白质框架进行设计,通过计算半胱氨酸侧链的氧化还原稳定性,确定了蛋白质支架内交联的最佳位置,随后成功改造出热稳定性和化学稳定性显著提高的环丙烷酶. ...
AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading
1
2010
... 然而,仅仅获得蛋白质的三维结构并不足以完成对其活性中心或突变热点的寻找,即使可以通过人工与已知同工酶或同源蛋白的序列的比对来推测其关键位点和保守序列[98 -100 ] ,但人工筛选的低效和可能存在的疏漏仍表明计算机辅助是更优的选择.常见的寻找酶活性中心的方法是通过序列比对预测(ClustalW[101 ] )和酶-底物对接模拟(AutoDock[102 ] 、Rosetta[93 ] )等[88 ] .其中,Rosetta是一个广泛使用且功能强大的软件,该软件自开发至今不过二十余年,却已经衍生出超过80种方法,其平台现在由来自约100所大学和实验室的开发人员和科学家贡献的代码组成(图5 ),功能包括但不限于蛋白质结构预测、蛋白质-蛋白质对接、配体对接、蛋白质设计等[93 ] .S. Khare课题组[103 ] 使用Rosetta对蛋白质框架进行设计,通过计算半胱氨酸侧链的氧化还原稳定性,确定了蛋白质支架内交联的最佳位置,随后成功改造出热稳定性和化学稳定性显著提高的环丙烷酶. ...
Enzyme stabilization via computationally guided protein stapling
1
2017
... 然而,仅仅获得蛋白质的三维结构并不足以完成对其活性中心或突变热点的寻找,即使可以通过人工与已知同工酶或同源蛋白的序列的比对来推测其关键位点和保守序列[98 -100 ] ,但人工筛选的低效和可能存在的疏漏仍表明计算机辅助是更优的选择.常见的寻找酶活性中心的方法是通过序列比对预测(ClustalW[101 ] )和酶-底物对接模拟(AutoDock[102 ] 、Rosetta[93 ] )等[88 ] .其中,Rosetta是一个广泛使用且功能强大的软件,该软件自开发至今不过二十余年,却已经衍生出超过80种方法,其平台现在由来自约100所大学和实验室的开发人员和科学家贡献的代码组成(图5 ),功能包括但不限于蛋白质结构预测、蛋白质-蛋白质对接、配体对接、蛋白质设计等[93 ] .S. Khare课题组[103 ] 使用Rosetta对蛋白质框架进行设计,通过计算半胱氨酸侧链的氧化还原稳定性,确定了蛋白质支架内交联的最佳位置,随后成功改造出热稳定性和化学稳定性显著提高的环丙烷酶. ...
An introduction to machine learning
1
2020
... 另外,随着计算机技术的迅速发展,机器学习技术在近几十年中迅速崛起.不同于早期的生物信息学算法和软件,机器学习基于大量具有不同特征的数据点的训练找到一种通用的模型,并套用该模型解决问题[104 ] .机器学习的两种主要类型为无监督学习和监督学习.监督学习即为算法提供一个或几个目标属性作为标签,使用标记的数据集,根据标签为数据进行分类分析;而无监督学习则不提供标签,由计算机在学习过程中对样本进行分类,在学习过程中通常将高尺度的数据压缩或转换为低尺度的数据,以数学方法对特定信息进行分析[105 ] .此外,这两种类型结合的方法被称为半监督学习. ...
Machine learning in enzyme engineering
2
2020
... 另外,随着计算机技术的迅速发展,机器学习技术在近几十年中迅速崛起.不同于早期的生物信息学算法和软件,机器学习基于大量具有不同特征的数据点的训练找到一种通用的模型,并套用该模型解决问题[104 ] .机器学习的两种主要类型为无监督学习和监督学习.监督学习即为算法提供一个或几个目标属性作为标签,使用标记的数据集,根据标签为数据进行分类分析;而无监督学习则不提供标签,由计算机在学习过程中对样本进行分类,在学习过程中通常将高尺度的数据压缩或转换为低尺度的数据,以数学方法对特定信息进行分析[105 ] .此外,这两种类型结合的方法被称为半监督学习. ...
... 目前机器学习已经被广泛应用于蛋白质结构预测[106 ] 、代谢工程[107 ] 、酶工程[105 ] 、定向进化[108 ] 等领域,其在定向进化领域中的应用尤其重要.随着基因组数据的挖掘,更多新的蛋白质序列被发现,然而其功能常常难以准确界定,而且基于蛋白质全序列突变产生的巨大突变文库往往难以通过单纯的实验技术完成覆盖鉴定,同时部分优化方向难以开发合适的高通量筛选方式.以上原因促使研究人员使用计算机技术对蛋白质及其序列进行功能预测分析,借助其快速运算的能力提高实验效率[109 ] .G. Church课题组最新开发的Unirep模型,利用循环神经网络从2400万个蛋白质序列中深度学习,将蛋白质序列提炼成包含结构、进化等生物学信息的固定长度的向量[110 ] ,拓宽了蛋白质原始序列有益信息的挖掘,实现了更丰富更有效突变体库的构建,为蛋白质设计、功能改造提供了更多可能. ...
Machine learning in protein structure prediction
4
2021
... 目前机器学习已经被广泛应用于蛋白质结构预测[106 ] 、代谢工程[107 ] 、酶工程[105 ] 、定向进化[108 ] 等领域,其在定向进化领域中的应用尤其重要.随着基因组数据的挖掘,更多新的蛋白质序列被发现,然而其功能常常难以准确界定,而且基于蛋白质全序列突变产生的巨大突变文库往往难以通过单纯的实验技术完成覆盖鉴定,同时部分优化方向难以开发合适的高通量筛选方式.以上原因促使研究人员使用计算机技术对蛋白质及其序列进行功能预测分析,借助其快速运算的能力提高实验效率[109 ] .G. Church课题组最新开发的Unirep模型,利用循环神经网络从2400万个蛋白质序列中深度学习,将蛋白质序列提炼成包含结构、进化等生物学信息的固定长度的向量[110 ] ,拓宽了蛋白质原始序列有益信息的挖掘,实现了更丰富更有效突变体库的构建,为蛋白质设计、功能改造提供了更多可能. ...
... 除活性、稳定性、选择性外,另一个限制酶的应用范围的因素为酶催化的底物类型、范围非常局限.因此如何拓宽酶的催化底物范围及反应类型为该领域更前沿、更具挑战性的研究课题.对此,F. Arnold课题组致力于对细胞色素P450家族进行改造,赋予其新的催化活性(图9 )[121 -122 ] .细胞色素P450是一种单加氧酶,属于亚铁血红素蛋白酶(Heme)家族,因其在450 nm处存在特异吸收峰而得名.其野生型作用为催化底物的羟基化、环氧化及氧化脱氢等.F. Arnold开发了一系列P450,用以催化重氮化合物与烯烃的环丙化,合成光学纯的环丙烷衍生物,这些方案的共同之处在于利用重氮化合物的氮气逃逸生成卡宾中间体,进而发生烯烃的卡宾插入,得到环丙烷衍生物[123 ] .除烯烃外,该卡宾中间体还可以Si—H、B—H键插入的途径构建C—Si键[124 ] 与C—B键[125 ] ,得到相应的硅烷及硼烷化合物.相比于上述Si—H与B—H键,C—H键更为稳定,因此C—H键的官能化便是一项巨大的挑战,对此常见的策略为过渡金属催化的C—H键活化.F. Arnold则创新性地开发了P411催化叠氮化合物形成氮卡宾,以氮卡宾转移的方式构建C—N键,实现了无金属催化的C—H官能化[126 ] .他们以P411Diane1 为亲本,通过位点饱和诱变引入3个突变氨基酸得到P411Diane2 ,可成功催化叠氮化合物以氮气逃逸的方式形成氮卡宾,选择性插入苄基C—H[106 ] ,进行化学催化难以进行的不对称伯、仲、叔C(sp3 )—H氨基化(图10 ),具有极佳的对映选择性(99.9% ee )与高转化数(72000).值得注意的是,F. Arnold课题组对P450进行的改造,不仅使其获得相较于化学催化更佳的对映选择性,还可实现化学催化难以实现的反应类型.如以P411-S1为亲本,通过定点饱和突变,改变2个氨基酸的P411-E10可催化苯乙炔与重氮乙酸乙酯EDA发生两次连续的卡宾转移(图11 ),得到具有高环张力的双丁烷[127 ] . ...
... [
106 ]
Asymmetric synthesis catalyzed by P450 variants<sup>[<xref ref-type="bibr" rid="R106">106</xref>]</sup> Fig. 9 ![]()
图10 P411<sub>Diane2</sub>催化的不对称C(sp<sup>3</sup>)—H氨基化 P411<sub>Diane2</sub> catalyzed asymmetric amination of primary, secondary and tertiary C(sp<sup>3</sup>)—H bonds via nitrene insertion Fig. 10 ![]()
图11 P411-E10催化的苯乙炔与重氮乙酸乙酯的双卡宾转移 P411-E10 catalyzed double carbene transfer of phenylacetylene with EDA Fig. 11 ![]()
P411除具备催化叠氮化合物,以氮卡宾插入的途径构建C—N键能力外,Y. Yang还报道了以P411BM3 -CIS T438S(P)为亲本,通过位点饱和诱变引入5个突变氨基酸得到P450ATRCase1 ,可催化分子内的原子转移自由基环化(atom transfer radical cyclization,ATRC)[128 ] .对于末端取代烯烃,他们又以P450ATRCase1 为亲本,通过位点饱和诱变引入4个突变氨基酸得到P450ATRCase3 ,可一步得到具有连续两个手性中心的N -取代四氢吡咯衍生物(图12 ). ...
