The coming of age of de novo protein design
1
2016
... 蛋白质的氨基酸序列排布方式决定了其折叠后结构和活性功能.对于一个链长为100个氨基酸的蛋白质,其可能的氨基酸序列组合有20100 种.在如此广大的序列空间内进行氨基酸序列的优化搜索面临着巨大的困难[1 ] .蛋白计算设计避免了相对随机的突变策略,并提供了基于蛋白质的生物物理和生物化学原理的指导性设计蓝图.计算蛋白质设计的目标是设计一个能够折叠成预定义的结构且具有所需功能的氨基酸序列,通常会从一个已知的结构出发,保留活性位点,并修改部分序列以提高所设计蛋白质的稳定性或实现新的功能[2 , 3 ] . ...
Automated design of efficient and functionally diverse enzyme repertoires
1
2018
... 蛋白质的氨基酸序列排布方式决定了其折叠后结构和活性功能.对于一个链长为100个氨基酸的蛋白质,其可能的氨基酸序列组合有20100 种.在如此广大的序列空间内进行氨基酸序列的优化搜索面临着巨大的困难[1 ] .蛋白计算设计避免了相对随机的突变策略,并提供了基于蛋白质的生物物理和生物化学原理的指导性设计蓝图.计算蛋白质设计的目标是设计一个能够折叠成预定义的结构且具有所需功能的氨基酸序列,通常会从一个已知的结构出发,保留活性位点,并修改部分序列以提高所设计蛋白质的稳定性或实现新的功能[2 , 3 ] . ...
Computational design of a modular protein sense-response system
2
2019
... 蛋白质的氨基酸序列排布方式决定了其折叠后结构和活性功能.对于一个链长为100个氨基酸的蛋白质,其可能的氨基酸序列组合有20100 种.在如此广大的序列空间内进行氨基酸序列的优化搜索面临着巨大的困难[1 ] .蛋白计算设计避免了相对随机的突变策略,并提供了基于蛋白质的生物物理和生物化学原理的指导性设计蓝图.计算蛋白质设计的目标是设计一个能够折叠成预定义的结构且具有所需功能的氨基酸序列,通常会从一个已知的结构出发,保留活性位点,并修改部分序列以提高所设计蛋白质的稳定性或实现新的功能[2 , 3 ] . ...
... 2022年许锦波组提出了一种直接在三维坐标空间中对蛋白质结构进行建模的、基于VAE的模型[95 ] ,相比于先前提出的直接坐标生成模型[3 ] 其应用仅限于固定长度的蛋白质,新提出的模型通过提取蛋白质几何形状的不变表示(Invariant representations),并使用局部对齐的坐标损失函数直接在坐标空间上执行梯度优化,解决了输入和输出空间中的旋转和平移等方差,因此可以直接、灵活地对三维结构进行建模. ...
Principles that govern the folding of protein chains
1
1973
... 依据Anfinsen的折叠热力学假说[4 ] ,蛋白质折叠到最低自由能状态,其3D结构由氨基酸序列决定.然而,在折叠过程中最重要的不是折叠态的绝对能量,而是折叠态与最低的备选态之间的能量差.这种计算不仅涉及到所有可能的氨基酸序列,而且涉及到所有可能的结构,因此多数现有的方法都集中在寻找所需结构的最低能量氨基酸序列这个更容易处理的问题上.目前广泛使用的方法仍然是基于能量函数和启发式采样方法的算法[5 ] .RosettaDesign[6 ] 、FoldX[7 ] 、EvoDesign / EvoEF2[8 ] 等设计方法使用使用蛋白质结构参数化的打分项来量化氨基酸序列和特定三维结构之间的匹配度,其中RosettaDesign是目前使用最为广泛的方法之一.RosettaDesign采用能量函数[9 ] 来捕捉序列-结构关系,对结构中每个残基侧链的氨基酸种类和构象进行采样,并使用蒙特卡洛模拟退火等方法进行优化以获得低能序列和构象.在过去的三十年中,基于能量函数的蛋白计算设计取得了巨大的进展,包括设计新颖的3D折叠[10 ] 、酶[11 ] 和复合物[11 ] ,更包括免疫信号[12 -13 ] 、靶向治疗蛋白[14 -15 ] 、蛋白质开关[16 -17 ] 、自组装蛋白[18 -19 ] 等.尽管取得了这些成功,但是基于能量函数的蛋白质设计方法准确度仍然较低,在没有多轮实验试错的情况下无法可靠使用,导致蛋白设计实验成功率难以提升[20 ] . ...