... [
106 ]
Fig. 9 ![]()
图10 P411<sub>Diane2</sub>催化的不对称C(sp<sup>3</sup>)—H氨基化 P411<sub>Diane2</sub> catalyzed asymmetric amination of primary, secondary and tertiary C(sp<sup>3</sup>)—H bonds via nitrene insertion Fig. 10 ![]()
图11 P411-E10催化的苯乙炔与重氮乙酸乙酯的双卡宾转移 P411-E10 catalyzed double carbene transfer of phenylacetylene with EDA Fig. 11 ![]()
P411除具备催化叠氮化合物,以氮卡宾插入的途径构建C—N键能力外,Y. Yang还报道了以P411BM3 -CIS T438S(P)为亲本,通过位点饱和诱变引入5个突变氨基酸得到P450ATRCase1 ,可催化分子内的原子转移自由基环化(atom transfer radical cyclization,ATRC)[128 ] .对于末端取代烯烃,他们又以P450ATRCase1 为亲本,通过位点饱和诱变引入4个突变氨基酸得到P450ATRCase3 ,可一步得到具有连续两个手性中心的N -取代四氢吡咯衍生物(图12 ). ...
Systems metabolic engineering meets machine learning: a new era for data-driven metabolic engineering
1
2019
... 目前机器学习已经被广泛应用于蛋白质结构预测[106 ] 、代谢工程[107 ] 、酶工程[105 ] 、定向进化[108 ] 等领域,其在定向进化领域中的应用尤其重要.随着基因组数据的挖掘,更多新的蛋白质序列被发现,然而其功能常常难以准确界定,而且基于蛋白质全序列突变产生的巨大突变文库往往难以通过单纯的实验技术完成覆盖鉴定,同时部分优化方向难以开发合适的高通量筛选方式.以上原因促使研究人员使用计算机技术对蛋白质及其序列进行功能预测分析,借助其快速运算的能力提高实验效率[109 ] .G. Church课题组最新开发的Unirep模型,利用循环神经网络从2400万个蛋白质序列中深度学习,将蛋白质序列提炼成包含结构、进化等生物学信息的固定长度的向量[110 ] ,拓宽了蛋白质原始序列有益信息的挖掘,实现了更丰富更有效突变体库的构建,为蛋白质设计、功能改造提供了更多可能. ...
Advances in machine learning for directed evolution
2
2021
... 目前机器学习已经被广泛应用于蛋白质结构预测[106 ] 、代谢工程[107 ] 、酶工程[105 ] 、定向进化[108 ] 等领域,其在定向进化领域中的应用尤其重要.随着基因组数据的挖掘,更多新的蛋白质序列被发现,然而其功能常常难以准确界定,而且基于蛋白质全序列突变产生的巨大突变文库往往难以通过单纯的实验技术完成覆盖鉴定,同时部分优化方向难以开发合适的高通量筛选方式.以上原因促使研究人员使用计算机技术对蛋白质及其序列进行功能预测分析,借助其快速运算的能力提高实验效率[109 ] .G. Church课题组最新开发的Unirep模型,利用循环神经网络从2400万个蛋白质序列中深度学习,将蛋白质序列提炼成包含结构、进化等生物学信息的固定长度的向量[110 ] ,拓宽了蛋白质原始序列有益信息的挖掘,实现了更丰富更有效突变体库的构建,为蛋白质设计、功能改造提供了更多可能. ...
... 传统的定向进化可以概括为对目的蛋白质反复的诱变和筛选,使用每轮中的最佳变体作为下一轮突变的起点,直至达到功能目标,这种实验思路是有效但烦琐的,而机器学习通过在学习过程中的每个进化周期内对大量蛋白质进行计算评估,实现比实验室筛选更加深入的探索[108 ] .例如J. Peng和H. Zhao课题组[111 ] 最新发明的进化环境集成神经网络(evolutionary context-integrated neural network,ECNet)深度学习算法,利用进化环境来预测蛋白质的功能适应性.该算法将来自同源序列的局部进化环境与编码大型蛋白质序列的丰富结构特征的全局进化背景相结合,实现了从序列到功能的映射.目前已有部分研究使用机器学习进行定向进化,例如F. Arnold课题组对海红藻来源能够催化碳硅键的生成的一氧化氮双加氧酶(nitric oxide dioxygenase,NOD)进行预测和改造,通过机器学习的辅助,以典型的监督学习的方式,对NOD的突变体库进行筛选和缩减,仅通过两轮进化即使野生型NOD催化硅烷和重氮乙酸乙酯经卡宾插入途径(如图6 所示)得到S -2-[二甲基(苯基)甲硅烷基]丙酸乙酯(76% ee )转变为S 构型(93% ee ),此外他们还发现VCHV-49P,51R,53L突变体可催化得到R -2-[二甲基(苯基)甲硅烷基]丙酸乙酯(79% ee )[15 ] . ...
Machine-learning-guided directed evolution for protein engineering
3
2019
... 目前机器学习已经被广泛应用于蛋白质结构预测[106 ] 、代谢工程[107 ] 、酶工程[105 ] 、定向进化[108 ] 等领域,其在定向进化领域中的应用尤其重要.随着基因组数据的挖掘,更多新的蛋白质序列被发现,然而其功能常常难以准确界定,而且基于蛋白质全序列突变产生的巨大突变文库往往难以通过单纯的实验技术完成覆盖鉴定,同时部分优化方向难以开发合适的高通量筛选方式.以上原因促使研究人员使用计算机技术对蛋白质及其序列进行功能预测分析,借助其快速运算的能力提高实验效率[109 ] .G. Church课题组最新开发的Unirep模型,利用循环神经网络从2400万个蛋白质序列中深度学习,将蛋白质序列提炼成包含结构、进化等生物学信息的固定长度的向量[110 ] ,拓宽了蛋白质原始序列有益信息的挖掘,实现了更丰富更有效突变体库的构建,为蛋白质设计、功能改造提供了更多可能. ...
... 除天然酶外,定向进化也可用于改造人工酶[130 -131 ] .人工酶的构建通常经由两种不同的策略实现,其一是计算机辅助的蛋白质从头设计(de novo )[132 ] ,再则是设计新型人工辅因子(artificial cofactor)制备人工金属酶(artificial metalloenzyme,ArM)[109 ] .通过de novo 设计的人工酶,往往在催化活性与选择性方面表现不佳[111 ] ;而通过替换辅因子的策略制备人工酶的难题则是人工辅因子在酶活性位点的精准定位,同时蛋白骨架对辅因子的兼容性也是一大挑战[133 ] ,定向进化能有效改善上述问题. ...
... 人工金属酶(artificial metalloenzyme,ArM)将具备催化活性的金属辅因子与蛋白质骨架相结合,赋予蛋白催化活性,即构建了新型酶;同时也解决了金属催化剂在水相体系中的失活问题.ArM的设计难点在于如何将金属有效地结合到酶活性位点部位,即金属辅因子的定位[109 ] .幸运的是,目前已有多种方法被报道,其中运用最广泛同时也最成熟的方法为基于生物素-亲和素(biotin-avidin)系统的特异性结合[137 ] 与制备人工金属辅因子进行替换[112 ] . ...
Unified rational protein engineering with sequence-based deep representation learning
3
2019
... 目前机器学习已经被广泛应用于蛋白质结构预测[106 ] 、代谢工程[107 ] 、酶工程[105 ] 、定向进化[108 ] 等领域,其在定向进化领域中的应用尤其重要.随着基因组数据的挖掘,更多新的蛋白质序列被发现,然而其功能常常难以准确界定,而且基于蛋白质全序列突变产生的巨大突变文库往往难以通过单纯的实验技术完成覆盖鉴定,同时部分优化方向难以开发合适的高通量筛选方式.以上原因促使研究人员使用计算机技术对蛋白质及其序列进行功能预测分析,借助其快速运算的能力提高实验效率[109 ] .G. Church课题组最新开发的Unirep模型,利用循环神经网络从2400万个蛋白质序列中深度学习,将蛋白质序列提炼成包含结构、进化等生物学信息的固定长度的向量[110 ] ,拓宽了蛋白质原始序列有益信息的挖掘,实现了更丰富更有效突变体库的构建,为蛋白质设计、功能改造提供了更多可能. ...
... 除单个金属辅因子外,由于其为四元结构的二聚体,链霉亲和素允许精确定位两个金属辅因子以激活单个底物,从而扩大了人工金属酶的反应范围.近期,T. Ward设计了一种嵌合链霉亲和素,该嵌合链霉亲和素配有一个疏水盖,可保护其活性位点,从而加强了两种协同生物素化金辅助因子的有利定位[110 ] .经过三轮诱变,他们发现Sav-SOD S112N N118G K121G S122G(N-GGG)可催化乙炔脲的分子内环化,以96%的区域选择性得到5-endo-dig 反马氏加成产物吲哚甲酰胺,而Sav-SOD S112T N118S K121F S122G(T-SFG)则可以99%的区域选择性得到6-exo-dig 产物苯基-二氢喹唑啉酮[110 ] (图16 ).引起区域选择性改变的原因在于不同的蛋白质环境改变了Au对炔烃的活化方式(σ,π活化或π活化). ...
... [110 ](图16 ).引起区域选择性改变的原因在于不同的蛋白质环境改变了Au对炔烃的活化方式(σ,π活化或π活化). ...
ECNet is an evolutionary context-integrated deep learning framework for protein engineering
3
2021
... 传统的定向进化可以概括为对目的蛋白质反复的诱变和筛选,使用每轮中的最佳变体作为下一轮突变的起点,直至达到功能目标,这种实验思路是有效但烦琐的,而机器学习通过在学习过程中的每个进化周期内对大量蛋白质进行计算评估,实现比实验室筛选更加深入的探索[108 ] .例如J. Peng和H. Zhao课题组[111 ] 最新发明的进化环境集成神经网络(evolutionary context-integrated neural network,ECNet)深度学习算法,利用进化环境来预测蛋白质的功能适应性.该算法将来自同源序列的局部进化环境与编码大型蛋白质序列的丰富结构特征的全局进化背景相结合,实现了从序列到功能的映射.目前已有部分研究使用机器学习进行定向进化,例如F. Arnold课题组对海红藻来源能够催化碳硅键的生成的一氧化氮双加氧酶(nitric oxide dioxygenase,NOD)进行预测和改造,通过机器学习的辅助,以典型的监督学习的方式,对NOD的突变体库进行筛选和缩减,仅通过两轮进化即使野生型NOD催化硅烷和重氮乙酸乙酯经卡宾插入途径(如图6 所示)得到S -2-[二甲基(苯基)甲硅烷基]丙酸乙酯(76% ee )转变为S 构型(93% ee ),此外他们还发现VCHV-49P,51R,53L突变体可催化得到R -2-[二甲基(苯基)甲硅烷基]丙酸乙酯(79% ee )[15 ] . ...