Scientific benchmarks for guiding macromolecular energy function improvement
1
2013
... 依据Anfinsen的折叠热力学假说[4 ] ,蛋白质折叠到最低自由能状态,其3D结构由氨基酸序列决定.然而,在折叠过程中最重要的不是折叠态的绝对能量,而是折叠态与最低的备选态之间的能量差.这种计算不仅涉及到所有可能的氨基酸序列,而且涉及到所有可能的结构,因此多数现有的方法都集中在寻找所需结构的最低能量氨基酸序列这个更容易处理的问题上.目前广泛使用的方法仍然是基于能量函数和启发式采样方法的算法[5 ] .RosettaDesign[6 ] 、FoldX[7 ] 、EvoDesign / EvoEF2[8 ] 等设计方法使用使用蛋白质结构参数化的打分项来量化氨基酸序列和特定三维结构之间的匹配度,其中RosettaDesign是目前使用最为广泛的方法之一.RosettaDesign采用能量函数[9 ] 来捕捉序列-结构关系,对结构中每个残基侧链的氨基酸种类和构象进行采样,并使用蒙特卡洛模拟退火等方法进行优化以获得低能序列和构象.在过去的三十年中,基于能量函数的蛋白计算设计取得了巨大的进展,包括设计新颖的3D折叠[10 ] 、酶[11 ] 和复合物[11 ] ,更包括免疫信号[12 -13 ] 、靶向治疗蛋白[14 -15 ] 、蛋白质开关[16 -17 ] 、自组装蛋白[18 -19 ] 等.尽管取得了这些成功,但是基于能量函数的蛋白质设计方法准确度仍然较低,在没有多轮实验试错的情况下无法可靠使用,导致蛋白设计实验成功率难以提升[20 ] . ...
Macromolecular modeling and design in Rosetta: Recent methods and frameworks
1
2020
... 依据Anfinsen的折叠热力学假说[4 ] ,蛋白质折叠到最低自由能状态,其3D结构由氨基酸序列决定.然而,在折叠过程中最重要的不是折叠态的绝对能量,而是折叠态与最低的备选态之间的能量差.这种计算不仅涉及到所有可能的氨基酸序列,而且涉及到所有可能的结构,因此多数现有的方法都集中在寻找所需结构的最低能量氨基酸序列这个更容易处理的问题上.目前广泛使用的方法仍然是基于能量函数和启发式采样方法的算法[5 ] .RosettaDesign[6 ] 、FoldX[7 ] 、EvoDesign / EvoEF2[8 ] 等设计方法使用使用蛋白质结构参数化的打分项来量化氨基酸序列和特定三维结构之间的匹配度,其中RosettaDesign是目前使用最为广泛的方法之一.RosettaDesign采用能量函数[9 ] 来捕捉序列-结构关系,对结构中每个残基侧链的氨基酸种类和构象进行采样,并使用蒙特卡洛模拟退火等方法进行优化以获得低能序列和构象.在过去的三十年中,基于能量函数的蛋白计算设计取得了巨大的进展,包括设计新颖的3D折叠[10 ] 、酶[11 ] 和复合物[11 ] ,更包括免疫信号[12 -13 ] 、靶向治疗蛋白[14 -15 ] 、蛋白质开关[16 -17 ] 、自组装蛋白[18 -19 ] 等.尽管取得了这些成功,但是基于能量函数的蛋白质设计方法准确度仍然较低,在没有多轮实验试错的情况下无法可靠使用,导致蛋白设计实验成功率难以提升[20 ] . ...
Chapter one-DNA-binding specificity prediction with FoldX
1
2011
... 依据Anfinsen的折叠热力学假说[4 ] ,蛋白质折叠到最低自由能状态,其3D结构由氨基酸序列决定.然而,在折叠过程中最重要的不是折叠态的绝对能量,而是折叠态与最低的备选态之间的能量差.这种计算不仅涉及到所有可能的氨基酸序列,而且涉及到所有可能的结构,因此多数现有的方法都集中在寻找所需结构的最低能量氨基酸序列这个更容易处理的问题上.目前广泛使用的方法仍然是基于能量函数和启发式采样方法的算法[5 ] .RosettaDesign[6 ] 、FoldX[7 ] 、EvoDesign / EvoEF2[8 ] 等设计方法使用使用蛋白质结构参数化的打分项来量化氨基酸序列和特定三维结构之间的匹配度,其中RosettaDesign是目前使用最为广泛的方法之一.RosettaDesign采用能量函数[9 ] 来捕捉序列-结构关系,对结构中每个残基侧链的氨基酸种类和构象进行采样,并使用蒙特卡洛模拟退火等方法进行优化以获得低能序列和构象.在过去的三十年中,基于能量函数的蛋白计算设计取得了巨大的进展,包括设计新颖的3D折叠[10 ] 、酶[11 ] 和复合物[11 ] ,更包括免疫信号[12 -13 ] 、靶向治疗蛋白[14 -15 ] 、蛋白质开关[16 -17 ] 、自组装蛋白[18 -19 ] 等.尽管取得了这些成功,但是基于能量函数的蛋白质设计方法准确度仍然较低,在没有多轮实验试错的情况下无法可靠使用,导致蛋白设计实验成功率难以提升[20 ] . ...
EvoEF2: accurate and fast energy function for computational protein design
1
2020
... 依据Anfinsen的折叠热力学假说[4 ] ,蛋白质折叠到最低自由能状态,其3D结构由氨基酸序列决定.然而,在折叠过程中最重要的不是折叠态的绝对能量,而是折叠态与最低的备选态之间的能量差.这种计算不仅涉及到所有可能的氨基酸序列,而且涉及到所有可能的结构,因此多数现有的方法都集中在寻找所需结构的最低能量氨基酸序列这个更容易处理的问题上.目前广泛使用的方法仍然是基于能量函数和启发式采样方法的算法[5 ] .RosettaDesign[6 ] 、FoldX[7 ] 、EvoDesign / EvoEF2[8 ] 等设计方法使用使用蛋白质结构参数化的打分项来量化氨基酸序列和特定三维结构之间的匹配度,其中RosettaDesign是目前使用最为广泛的方法之一.RosettaDesign采用能量函数[9 ] 来捕捉序列-结构关系,对结构中每个残基侧链的氨基酸种类和构象进行采样,并使用蒙特卡洛模拟退火等方法进行优化以获得低能序列和构象.在过去的三十年中,基于能量函数的蛋白计算设计取得了巨大的进展,包括设计新颖的3D折叠[10 ] 、酶[11 ] 和复合物[11 ] ,更包括免疫信号[12 -13 ] 、靶向治疗蛋白[14 -15 ] 、蛋白质开关[16 -17 ] 、自组装蛋白[18 -19 ] 等.尽管取得了这些成功,但是基于能量函数的蛋白质设计方法准确度仍然较低,在没有多轮实验试错的情况下无法可靠使用,导致蛋白设计实验成功率难以提升[20 ] . ...
The Rosetta all-atom energy function for macromolecular modeling and design
1
2017
... 依据Anfinsen的折叠热力学假说[4 ] ,蛋白质折叠到最低自由能状态,其3D结构由氨基酸序列决定.然而,在折叠过程中最重要的不是折叠态的绝对能量,而是折叠态与最低的备选态之间的能量差.这种计算不仅涉及到所有可能的氨基酸序列,而且涉及到所有可能的结构,因此多数现有的方法都集中在寻找所需结构的最低能量氨基酸序列这个更容易处理的问题上.目前广泛使用的方法仍然是基于能量函数和启发式采样方法的算法[5 ] .RosettaDesign[6 ] 、FoldX[7 ] 、EvoDesign / EvoEF2[8 ] 等设计方法使用使用蛋白质结构参数化的打分项来量化氨基酸序列和特定三维结构之间的匹配度,其中RosettaDesign是目前使用最为广泛的方法之一.RosettaDesign采用能量函数[9 ] 来捕捉序列-结构关系,对结构中每个残基侧链的氨基酸种类和构象进行采样,并使用蒙特卡洛模拟退火等方法进行优化以获得低能序列和构象.在过去的三十年中,基于能量函数的蛋白计算设计取得了巨大的进展,包括设计新颖的3D折叠[10 ] 、酶[11 ] 和复合物[11 ] ,更包括免疫信号[12 -13 ] 、靶向治疗蛋白[14 -15 ] 、蛋白质开关[16 -17 ] 、自组装蛋白[18 -19 ] 等.尽管取得了这些成功,但是基于能量函数的蛋白质设计方法准确度仍然较低,在没有多轮实验试错的情况下无法可靠使用,导致蛋白设计实验成功率难以提升[20 ] . ...