... 除天然酶外,定向进化也可用于改造人工酶[130 -131 ] .人工酶的构建通常经由两种不同的策略实现,其一是计算机辅助的蛋白质从头设计(de novo )[132 ] ,再则是设计新型人工辅因子(artificial cofactor)制备人工金属酶(artificial metalloenzyme,ArM)[109 ] .通过de novo 设计的人工酶,往往在催化活性与选择性方面表现不佳[111 ] ;而通过替换辅因子的策略制备人工酶的难题则是人工辅因子在酶活性位点的精准定位,同时蛋白骨架对辅因子的兼容性也是一大挑战[133 ] ,定向进化能有效改善上述问题. ...
... 目前蛋白质结构和蛋白质折叠的原理已基本建立,许多稳定的蛋白质结构和组件也都能够以原子级精度设计.计算机设计蛋白质可以改变天然蛋白质骨架,随之改变其功能和用途.蛋白质从头设计有着诸多好处,但也存在着局限性.当利用大肠杆菌生产新设计的蛋白时,只有一小部分蛋白保持与设计相同的折叠结构.因此可采用定向进化技术对de novo 设计出的酶进行改造,提高其稳定性与催化活性[111 ] .该方向的代表性工作为羟醛缩合酶(aldolase)与Diels-Alder酶的设计. ...
A backbone-centred energy function of neural networks for protein design
4
2022
... 目前,国际主流的蛋白质人工设计的方法为采用天然蛋白质结构为模板来拼接产生人工结构,但这类方法有其局限性,包括设计结果单一、模板依赖度高等,从而限制了设计主链结构的多样性和可变性.2022年,H. Liu和Q. Chen课题组发展了一种能在氨基酸序列待定时从头设计全新主链结构的模型SCUBA(side chain-unknown backbone arrangement).该模型采用了一种新的统计学习策略,即包括核密度估计和神经网络训练的两步学习法,能够高保真地表示实际蛋白质结构数据的复杂性和高度相关性.该模型的另一个显著特点是,SCUBA模型不需要用已有结构片段来拼接产生新结构,能够显著扩展从头设计蛋白的结构多样性,设计出不同于已知天然蛋白的新颖结构.SCUBA模型及其算法还在此次研究中得到了实验验证,文章报告了9个新蛋白质的晶体结构,骨架均使用SCUBA模型进行设计,其中产生的新型结构的蛋白质清楚地表明了这种全新的蛋白质从头设计路线可以极大扩展新蛋白质设计的结构多样性和功能[112 ] . ...
... 人工金属酶(artificial metalloenzyme,ArM)将具备催化活性的金属辅因子与蛋白质骨架相结合,赋予蛋白催化活性,即构建了新型酶;同时也解决了金属催化剂在水相体系中的失活问题.ArM的设计难点在于如何将金属有效地结合到酶活性位点部位,即金属辅因子的定位[109 ] .幸运的是,目前已有多种方法被报道,其中运用最广泛同时也最成熟的方法为基于生物素-亲和素(biotin-avidin)系统的特异性结合[137 ] 与制备人工金属辅因子进行替换[112 ] . ...
... 天然金属酶催化的反应范围受到金属中心固有反应性的限制,为了克服这一局限性,J. Hartwig课题组[112 ] 开发了金属替代方法,体外合成铱卟啉以替代铁卟啉作为抹香鲸来源肌红蛋白的辅因子,构建了新型人造血红素蛋白.此前金属替代铁卟啉的方法多需要利用蛋白体外变性,同时加入抑制剂竞争性结合铁卟啉,该过程需要多步的蛋白纯化步骤[112 ] .近期,J. Hartwig课题组[139 ] 实现了通过转运蛋白将铱卟啉辅因子转运入胞内,与蛋白骨架Cyp119进行装配,这为快速进化通过辅酶因子替换而得的金属酶提供了可能.他们通过位点饱和诱变,引入两个突变氨基酸(R256W,V254A),得到Ir-Cyp119-P/R256W/V254A,并创造性地将柠檬烯的生物合成途径与Ir催化的卡宾转移相结合,实现了环丙基柠檬烯的生物合成[图17 (a)],为代谢通路的改造提供了可能.此外,他们还实现了胞内Ir-Cyp119催化的对映选择性、区域选择性C—H活化[图17 (b)]. ...
... [112 ].近期,J. Hartwig课题组[139 ] 实现了通过转运蛋白将铱卟啉辅因子转运入胞内,与蛋白骨架Cyp119进行装配,这为快速进化通过辅酶因子替换而得的金属酶提供了可能.他们通过位点饱和诱变,引入两个突变氨基酸(R256W,V254A),得到Ir-Cyp119-P/R256W/V254A,并创造性地将柠檬烯的生物合成途径与Ir催化的卡宾转移相结合,实现了环丙基柠檬烯的生物合成[图17 (a)],为代谢通路的改造提供了可能.此外,他们还实现了胞内Ir-Cyp119催化的对映选择性、区域选择性C—H活化[图17 (b)]. ...
Tuning the activity of an enzyme for unusual environments: sequential random mutagenesis of subtilisin E for catalysis in dimethylformamide
1
1993
... 早期,定向进化有效地提高了天然酶对有机溶剂的耐受性,这对于改善底物在溶剂中的溶解度以进行大规模工艺制造十分必要[7 ] .蛋白酶subtilisin E可用于水解酪蛋白(casein),但其在有机溶剂N ,N- 二甲基甲酰胺(DMF)中的稳定性极差,导致其在60% DMF溶液中的催化活性还不到其在水溶液中活性的0.5%.F. Arnold课题组[113 -114 ] 结合随机突变和定点突变,经过三轮诱变和筛选,引入10个氨基酸突变(D60N,D97G,Q103R,N218S,G131D,E156G,N181S,S182G,S188P和T255A),得到了在60% DMF溶液中的催化效率提高了256倍的subtilisin E-PC3,并且PC3的活性水平与野生型subtilisin E在水溶液中的活性水平相当.此研究展示了“定向进化”能有效提高酶在体外环境中稳定性以及催化活性. ...
Directed evolution of subtilisin E in Bacillus subtilis to enhance total activity in aqueous dimethylformamide
1
1996
... 早期,定向进化有效地提高了天然酶对有机溶剂的耐受性,这对于改善底物在溶剂中的溶解度以进行大规模工艺制造十分必要[7 ] .蛋白酶subtilisin E可用于水解酪蛋白(casein),但其在有机溶剂N ,N- 二甲基甲酰胺(DMF)中的稳定性极差,导致其在60% DMF溶液中的催化活性还不到其在水溶液中活性的0.5%.F. Arnold课题组[113 -114 ] 结合随机突变和定点突变,经过三轮诱变和筛选,引入10个氨基酸突变(D60N,D97G,Q103R,N218S,G131D,E156G,N181S,S182G,S188P和T255A),得到了在60% DMF溶液中的催化效率提高了256倍的subtilisin E-PC3,并且PC3的活性水平与野生型subtilisin E在水溶液中的活性水平相当.此研究展示了“定向进化”能有效提高酶在体外环境中稳定性以及催化活性. ...
Directed evolution of a para-nitrobenzyl esterase for aqueous-organic solvents
2
1996
... 催化活性的另一大重要体现为热稳定性.众所周知,高温易导致酶迅速变性失活.但某些情况下往往需要高温来增加底物溶解度,这极大地限制了酶在工业化中的应用.为解决这一难题,F. Arnold课题组[115 -116 ] 采用随机诱变与重组结合的策略,对枯草芽孢杆菌中p -NB酶进行改造,引入9个氨基酸突变(I60V,L144M,L313F,H322Y,A343V,M358V,Y370F,G412E,I437T),得到p -NB-6sF9.成功将其熔解温度T m 提高了14 ℃(相比于野生型).值得注意的是,耐受高温的变体在低温时往往活性很低,但提高p -NB-6sF9热稳定性的同时,并未牺牲其在低温下的催化活性. ...
... 双烯体与亲双烯体的[4+2]环加成,得到六元环的反应称为Diels-Alder(D-A)反应,是有机合成中非常重要的构建C—C键的手段之一,广泛运用于各种药物的全合成.该反应有丰富的立体选择性,可一次生成两个C—C键和最多四个相邻的手性碳.近年来,该领域最前沿的方向即为通过杂D-A反应[135 ] 构建药物优势骨架六元杂环.D-A反应通常由Lewis酸所催化,在高温下进行.此外,天然酶未被证明可催化该反应[136 ] .D. Hilvert课题组创制了可在温和条件下催化D-A反应的人工酶DA_20_10,并结合靶向诱变、计算改进和epPCR,经过8轮连续定向进化,在DA_20_10上引入5个氨基酸突变(R50H,V96I,T197R,E288D和L309S),最终得到了具备更高催化活性的DA_20_20,高效地催化了1,3-丁二烯氨基甲酸酯与二甲基丙烯酰胺(图14 )的[4+2]环加成[115 ] ,以高产率得到环己烯衍生物. ...
Directed evolution converts subtilisin E into a functional equivalent of thermitase
2
1999
... 催化活性的另一大重要体现为热稳定性.众所周知,高温易导致酶迅速变性失活.但某些情况下往往需要高温来增加底物溶解度,这极大地限制了酶在工业化中的应用.为解决这一难题,F. Arnold课题组[115 -116 ] 采用随机诱变与重组结合的策略,对枯草芽孢杆菌中p -NB酶进行改造,引入9个氨基酸突变(I60V,L144M,L313F,H322Y,A343V,M358V,Y370F,G412E,I437T),得到p -NB-6sF9.成功将其熔解温度T m 提高了14 ℃(相比于野生型).值得注意的是,耐受高温的变体在低温时往往活性很低,但提高p -NB-6sF9热稳定性的同时,并未牺牲其在低温下的催化活性. ...
... 金属催化的烯烃间C=C切断并重新结合的烯烃复分解反应,广泛应用于药物的研发和先进聚合物材料的制备.其中R. Grubbs[138 ] 开发了钌-卡宾复合物体系,成为应用最广泛的烯烃复分解催化剂,即Grubbs催化剂.虽然Grubbs催化剂在体系中存在少量水、醇、酸的情况下仍可保持催化活性,但水相中的烯烃复分解反应却鲜有报道.同时自然界中并不存在该类反应,因此无法利用天然酶成功催化该反应.T. Ward课题组用采用生物素-链霉亲和素(Biotin-Streptavidin)方法,将钌(Ru)锚定到链霉亲和素(streptavidin,SAV)支架中,制备了Biot-Ru-SAV,实现了在水相溶剂中催化烯烃复分解反应.同时,他们还通过多轮饱和突变,引入20个氨基酸突变,得到与Ru离子结合更稳定的SAVmut,在赋予酶新功能的同时,解决了金属催化剂在水和细胞环境下表现不佳的问题.值得一提的是,利用SAV-Biot方法进行制备ArM时,金属辅因子的活性可能被细胞质中成分抑制,因此通常需对蛋白支架SAV进行纯化、体外组装Biot-Ru-SAV,该过程烦琐,不利于高通量筛选Biot-Ru-SAV突变体库.T. Ward[116 ] 创新性地在细胞周质中表达SAV,随后通过自组装的方式制备Biot-Ru-SAV,并在体内实现了闭环烯烃复分解(图15 ),该方法不但加速了Biot-Ru-SAV突变库筛选,还为构造非天然的代谢通路提供了可能. ...
Catalytic asymmetric umpolung reactions of imines
1
2015
... 再则便是对于选择性的优化,在化学合成的众多选择性中(如化学选择性、区域选择性、对映选择性、立体选择性),最为重要的便是立体选择性与对映选择性,即手性分子与不对称合成[117 ] .1957年,沙利度胺在欧洲上市,不计其数的孕妇使用该药物缓解妊娠反应,因此又名“反应停”.但随后人们发现,沙利度胺可致婴儿畸形.而究其原因则在于沙利度胺为手性分子,存在R 与S 两种构型:其中R 构型具有镇静作用,S 构型则会引发畸形.该事件后,人们意识到手性分子在化学纯的基础上还存在着更高的纯度级别,即光学纯.随后FDA规定药品若是手性分子,则需分别对其两种对映异构体进行药理毒理学研究.因此,工业界的需求推动了合成方法学对于不对称合成的研究.直至目前,不对称合成与催化仍是有机化学领域最前沿、最具挑战性的研究方向之一.相较于化学催化方法使用昂贵的手性配体以控制产物的对映选择性,酶由于其活性中心存在立体构象,只有与活性中心相契合的底物才可与酶相结合.因此,通过定向进化提高酶活性中心与底物的契合度,可简便高效地获得高对映选择性产物.如手性胺是许多手性药物的重要合成中间体,手性胺的制备也是有机合成领域极具挑战性的课题[118 ] .在对西格列汀(sitagliptin)合成工艺优化过程中[119 ] ,Merck和Codexis公司合作,选择了一种天然的转氨酶催化甲基酮和小分子环酮的R -特异性转氨化作为起点,使用计算模型和体外协同进化方法生成了一种活性较弱的转氨酶变体.之后再经过11轮随机和定点饱和突变,得到了一个含有27个突变位点的转氨酶,与野生型相比,其最终催化活性提高了27000倍,并且可以在200 g/L的规模下生产>99.95% ee 的西格列汀(图7 ).通过该策略,可直接将酮转化为所需的手性胺,避免了高压反应体系和过渡金属催化剂铑(Rh)的使用[99 ] ,也是“绿色化学”在现代药物合成中应用的典范案例.与化学催化方法相比,进化后的转氨酶用于“西格列汀”的合成在产率上提升了10%~13%,立体选择性几乎完美(99.95% ee ),日产量增长了53%,工业废料减少了19%,避免了重金属的使用,缩短了反应步骤,无需高压条件以及高压设备,大幅降低了工业生产成本. ...
Enantioselective synthesis of ammonium cations
1
2021
... 再则便是对于选择性的优化,在化学合成的众多选择性中(如化学选择性、区域选择性、对映选择性、立体选择性),最为重要的便是立体选择性与对映选择性,即手性分子与不对称合成[117 ] .1957年,沙利度胺在欧洲上市,不计其数的孕妇使用该药物缓解妊娠反应,因此又名“反应停”.但随后人们发现,沙利度胺可致婴儿畸形.而究其原因则在于沙利度胺为手性分子,存在R 与S 两种构型:其中R 构型具有镇静作用,S 构型则会引发畸形.该事件后,人们意识到手性分子在化学纯的基础上还存在着更高的纯度级别,即光学纯.随后FDA规定药品若是手性分子,则需分别对其两种对映异构体进行药理毒理学研究.因此,工业界的需求推动了合成方法学对于不对称合成的研究.直至目前,不对称合成与催化仍是有机化学领域最前沿、最具挑战性的研究方向之一.相较于化学催化方法使用昂贵的手性配体以控制产物的对映选择性,酶由于其活性中心存在立体构象,只有与活性中心相契合的底物才可与酶相结合.因此,通过定向进化提高酶活性中心与底物的契合度,可简便高效地获得高对映选择性产物.如手性胺是许多手性药物的重要合成中间体,手性胺的制备也是有机合成领域极具挑战性的课题[118 ] .在对西格列汀(sitagliptin)合成工艺优化过程中[119 ] ,Merck和Codexis公司合作,选择了一种天然的转氨酶催化甲基酮和小分子环酮的R -特异性转氨化作为起点,使用计算模型和体外协同进化方法生成了一种活性较弱的转氨酶变体.之后再经过11轮随机和定点饱和突变,得到了一个含有27个突变位点的转氨酶,与野生型相比,其最终催化活性提高了27000倍,并且可以在200 g/L的规模下生产>99.95% ee 的西格列汀(图7 ).通过该策略,可直接将酮转化为所需的手性胺,避免了高压反应体系和过渡金属催化剂铑(Rh)的使用[99 ] ,也是“绿色化学”在现代药物合成中应用的典范案例.与化学催化方法相比,进化后的转氨酶用于“西格列汀”的合成在产率上提升了10%~13%,立体选择性几乎完美(99.95% ee ),日产量增长了53%,工业废料减少了19%,避免了重金属的使用,缩短了反应步骤,无需高压条件以及高压设备,大幅降低了工业生产成本. ...
Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture
2
2010
... 再则便是对于选择性的优化,在化学合成的众多选择性中(如化学选择性、区域选择性、对映选择性、立体选择性),最为重要的便是立体选择性与对映选择性,即手性分子与不对称合成[117 ] .1957年,沙利度胺在欧洲上市,不计其数的孕妇使用该药物缓解妊娠反应,因此又名“反应停”.但随后人们发现,沙利度胺可致婴儿畸形.而究其原因则在于沙利度胺为手性分子,存在R 与S 两种构型:其中R 构型具有镇静作用,S 构型则会引发畸形.该事件后,人们意识到手性分子在化学纯的基础上还存在着更高的纯度级别,即光学纯.随后FDA规定药品若是手性分子,则需分别对其两种对映异构体进行药理毒理学研究.因此,工业界的需求推动了合成方法学对于不对称合成的研究.直至目前,不对称合成与催化仍是有机化学领域最前沿、最具挑战性的研究方向之一.相较于化学催化方法使用昂贵的手性配体以控制产物的对映选择性,酶由于其活性中心存在立体构象,只有与活性中心相契合的底物才可与酶相结合.因此,通过定向进化提高酶活性中心与底物的契合度,可简便高效地获得高对映选择性产物.如手性胺是许多手性药物的重要合成中间体,手性胺的制备也是有机合成领域极具挑战性的课题[118 ] .在对西格列汀(sitagliptin)合成工艺优化过程中[119 ] ,Merck和Codexis公司合作,选择了一种天然的转氨酶催化甲基酮和小分子环酮的R -特异性转氨化作为起点,使用计算模型和体外协同进化方法生成了一种活性较弱的转氨酶变体.之后再经过11轮随机和定点饱和突变,得到了一个含有27个突变位点的转氨酶,与野生型相比,其最终催化活性提高了27000倍,并且可以在200 g/L的规模下生产>99.95% ee 的西格列汀(图7 ).通过该策略,可直接将酮转化为所需的手性胺,避免了高压反应体系和过渡金属催化剂铑(Rh)的使用[99 ] ,也是“绿色化学”在现代药物合成中应用的典范案例.与化学催化方法相比,进化后的转氨酶用于“西格列汀”的合成在产率上提升了10%~13%,立体选择性几乎完美(99.95% ee ),日产量增长了53%,工业废料减少了19%,避免了重金属的使用,缩短了反应步骤,无需高压条件以及高压设备,大幅降低了工业生产成本. ...
... 相比于天然酶,利用该方法进行构建新型人工金属酶时,涉及到游离的天然辅因子的去除以及人工辅因子的引入[140 ] .而单个宿主蛋白的纯化十分耗时,因此辅因子和蛋白的体外重组限制了高通量筛选和定向进化的速率.对此,J. Hartwig课题组[119 ] 在原有研究基础上,将辅因子转移到细胞质中,实现了全细胞中人工金属酶的构建. ...