Design of a novel globular protein fold with atomic-level accuracy
2
2003
... 依据Anfinsen的折叠热力学假说[4 ] ,蛋白质折叠到最低自由能状态,其3D结构由氨基酸序列决定.然而,在折叠过程中最重要的不是折叠态的绝对能量,而是折叠态与最低的备选态之间的能量差.这种计算不仅涉及到所有可能的氨基酸序列,而且涉及到所有可能的结构,因此多数现有的方法都集中在寻找所需结构的最低能量氨基酸序列这个更容易处理的问题上.目前广泛使用的方法仍然是基于能量函数和启发式采样方法的算法[5 ] .RosettaDesign[6 ] 、FoldX[7 ] 、EvoDesign / EvoEF2[8 ] 等设计方法使用使用蛋白质结构参数化的打分项来量化氨基酸序列和特定三维结构之间的匹配度,其中RosettaDesign是目前使用最为广泛的方法之一.RosettaDesign采用能量函数[9 ] 来捕捉序列-结构关系,对结构中每个残基侧链的氨基酸种类和构象进行采样,并使用蒙特卡洛模拟退火等方法进行优化以获得低能序列和构象.在过去的三十年中,基于能量函数的蛋白计算设计取得了巨大的进展,包括设计新颖的3D折叠[10 ] 、酶[11 ] 和复合物[11 ] ,更包括免疫信号[12 -13 ] 、靶向治疗蛋白[14 -15 ] 、蛋白质开关[16 -17 ] 、自组装蛋白[18 -19 ] 等.尽管取得了这些成功,但是基于能量函数的蛋白质设计方法准确度仍然较低,在没有多轮实验试错的情况下无法可靠使用,导致蛋白设计实验成功率难以提升[20 ] . ...
... 在模板未知条件下,使用神经网络形式的能量项模型SCUBA 驱动的随机动力学(Stochastic dynamics)和模拟退火算法(Simulated annealing)来生成可设计的新蛋白质主链骨架,再使用前文中提到的ABACUS2[69 ] 对主链骨架序列进行序列优化和骨架松弛[10 ] 设计的迭代,从而完成对蛋白质的可变骨架从头设计任务.在9种用SCUBA设计的高精度骨架蛋白结构中,其中有4种具有新颖的非天然结构.对于这一结果充分展示了SCUBA在蛋白设计中的实用性,特别是在设计功能蛋白时,能量函数驱动的骨架采样和优化可以很容易地进行定制,以促进对结构空间的广泛探索.另外,SCUBA+ ABACUS2[87 ] 策略所设计的蛋白质具有高于天然蛋白质骨架的热稳定性,设计成功率为~ 42 % ( 38个经实验验证的蛋白质中有16个成功折叠,14个H2E4蛋白质和4个H4蛋白质),设计的骨架与实验获得的结构一致,具有原子精度,同时设计的H2E4和H4蛋白与具有相似结构的已知天然蛋白质具有低序列同一性(平均同一性14%). ...
Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction
2
2010
... 依据Anfinsen的折叠热力学假说[4 ] ,蛋白质折叠到最低自由能状态,其3D结构由氨基酸序列决定.然而,在折叠过程中最重要的不是折叠态的绝对能量,而是折叠态与最低的备选态之间的能量差.这种计算不仅涉及到所有可能的氨基酸序列,而且涉及到所有可能的结构,因此多数现有的方法都集中在寻找所需结构的最低能量氨基酸序列这个更容易处理的问题上.目前广泛使用的方法仍然是基于能量函数和启发式采样方法的算法[5 ] .RosettaDesign[6 ] 、FoldX[7 ] 、EvoDesign / EvoEF2[8 ] 等设计方法使用使用蛋白质结构参数化的打分项来量化氨基酸序列和特定三维结构之间的匹配度,其中RosettaDesign是目前使用最为广泛的方法之一.RosettaDesign采用能量函数[9 ] 来捕捉序列-结构关系,对结构中每个残基侧链的氨基酸种类和构象进行采样,并使用蒙特卡洛模拟退火等方法进行优化以获得低能序列和构象.在过去的三十年中,基于能量函数的蛋白计算设计取得了巨大的进展,包括设计新颖的3D折叠[10 ] 、酶[11 ] 和复合物[11 ] ,更包括免疫信号[12 -13 ] 、靶向治疗蛋白[14 -15 ] 、蛋白质开关[16 -17 ] 、自组装蛋白[18 -19 ] 等.尽管取得了这些成功,但是基于能量函数的蛋白质设计方法准确度仍然较低,在没有多轮实验试错的情况下无法可靠使用,导致蛋白设计实验成功率难以提升[20 ] . ...