Design of an in vitro biocatalytic cascade for the manufacture of islatravir
1
2019
... 除了对单个酶进行改造外,定向进化还可同时对多个酶进行改造,构建多酶级联催化体系,实现酶促的药物全合成.酶级联实现药物全合成中的挑战性在于如何高效实现上一步的产物作为下一步的底物这一串联过程,这对酶的催化底物适用性以及催化活性提出要求.对此,Merck公司的研究人员通过定向进化策略优化了五种酶,使其与非天然底物兼容,并在每个酶促步骤中都以高对映选择性得到目标产物,最终以高原子利用率实现了抗HIV药物依拉曲韦(islatravir)的体外生物催化级联合成[120 ] .他们通过epPCR,经过12轮进化,改变了34个氨基酸,得到了比野生型半乳糖氧化酶GOase具备更优异立体选择性以及催化活性的GOaseRd13 ;经过2轮进化,改变了11个氨基酸,得到了比野生型脱氧核糖5-磷酸醛缩酶DERA更耐受乙醛的DERARd3 以及分别经过3轮、2轮、4轮突变,改变了10个、5个、7个氨基酸,具备更高催化活性的泛酸激酶PanKRd4 、磷酸戊醇变位酶PPMRd3 和嘌呤核苷磷酸化酶PNPRd5 .此外,他们将上述改造后的酶与四种辅酶(辣根过氧化物酶HRP,过氧化氢酶CAT,乙酸激酶ACK和蔗糖磷酸化酶SP)相结合,成功实现以2-乙炔甘油为底物,三模块、九酶级联催化合成依拉曲韦(图8 ).该方案避免了提纯中间体,并通过级联策略克服不利的平衡和避免不稳定或抑制性中间体的积累,并以51%的总收率得到依拉曲韦.与之前化学合成法中涉及的多步骤合成、纯化和基团保护步骤的技术相比,此策略显然是一项巨大进步. ...
Navigating the unnatural reaction space: directed evolution of heme proteins for selective carbene and nitrene transfer
1
2021
... 除活性、稳定性、选择性外,另一个限制酶的应用范围的因素为酶催化的底物类型、范围非常局限.因此如何拓宽酶的催化底物范围及反应类型为该领域更前沿、更具挑战性的研究课题.对此,F. Arnold课题组致力于对细胞色素P450家族进行改造,赋予其新的催化活性(图9 )[121 -122 ] .细胞色素P450是一种单加氧酶,属于亚铁血红素蛋白酶(Heme)家族,因其在450 nm处存在特异吸收峰而得名.其野生型作用为催化底物的羟基化、环氧化及氧化脱氢等.F. Arnold开发了一系列P450,用以催化重氮化合物与烯烃的环丙化,合成光学纯的环丙烷衍生物,这些方案的共同之处在于利用重氮化合物的氮气逃逸生成卡宾中间体,进而发生烯烃的卡宾插入,得到环丙烷衍生物[123 ] .除烯烃外,该卡宾中间体还可以Si—H、B—H键插入的途径构建C—Si键[124 ] 与C—B键[125 ] ,得到相应的硅烷及硼烷化合物.相比于上述Si—H与B—H键,C—H键更为稳定,因此C—H键的官能化便是一项巨大的挑战,对此常见的策略为过渡金属催化的C—H键活化.F. Arnold则创新性地开发了P411催化叠氮化合物形成氮卡宾,以氮卡宾转移的方式构建C—N键,实现了无金属催化的C—H官能化[126 ] .他们以P411Diane1 为亲本,通过位点饱和诱变引入3个突变氨基酸得到P411Diane2 ,可成功催化叠氮化合物以氮气逃逸的方式形成氮卡宾,选择性插入苄基C—H[106 ] ,进行化学催化难以进行的不对称伯、仲、叔C(sp3 )—H氨基化(图10 ),具有极佳的对映选择性(99.9% ee )与高转化数(72000).值得注意的是,F. Arnold课题组对P450进行的改造,不仅使其获得相较于化学催化更佳的对映选择性,还可实现化学催化难以实现的反应类型.如以P411-S1为亲本,通过定点饱和突变,改变2个氨基酸的P411-E10可催化苯乙炔与重氮乙酸乙酯EDA发生两次连续的卡宾转移(图11 ),得到具有高环张力的双丁烷[127 ] . ...
Engineering new catalytic activities in enzymes
1
2020
... 除活性、稳定性、选择性外,另一个限制酶的应用范围的因素为酶催化的底物类型、范围非常局限.因此如何拓宽酶的催化底物范围及反应类型为该领域更前沿、更具挑战性的研究课题.对此,F. Arnold课题组致力于对细胞色素P450家族进行改造,赋予其新的催化活性(图9 )[121 -122 ] .细胞色素P450是一种单加氧酶,属于亚铁血红素蛋白酶(Heme)家族,因其在450 nm处存在特异吸收峰而得名.其野生型作用为催化底物的羟基化、环氧化及氧化脱氢等.F. Arnold开发了一系列P450,用以催化重氮化合物与烯烃的环丙化,合成光学纯的环丙烷衍生物,这些方案的共同之处在于利用重氮化合物的氮气逃逸生成卡宾中间体,进而发生烯烃的卡宾插入,得到环丙烷衍生物[123 ] .除烯烃外,该卡宾中间体还可以Si—H、B—H键插入的途径构建C—Si键[124 ] 与C—B键[125 ] ,得到相应的硅烷及硼烷化合物.相比于上述Si—H与B—H键,C—H键更为稳定,因此C—H键的官能化便是一项巨大的挑战,对此常见的策略为过渡金属催化的C—H键活化.F. Arnold则创新性地开发了P411催化叠氮化合物形成氮卡宾,以氮卡宾转移的方式构建C—N键,实现了无金属催化的C—H官能化[126 ] .他们以P411Diane1 为亲本,通过位点饱和诱变引入3个突变氨基酸得到P411Diane2 ,可成功催化叠氮化合物以氮气逃逸的方式形成氮卡宾,选择性插入苄基C—H[106 ] ,进行化学催化难以进行的不对称伯、仲、叔C(sp3 )—H氨基化(图10 ),具有极佳的对映选择性(99.9% ee )与高转化数(72000).值得注意的是,F. Arnold课题组对P450进行的改造,不仅使其获得相较于化学催化更佳的对映选择性,还可实现化学催化难以实现的反应类型.如以P411-S1为亲本,通过定点饱和突变,改变2个氨基酸的P411-E10可催化苯乙炔与重氮乙酸乙酯EDA发生两次连续的卡宾转移(图11 ),得到具有高环张力的双丁烷[127 ] . ...
Olefin cyclopropanation via carbene transfer catalyzed by engineered cytochrome P450 enzymes
1
2013
... 除活性、稳定性、选择性外,另一个限制酶的应用范围的因素为酶催化的底物类型、范围非常局限.因此如何拓宽酶的催化底物范围及反应类型为该领域更前沿、更具挑战性的研究课题.对此,F. Arnold课题组致力于对细胞色素P450家族进行改造,赋予其新的催化活性(图9 )[121 -122 ] .细胞色素P450是一种单加氧酶,属于亚铁血红素蛋白酶(Heme)家族,因其在450 nm处存在特异吸收峰而得名.其野生型作用为催化底物的羟基化、环氧化及氧化脱氢等.F. Arnold开发了一系列P450,用以催化重氮化合物与烯烃的环丙化,合成光学纯的环丙烷衍生物,这些方案的共同之处在于利用重氮化合物的氮气逃逸生成卡宾中间体,进而发生烯烃的卡宾插入,得到环丙烷衍生物[123 ] .除烯烃外,该卡宾中间体还可以Si—H、B—H键插入的途径构建C—Si键[124 ] 与C—B键[125 ] ,得到相应的硅烷及硼烷化合物.相比于上述Si—H与B—H键,C—H键更为稳定,因此C—H键的官能化便是一项巨大的挑战,对此常见的策略为过渡金属催化的C—H键活化.F. Arnold则创新性地开发了P411催化叠氮化合物形成氮卡宾,以氮卡宾转移的方式构建C—N键,实现了无金属催化的C—H官能化[126 ] .他们以P411Diane1 为亲本,通过位点饱和诱变引入3个突变氨基酸得到P411Diane2 ,可成功催化叠氮化合物以氮气逃逸的方式形成氮卡宾,选择性插入苄基C—H[106 ] ,进行化学催化难以进行的不对称伯、仲、叔C(sp3 )—H氨基化(图10 ),具有极佳的对映选择性(99.9% ee )与高转化数(72000).值得注意的是,F. Arnold课题组对P450进行的改造,不仅使其获得相较于化学催化更佳的对映选择性,还可实现化学催化难以实现的反应类型.如以P411-S1为亲本,通过定点饱和突变,改变2个氨基酸的P411-E10可催化苯乙炔与重氮乙酸乙酯EDA发生两次连续的卡宾转移(图11 ),得到具有高环张力的双丁烷[127 ] . ...
Directed evolution of cytochrome c for carbon-silicon bond formation: bringing silicon to life
1
2016
... 除活性、稳定性、选择性外,另一个限制酶的应用范围的因素为酶催化的底物类型、范围非常局限.因此如何拓宽酶的催化底物范围及反应类型为该领域更前沿、更具挑战性的研究课题.对此,F. Arnold课题组致力于对细胞色素P450家族进行改造,赋予其新的催化活性(图9 )[121 -122 ] .细胞色素P450是一种单加氧酶,属于亚铁血红素蛋白酶(Heme)家族,因其在450 nm处存在特异吸收峰而得名.其野生型作用为催化底物的羟基化、环氧化及氧化脱氢等.F. Arnold开发了一系列P450,用以催化重氮化合物与烯烃的环丙化,合成光学纯的环丙烷衍生物,这些方案的共同之处在于利用重氮化合物的氮气逃逸生成卡宾中间体,进而发生烯烃的卡宾插入,得到环丙烷衍生物[123 ] .除烯烃外,该卡宾中间体还可以Si—H、B—H键插入的途径构建C—Si键[124 ] 与C—B键[125 ] ,得到相应的硅烷及硼烷化合物.相比于上述Si—H与B—H键,C—H键更为稳定,因此C—H键的官能化便是一项巨大的挑战,对此常见的策略为过渡金属催化的C—H键活化.F. Arnold则创新性地开发了P411催化叠氮化合物形成氮卡宾,以氮卡宾转移的方式构建C—N键,实现了无金属催化的C—H官能化[126 ] .他们以P411Diane1 为亲本,通过位点饱和诱变引入3个突变氨基酸得到P411Diane2 ,可成功催化叠氮化合物以氮气逃逸的方式形成氮卡宾,选择性插入苄基C—H[106 ] ,进行化学催化难以进行的不对称伯、仲、叔C(sp3 )—H氨基化(图10 ),具有极佳的对映选择性(99.9% ee )与高转化数(72000).值得注意的是,F. Arnold课题组对P450进行的改造,不仅使其获得相较于化学催化更佳的对映选择性,还可实现化学催化难以实现的反应类型.如以P411-S1为亲本,通过定点饱和突变,改变2个氨基酸的P411-E10可催化苯乙炔与重氮乙酸乙酯EDA发生两次连续的卡宾转移(图11 ),得到具有高环张力的双丁烷[127 ] . ...
A biocatalytic platform for synthesis of chiral α- trifluoromethylated organoborons
1
2019
... 除活性、稳定性、选择性外,另一个限制酶的应用范围的因素为酶催化的底物类型、范围非常局限.因此如何拓宽酶的催化底物范围及反应类型为该领域更前沿、更具挑战性的研究课题.对此,F. Arnold课题组致力于对细胞色素P450家族进行改造,赋予其新的催化活性(图9 )[121 -122 ] .细胞色素P450是一种单加氧酶,属于亚铁血红素蛋白酶(Heme)家族,因其在450 nm处存在特异吸收峰而得名.其野生型作用为催化底物的羟基化、环氧化及氧化脱氢等.F. Arnold开发了一系列P450,用以催化重氮化合物与烯烃的环丙化,合成光学纯的环丙烷衍生物,这些方案的共同之处在于利用重氮化合物的氮气逃逸生成卡宾中间体,进而发生烯烃的卡宾插入,得到环丙烷衍生物[123 ] .除烯烃外,该卡宾中间体还可以Si—H、B—H键插入的途径构建C—Si键[124 ] 与C—B键[125 ] ,得到相应的硅烷及硼烷化合物.相比于上述Si—H与B—H键,C—H键更为稳定,因此C—H键的官能化便是一项巨大的挑战,对此常见的策略为过渡金属催化的C—H键活化.F. Arnold则创新性地开发了P411催化叠氮化合物形成氮卡宾,以氮卡宾转移的方式构建C—N键,实现了无金属催化的C—H官能化[126 ] .他们以P411Diane1 为亲本,通过位点饱和诱变引入3个突变氨基酸得到P411Diane2 ,可成功催化叠氮化合物以氮气逃逸的方式形成氮卡宾,选择性插入苄基C—H[106 ] ,进行化学催化难以进行的不对称伯、仲、叔C(sp3 )—H氨基化(图10 ),具有极佳的对映选择性(99.9% ee )与高转化数(72000).值得注意的是,F. Arnold课题组对P450进行的改造,不仅使其获得相较于化学催化更佳的对映选择性,还可实现化学催化难以实现的反应类型.如以P411-S1为亲本,通过定点饱和突变,改变2个氨基酸的P411-E10可催化苯乙炔与重氮乙酸乙酯EDA发生两次连续的卡宾转移(图11 ),得到具有高环张力的双丁烷[127 ] . ...
An enzymatic platform for the asymmetric amination of primary, secondary and tertiary C(sp3 )-H bonds
1
2019
... 除活性、稳定性、选择性外,另一个限制酶的应用范围的因素为酶催化的底物类型、范围非常局限.因此如何拓宽酶的催化底物范围及反应类型为该领域更前沿、更具挑战性的研究课题.对此,F. Arnold课题组致力于对细胞色素P450家族进行改造,赋予其新的催化活性(图9 )[121 -122 ] .细胞色素P450是一种单加氧酶,属于亚铁血红素蛋白酶(Heme)家族,因其在450 nm处存在特异吸收峰而得名.其野生型作用为催化底物的羟基化、环氧化及氧化脱氢等.F. Arnold开发了一系列P450,用以催化重氮化合物与烯烃的环丙化,合成光学纯的环丙烷衍生物,这些方案的共同之处在于利用重氮化合物的氮气逃逸生成卡宾中间体,进而发生烯烃的卡宾插入,得到环丙烷衍生物[123 ] .除烯烃外,该卡宾中间体还可以Si—H、B—H键插入的途径构建C—Si键[124 ] 与C—B键[125 ] ,得到相应的硅烷及硼烷化合物.相比于上述Si—H与B—H键,C—H键更为稳定,因此C—H键的官能化便是一项巨大的挑战,对此常见的策略为过渡金属催化的C—H键活化.F. Arnold则创新性地开发了P411催化叠氮化合物形成氮卡宾,以氮卡宾转移的方式构建C—N键,实现了无金属催化的C—H官能化[126 ] .他们以P411Diane1 为亲本,通过位点饱和诱变引入3个突变氨基酸得到P411Diane2 ,可成功催化叠氮化合物以氮气逃逸的方式形成氮卡宾,选择性插入苄基C—H[106 ] ,进行化学催化难以进行的不对称伯、仲、叔C(sp3 )—H氨基化(图10 ),具有极佳的对映选择性(99.9% ee )与高转化数(72000).值得注意的是,F. Arnold课题组对P450进行的改造,不仅使其获得相较于化学催化更佳的对映选择性,还可实现化学催化难以实现的反应类型.如以P411-S1为亲本,通过定点饱和突变,改变2个氨基酸的P411-E10可催化苯乙炔与重氮乙酸乙酯EDA发生两次连续的卡宾转移(图11 ),得到具有高环张力的双丁烷[127 ] . ...
Enzymatic construction of highly strained carbocycles
1
2018
... 除活性、稳定性、选择性外,另一个限制酶的应用范围的因素为酶催化的底物类型、范围非常局限.因此如何拓宽酶的催化底物范围及反应类型为该领域更前沿、更具挑战性的研究课题.对此,F. Arnold课题组致力于对细胞色素P450家族进行改造,赋予其新的催化活性(图9 )[121 -122 ] .细胞色素P450是一种单加氧酶,属于亚铁血红素蛋白酶(Heme)家族,因其在450 nm处存在特异吸收峰而得名.其野生型作用为催化底物的羟基化、环氧化及氧化脱氢等.F. Arnold开发了一系列P450,用以催化重氮化合物与烯烃的环丙化,合成光学纯的环丙烷衍生物,这些方案的共同之处在于利用重氮化合物的氮气逃逸生成卡宾中间体,进而发生烯烃的卡宾插入,得到环丙烷衍生物[123 ] .除烯烃外,该卡宾中间体还可以Si—H、B—H键插入的途径构建C—Si键[124 ] 与C—B键[125 ] ,得到相应的硅烷及硼烷化合物.相比于上述Si—H与B—H键,C—H键更为稳定,因此C—H键的官能化便是一项巨大的挑战,对此常见的策略为过渡金属催化的C—H键活化.F. Arnold则创新性地开发了P411催化叠氮化合物形成氮卡宾,以氮卡宾转移的方式构建C—N键,实现了无金属催化的C—H官能化[126 ] .他们以P411Diane1 为亲本,通过位点饱和诱变引入3个突变氨基酸得到P411Diane2 ,可成功催化叠氮化合物以氮气逃逸的方式形成氮卡宾,选择性插入苄基C—H[106 ] ,进行化学催化难以进行的不对称伯、仲、叔C(sp3 )—H氨基化(图10 ),具有极佳的对映选择性(99.9% ee )与高转化数(72000).值得注意的是,F. Arnold课题组对P450进行的改造,不仅使其获得相较于化学催化更佳的对映选择性,还可实现化学催化难以实现的反应类型.如以P411-S1为亲本,通过定点饱和突变,改变2个氨基酸的P411-E10可催化苯乙炔与重氮乙酸乙酯EDA发生两次连续的卡宾转移(图11 ),得到具有高环张力的双丁烷[127 ] . ...
Stereodivergent atom-transfer radical cyclization by engineered cytochromes P450
1
2021
... P411除具备催化叠氮化合物,以氮卡宾插入的途径构建C—N键能力外,Y. Yang还报道了以P411BM3 -CIS T438S(P)为亲本,通过位点饱和诱变引入5个突变氨基酸得到P450ATRCase1 ,可催化分子内的原子转移自由基环化(atom transfer radical cyclization,ATRC)[128 ] .对于末端取代烯烃,他们又以P450ATRCase1 为亲本,通过位点饱和诱变引入4个突变氨基酸得到P450ATRCase3 ,可一步得到具有连续两个手性中心的N -取代四氢吡咯衍生物(图12 ). ...
Improved linalool production in Saccharomyces cerevisiae by combining directed evolution of linalool synthase and overexpression of the complete mevalonate pathway
1
2020
... 在合成生物学领域,定向进化常用来改善天然酶的催化性能,进一步实现目标产物前体物质的高效转化.周萍萍课题组[129 ] 致力于高产芳樟醇的酿酒酵母细胞工厂的构建,由于芳樟醇和番茄红素在合成途径中竞争相同的前体物质香叶基焦磷酸(geranyl pyrophosphate,GPP),因此,课题组利用番茄红素的颜色变化建立高通量筛选方法,对芳樟醇合成酶(t67OMcLIS)进行定向进化,最终筛选到了一株活性提高的突变体t670McLIS(E343D/E352H),利用该突变体构建的YLin-05菌株,相比于YLin-04产量提高了52.7%,达到了53.14 mg/L. ...
Artificial metalloenzymes: reaction scope and optimization strategies
1
2018
... 除天然酶外,定向进化也可用于改造人工酶[130 -131 ] .人工酶的构建通常经由两种不同的策略实现,其一是计算机辅助的蛋白质从头设计(de novo )[132 ] ,再则是设计新型人工辅因子(artificial cofactor)制备人工金属酶(artificial metalloenzyme,ArM)[109 ] .通过de novo 设计的人工酶,往往在催化活性与选择性方面表现不佳[111 ] ;而通过替换辅因子的策略制备人工酶的难题则是人工辅因子在酶活性位点的精准定位,同时蛋白骨架对辅因子的兼容性也是一大挑战[133 ] ,定向进化能有效改善上述问题. ...