... [11 ],更包括免疫信号[12 -13 ] 、靶向治疗蛋白[14 -15 ] 、蛋白质开关[16 -17 ] 、自组装蛋白[18 -19 ] 等.尽管取得了这些成功,但是基于能量函数的蛋白质设计方法准确度仍然较低,在没有多轮实验试错的情况下无法可靠使用,导致蛋白设计实验成功率难以提升[20 ] . ...
De novo design of potent and selective mimics of IL-2 and IL-15
1
2019
... 依据Anfinsen的折叠热力学假说[4 ] ,蛋白质折叠到最低自由能状态,其3D结构由氨基酸序列决定.然而,在折叠过程中最重要的不是折叠态的绝对能量,而是折叠态与最低的备选态之间的能量差.这种计算不仅涉及到所有可能的氨基酸序列,而且涉及到所有可能的结构,因此多数现有的方法都集中在寻找所需结构的最低能量氨基酸序列这个更容易处理的问题上.目前广泛使用的方法仍然是基于能量函数和启发式采样方法的算法[5 ] .RosettaDesign[6 ] 、FoldX[7 ] 、EvoDesign / EvoEF2[8 ] 等设计方法使用使用蛋白质结构参数化的打分项来量化氨基酸序列和特定三维结构之间的匹配度,其中RosettaDesign是目前使用最为广泛的方法之一.RosettaDesign采用能量函数[9 ] 来捕捉序列-结构关系,对结构中每个残基侧链的氨基酸种类和构象进行采样,并使用蒙特卡洛模拟退火等方法进行优化以获得低能序列和构象.在过去的三十年中,基于能量函数的蛋白计算设计取得了巨大的进展,包括设计新颖的3D折叠[10 ] 、酶[11 ] 和复合物[11 ] ,更包括免疫信号[12 -13 ] 、靶向治疗蛋白[14 -15 ] 、蛋白质开关[16 -17 ] 、自组装蛋白[18 -19 ] 等.尽管取得了这些成功,但是基于能量函数的蛋白质设计方法准确度仍然较低,在没有多轮实验试错的情况下无法可靠使用,导致蛋白设计实验成功率难以提升[20 ] . ...
Topological control of cytokine receptor signaling induces differential effects in hematopoiesis
1
2019
... 依据Anfinsen的折叠热力学假说[4 ] ,蛋白质折叠到最低自由能状态,其3D结构由氨基酸序列决定.然而,在折叠过程中最重要的不是折叠态的绝对能量,而是折叠态与最低的备选态之间的能量差.这种计算不仅涉及到所有可能的氨基酸序列,而且涉及到所有可能的结构,因此多数现有的方法都集中在寻找所需结构的最低能量氨基酸序列这个更容易处理的问题上.目前广泛使用的方法仍然是基于能量函数和启发式采样方法的算法[5 ] .RosettaDesign[6 ] 、FoldX[7 ] 、EvoDesign / EvoEF2[8 ] 等设计方法使用使用蛋白质结构参数化的打分项来量化氨基酸序列和特定三维结构之间的匹配度,其中RosettaDesign是目前使用最为广泛的方法之一.RosettaDesign采用能量函数[9 ] 来捕捉序列-结构关系,对结构中每个残基侧链的氨基酸种类和构象进行采样,并使用蒙特卡洛模拟退火等方法进行优化以获得低能序列和构象.在过去的三十年中,基于能量函数的蛋白计算设计取得了巨大的进展,包括设计新颖的3D折叠[10 ] 、酶[11 ] 和复合物[11 ] ,更包括免疫信号[12 -13 ] 、靶向治疗蛋白[14 -15 ] 、蛋白质开关[16 -17 ] 、自组装蛋白[18 -19 ] 等.尽管取得了这些成功,但是基于能量函数的蛋白质设计方法准确度仍然较低,在没有多轮实验试错的情况下无法可靠使用,导致蛋白设计实验成功率难以提升[20 ] . ...