Design and evolution of chimeric streptavidin for protein-enabled dual gold catalysis
1
2021
... 除天然酶外,定向进化也可用于改造人工酶[130 -131 ] .人工酶的构建通常经由两种不同的策略实现,其一是计算机辅助的蛋白质从头设计(de novo )[132 ] ,再则是设计新型人工辅因子(artificial cofactor)制备人工金属酶(artificial metalloenzyme,ArM)[109 ] .通过de novo 设计的人工酶,往往在催化活性与选择性方面表现不佳[111 ] ;而通过替换辅因子的策略制备人工酶的难题则是人工辅因子在酶活性位点的精准定位,同时蛋白骨架对辅因子的兼容性也是一大挑战[133 ] ,定向进化能有效改善上述问题. ...
De novo protein design by deep network hallucination
1
2021
... 除天然酶外,定向进化也可用于改造人工酶[130 -131 ] .人工酶的构建通常经由两种不同的策略实现,其一是计算机辅助的蛋白质从头设计(de novo )[132 ] ,再则是设计新型人工辅因子(artificial cofactor)制备人工金属酶(artificial metalloenzyme,ArM)[109 ] .通过de novo 设计的人工酶,往往在催化活性与选择性方面表现不佳[111 ] ;而通过替换辅因子的策略制备人工酶的难题则是人工辅因子在酶活性位点的精准定位,同时蛋白骨架对辅因子的兼容性也是一大挑战[133 ] ,定向进化能有效改善上述问题. ...
Abiological catalysis by artificial haem proteins containing noble metals in place of iron
1
2016
... 除天然酶外,定向进化也可用于改造人工酶[130 -131 ] .人工酶的构建通常经由两种不同的策略实现,其一是计算机辅助的蛋白质从头设计(de novo )[132 ] ,再则是设计新型人工辅因子(artificial cofactor)制备人工金属酶(artificial metalloenzyme,ArM)[109 ] .通过de novo 设计的人工酶,往往在催化活性与选择性方面表现不佳[111 ] ;而通过替换辅因子的策略制备人工酶的难题则是人工辅因子在酶活性位点的精准定位,同时蛋白骨架对辅因子的兼容性也是一大挑战[133 ] ,定向进化能有效改善上述问题. ...
Computational design of catalytic dyads and oxyanion holes for ester hydrolysis
1
2012
... 具有α-H的醛/酮,在酸或碱催化下与另一分子的醛/酮进行亲核加成,生成β-羟基醛/酮的反应,称为羟醛缩合(aldol)反应.该反应是一种重要的构建C—C键的策略,常用于实现碳链的增长.人工设计的Ⅰ类缩醛酶可通过活性位点赖氨酸和底物形成席夫碱,催化C—C键的可逆裂解,然而较低的活性与选择性无法使其成为有效的催化剂.D. Hilvert与D. Baker合作,将计算设计与盒式诱变相结合,引入12个氨基酸突变,得到了比先前设计的人工Ⅰ类醛缩酶活性提高了大于109 倍的DA7[134 ] ,且所得的酶催化可逆Aldol反应时具有优秀的立体选择性与良好的底物普适性(图13 ).他们对酶的生化结构进行了探究,发现该Aldol缩合酶的催化活性取决于在定向进化过程中出现在计算设计的疏水口袋附近的Lys-Tyr-Asn-Tyr四联体.该缩合酶的作用为通过碱基与羰基形成席夫碱从而激活底物,并促进质子转移,稳定多个过渡态,实现了高效的Aldol反应及可逆Aldol缩合. ...
The aza-hexadehydro-diels-alder reaction
1
2019
... 双烯体与亲双烯体的[4+2]环加成,得到六元环的反应称为Diels-Alder(D-A)反应,是有机合成中非常重要的构建C—C键的手段之一,广泛运用于各种药物的全合成.该反应有丰富的立体选择性,可一次生成两个C—C键和最多四个相邻的手性碳.近年来,该领域最前沿的方向即为通过杂D-A反应[135 ] 构建药物优势骨架六元杂环.D-A反应通常由Lewis酸所催化,在高温下进行.此外,天然酶未被证明可催化该反应[136 ] .D. Hilvert课题组创制了可在温和条件下催化D-A反应的人工酶DA_20_10,并结合靶向诱变、计算改进和epPCR,经过8轮连续定向进化,在DA_20_10上引入5个氨基酸突变(R50H,V96I,T197R,E288D和L309S),最终得到了具备更高催化活性的DA_20_20,高效地催化了1,3-丁二烯氨基甲酸酯与二甲基丙烯酰胺(图14 )的[4+2]环加成[115 ] ,以高产率得到环己烯衍生物. ...
Impact of scaffold rigidity on the design and evolution of an artificial Diels-Alderase
1
2014
... 双烯体与亲双烯体的[4+2]环加成,得到六元环的反应称为Diels-Alder(D-A)反应,是有机合成中非常重要的构建C—C键的手段之一,广泛运用于各种药物的全合成.该反应有丰富的立体选择性,可一次生成两个C—C键和最多四个相邻的手性碳.近年来,该领域最前沿的方向即为通过杂D-A反应[135 ] 构建药物优势骨架六元杂环.D-A反应通常由Lewis酸所催化,在高温下进行.此外,天然酶未被证明可催化该反应[136 ] .D. Hilvert课题组创制了可在温和条件下催化D-A反应的人工酶DA_20_10,并结合靶向诱变、计算改进和epPCR,经过8轮连续定向进化,在DA_20_10上引入5个氨基酸突变(R50H,V96I,T197R,E288D和L309S),最终得到了具备更高催化活性的DA_20_20,高效地催化了1,3-丁二烯氨基甲酸酯与二甲基丙烯酰胺(图14 )的[4+2]环加成[115 ] ,以高产率得到环己烯衍生物. ...
Directed evolution of artificial metalloenzymes for in vivo metathesis
1
2016
... 人工金属酶(artificial metalloenzyme,ArM)将具备催化活性的金属辅因子与蛋白质骨架相结合,赋予蛋白催化活性,即构建了新型酶;同时也解决了金属催化剂在水相体系中的失活问题.ArM的设计难点在于如何将金属有效地结合到酶活性位点部位,即金属辅因子的定位[109 ] .幸运的是,目前已有多种方法被报道,其中运用最广泛同时也最成熟的方法为基于生物素-亲和素(biotin-avidin)系统的特异性结合[137 ] 与制备人工金属辅因子进行替换[112 ] . ...
Synthesis of trisubstituted alkenes via olefin cross-metathesis
1
1999
... 金属催化的烯烃间C=C切断并重新结合的烯烃复分解反应,广泛应用于药物的研发和先进聚合物材料的制备.其中R. Grubbs[138 ] 开发了钌-卡宾复合物体系,成为应用最广泛的烯烃复分解催化剂,即Grubbs催化剂.虽然Grubbs催化剂在体系中存在少量水、醇、酸的情况下仍可保持催化活性,但水相中的烯烃复分解反应却鲜有报道.同时自然界中并不存在该类反应,因此无法利用天然酶成功催化该反应.T. Ward课题组用采用生物素-链霉亲和素(Biotin-Streptavidin)方法,将钌(Ru)锚定到链霉亲和素(streptavidin,SAV)支架中,制备了Biot-Ru-SAV,实现了在水相溶剂中催化烯烃复分解反应.同时,他们还通过多轮饱和突变,引入20个氨基酸突变,得到与Ru离子结合更稳定的SAVmut,在赋予酶新功能的同时,解决了金属催化剂在水和细胞环境下表现不佳的问题.值得一提的是,利用SAV-Biot方法进行制备ArM时,金属辅因子的活性可能被细胞质中成分抑制,因此通常需对蛋白支架SAV进行纯化、体外组装Biot-Ru-SAV,该过程烦琐,不利于高通量筛选Biot-Ru-SAV突变体库.T. Ward[116 ] 创新性地在细胞周质中表达SAV,随后通过自组装的方式制备Biot-Ru-SAV,并在体内实现了闭环烯烃复分解(图15 ),该方法不但加速了Biot-Ru-SAV突变库筛选,还为构造非天然的代谢通路提供了可能. ...
Unnatural biosynthesis by an engineered microorganism with heterologously expressed natural enzymes and an artificial metalloenzyme
1
2021
... 天然金属酶催化的反应范围受到金属中心固有反应性的限制,为了克服这一局限性,J. Hartwig课题组[112 ] 开发了金属替代方法,体外合成铱卟啉以替代铁卟啉作为抹香鲸来源肌红蛋白的辅因子,构建了新型人造血红素蛋白.此前金属替代铁卟啉的方法多需要利用蛋白体外变性,同时加入抑制剂竞争性结合铁卟啉,该过程需要多步的蛋白纯化步骤[112 ] .近期,J. Hartwig课题组[139 ] 实现了通过转运蛋白将铱卟啉辅因子转运入胞内,与蛋白骨架Cyp119进行装配,这为快速进化通过辅酶因子替换而得的金属酶提供了可能.他们通过位点饱和诱变,引入两个突变氨基酸(R256W,V254A),得到Ir-Cyp119-P/R256W/V254A,并创造性地将柠檬烯的生物合成途径与Ir催化的卡宾转移相结合,实现了环丙基柠檬烯的生物合成[图17 (a)],为代谢通路的改造提供了可能.此外,他们还实现了胞内Ir-Cyp119催化的对映选择性、区域选择性C—H活化[图17 (b)]. ...
Directed evolution of artificial metalloenzymes in whole cells
1
2022
... 相比于天然酶,利用该方法进行构建新型人工金属酶时,涉及到游离的天然辅因子的去除以及人工辅因子的引入[140 ] .而单个宿主蛋白的纯化十分耗时,因此辅因子和蛋白的体外重组限制了高通量筛选和定向进化的速率.对此,J. Hartwig课题组[119 ] 在原有研究基础上,将辅因子转移到细胞质中,实现了全细胞中人工金属酶的构建. ...
Advances in ultrahigh-throughput screening for directed enzyme evolution
1
2020
... 定向进化过程中亟待解决的问题是筛选和选择的通量,随机突变技术的突变范围通常包括整个蛋白质序列,面对其产生的极为庞大的突变文库,开发自动化、高通量筛选技术对缩短实验周期和提高突变覆盖率来说都必不可少[141 ] .现今已开发的多种高通量筛选技术中,多以光谱和质谱为检测手段.FACS是其中应用较为广泛的一种,其优点在于极高的筛选速率,但筛选依赖光散射或荧光信号,在面对无光学特性的酶反应时通常需要偶联额外的过程以引入光信号的改变,即使如此FACS仍无法应对所有酶的筛选;而MALDI- MS虽然可以对样品进行无标记分析,但其样品制备通常较为复杂,限制了其在高通量筛选中的应用;另外一种常用技术——微流控技术,同样需要依赖基于荧光等信号的响应,这使得其无法应对所有酶的筛选.一些现有的研究方法揭示了高通量筛选技术发展方向,即将不同的方法进行结合,例如微流控技术和MALDI-MS结合等方法,这些结合的方法通常能够结合双方的优点,用于分析单个方法难以处理的样品. ...
Engineering biological systems using automated biofoundries
1
2017
... 另外,面对数据资源的快速增长,将自动化技术应用于“设计—构建—测试—学习”的循环以建立一个全自动生物合成平台,能够有效提高酶的进化效率.目前,已有部分自动化生物合成平台或方法被建立并投入使用,例如伊利诺伊大学的iBioFAB(illinois biological foundry for advanced biomanufacturing)[142 ] 、中国科学院天津工业生物技术研究所的MACBETH(multiplex automated Corynebacterium glutamicum base editing method)[68 ] 等,这些方法通过计算机与自动化设备实现了某些宿主或定向进化中某些步骤的自动化,然而这尚不能实现定向进化过程的完全自动.在未来,随着突变与筛选过程中的实验步骤自动化技术以及人工智能技术的不断精进,智能制造的理念将不断引入合成生物学,定向进化也终将走向全自动. ...
Deep diversification of an AAV capsid protein by machine learning
1
2021
... 除了提高通量,构建精准的突变体库也是提高定向进化效率的关键,其中最具代表性的是M. Reetz教授团队所发展的CSAT、ISM、FRISM等一系列方法,以及Twist Bioscience公司与M. Reetz教授合作,在2018年报道的依托高通量合成DNA的体外突变体库构建方法已经用于商业化[18 ] .得益于测序技术的发展,未被解析的氨基酸序列数量飞速增长,机器学习技术在预测蛋白结构、功能及活性位点等领域取得了一定成功[91 ] .然而,目前主流的两种机器学习方法仍然需要依赖庞大、多样且高质量数据点来完成学习并做出较为精确的预测[143 ] ,而蛋白质序列及折叠方式的复杂性也使得现有算法无法同时预测两个或以上的性质.AlphaFold 2将利用机器学习预测蛋白质结构的研究推向了一个新的巅峰,其已经成功预测了超过30万种蛋白质的结构,但并未能准确预测庞大的序列数据库对应的所有蛋白序列.另外,对于蛋白质功能的预测目前完全依赖基于样本的机器学习模型构建或同源分析,而现有算法虽可以通过缩小突变体库来辅助定向进化,但其精确性也较大依赖于基于对蛋白质结构及功能的理解所设定的限制条件或突变倾向[144 ] .因此,如何减少建模方法对人工设置的依赖和实现自主模型的构建、如何通过较少的样本库来达到较高的预测精确度、如何同时模拟预测多个性状都将是未来探索的重点方向. ...
Informed training set design enables efficient machine learning-assisted directed protein evolution
1
2021
... 除了提高通量,构建精准的突变体库也是提高定向进化效率的关键,其中最具代表性的是M. Reetz教授团队所发展的CSAT、ISM、FRISM等一系列方法,以及Twist Bioscience公司与M. Reetz教授合作,在2018年报道的依托高通量合成DNA的体外突变体库构建方法已经用于商业化[18 ] .得益于测序技术的发展,未被解析的氨基酸序列数量飞速增长,机器学习技术在预测蛋白结构、功能及活性位点等领域取得了一定成功[91 ] .然而,目前主流的两种机器学习方法仍然需要依赖庞大、多样且高质量数据点来完成学习并做出较为精确的预测[143 ] ,而蛋白质序列及折叠方式的复杂性也使得现有算法无法同时预测两个或以上的性质.AlphaFold 2将利用机器学习预测蛋白质结构的研究推向了一个新的巅峰,其已经成功预测了超过30万种蛋白质的结构,但并未能准确预测庞大的序列数据库对应的所有蛋白序列.另外,对于蛋白质功能的预测目前完全依赖基于样本的机器学习模型构建或同源分析,而现有算法虽可以通过缩小突变体库来辅助定向进化,但其精确性也较大依赖于基于对蛋白质结构及功能的理解所设定的限制条件或突变倾向[144 ] .因此,如何减少建模方法对人工设置的依赖和实现自主模型的构建、如何通过较少的样本库来达到较高的预测精确度、如何同时模拟预测多个性状都将是未来探索的重点方向. ...



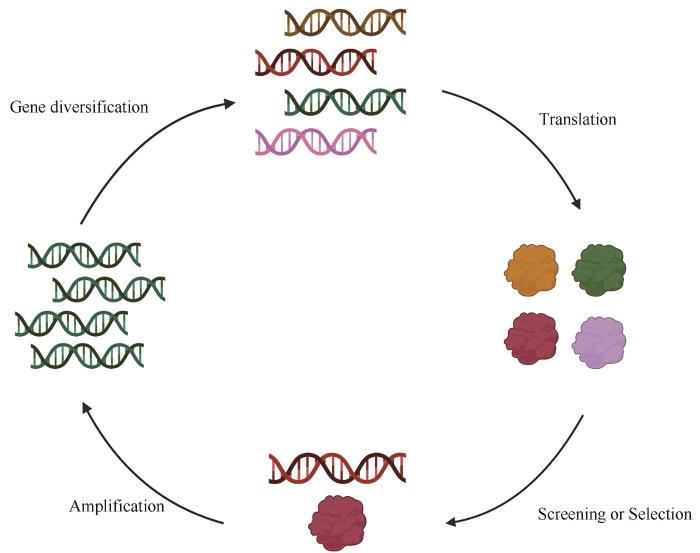

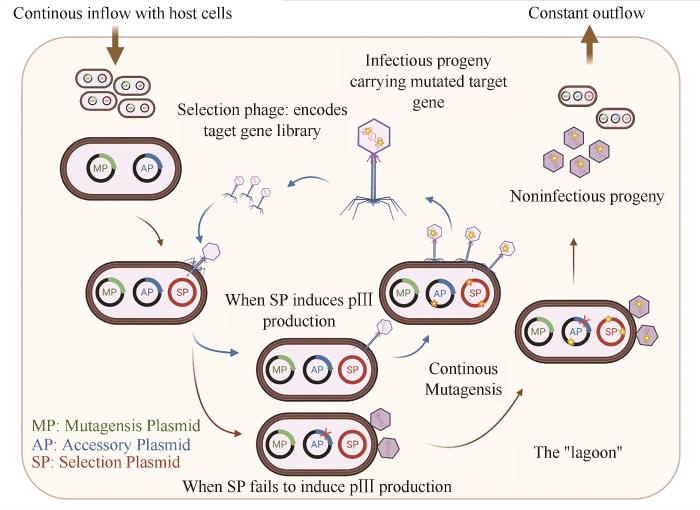

 73]
73]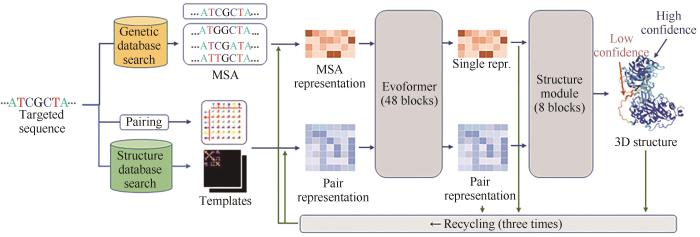

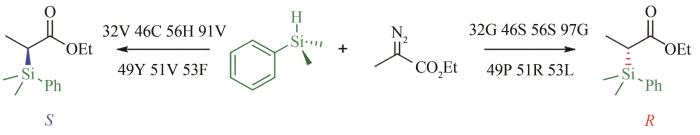
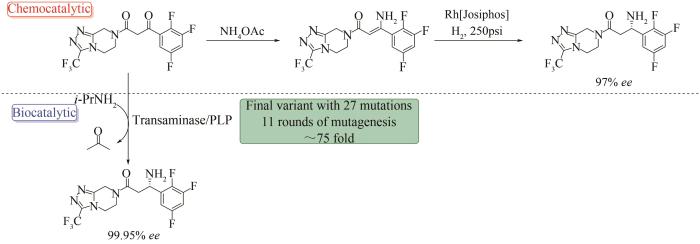




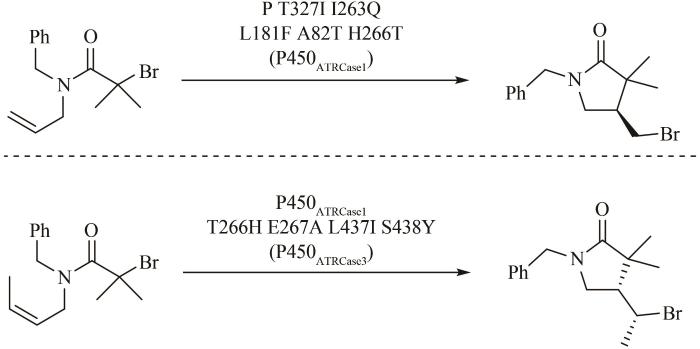
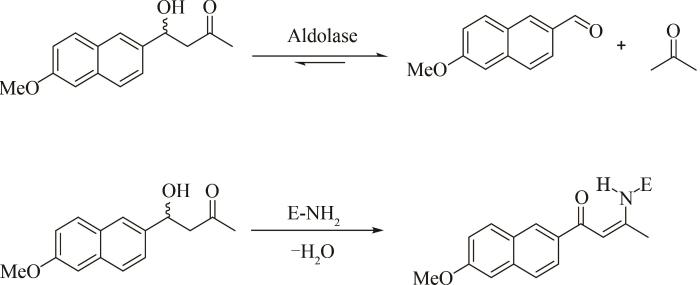



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}