Massively parallel de novo protein design for targeted therapeutics
1
2017
... 依据Anfinsen的折叠热力学假说[4 ] ,蛋白质折叠到最低自由能状态,其3D结构由氨基酸序列决定.然而,在折叠过程中最重要的不是折叠态的绝对能量,而是折叠态与最低的备选态之间的能量差.这种计算不仅涉及到所有可能的氨基酸序列,而且涉及到所有可能的结构,因此多数现有的方法都集中在寻找所需结构的最低能量氨基酸序列这个更容易处理的问题上.目前广泛使用的方法仍然是基于能量函数和启发式采样方法的算法[5 ] .RosettaDesign[6 ] 、FoldX[7 ] 、EvoDesign / EvoEF2[8 ] 等设计方法使用使用蛋白质结构参数化的打分项来量化氨基酸序列和特定三维结构之间的匹配度,其中RosettaDesign是目前使用最为广泛的方法之一.RosettaDesign采用能量函数[9 ] 来捕捉序列-结构关系,对结构中每个残基侧链的氨基酸种类和构象进行采样,并使用蒙特卡洛模拟退火等方法进行优化以获得低能序列和构象.在过去的三十年中,基于能量函数的蛋白计算设计取得了巨大的进展,包括设计新颖的3D折叠[10 ] 、酶[11 ] 和复合物[11 ] ,更包括免疫信号[12 -13 ] 、靶向治疗蛋白[14 -15 ] 、蛋白质开关[16 -17 ] 、自组装蛋白[18 -19 ] 等.尽管取得了这些成功,但是基于能量函数的蛋白质设计方法准确度仍然较低,在没有多轮实验试错的情况下无法可靠使用,导致蛋白设计实验成功率难以提升[20 ] . ...
De novo design of picomolar SARS-CoV-2 miniprotein inhibitors
1
2020
... 依据Anfinsen的折叠热力学假说[4 ] ,蛋白质折叠到最低自由能状态,其3D结构由氨基酸序列决定.然而,在折叠过程中最重要的不是折叠态的绝对能量,而是折叠态与最低的备选态之间的能量差.这种计算不仅涉及到所有可能的氨基酸序列,而且涉及到所有可能的结构,因此多数现有的方法都集中在寻找所需结构的最低能量氨基酸序列这个更容易处理的问题上.目前广泛使用的方法仍然是基于能量函数和启发式采样方法的算法[5 ] .RosettaDesign[6 ] 、FoldX[7 ] 、EvoDesign / EvoEF2[8 ] 等设计方法使用使用蛋白质结构参数化的打分项来量化氨基酸序列和特定三维结构之间的匹配度,其中RosettaDesign是目前使用最为广泛的方法之一.RosettaDesign采用能量函数[9 ] 来捕捉序列-结构关系,对结构中每个残基侧链的氨基酸种类和构象进行采样,并使用蒙特卡洛模拟退火等方法进行优化以获得低能序列和构象.在过去的三十年中,基于能量函数的蛋白计算设计取得了巨大的进展,包括设计新颖的3D折叠[10 ] 、酶[11 ] 和复合物[11 ] ,更包括免疫信号[12 -13 ] 、靶向治疗蛋白[14 -15 ] 、蛋白质开关[16 -17 ] 、自组装蛋白[18 -19 ] 等.尽管取得了这些成功,但是基于能量函数的蛋白质设计方法准确度仍然较低,在没有多轮实验试错的情况下无法可靠使用,导致蛋白设计实验成功率难以提升[20 ] . ...
De novo design of bioactive protein switches
1
2019
... 依据Anfinsen的折叠热力学假说[4 ] ,蛋白质折叠到最低自由能状态,其3D结构由氨基酸序列决定.然而,在折叠过程中最重要的不是折叠态的绝对能量,而是折叠态与最低的备选态之间的能量差.这种计算不仅涉及到所有可能的氨基酸序列,而且涉及到所有可能的结构,因此多数现有的方法都集中在寻找所需结构的最低能量氨基酸序列这个更容易处理的问题上.目前广泛使用的方法仍然是基于能量函数和启发式采样方法的算法[5 ] .RosettaDesign[6 ] 、FoldX[7 ] 、EvoDesign / EvoEF2[8 ] 等设计方法使用使用蛋白质结构参数化的打分项来量化氨基酸序列和特定三维结构之间的匹配度,其中RosettaDesign是目前使用最为广泛的方法之一.RosettaDesign采用能量函数[9 ] 来捕捉序列-结构关系,对结构中每个残基侧链的氨基酸种类和构象进行采样,并使用蒙特卡洛模拟退火等方法进行优化以获得低能序列和构象.在过去的三十年中,基于能量函数的蛋白计算设计取得了巨大的进展,包括设计新颖的3D折叠[10 ] 、酶[11 ] 和复合物[11 ] ,更包括免疫信号[12 -13 ] 、靶向治疗蛋白[14 -15 ] 、蛋白质开关[16 -17 ] 、自组装蛋白[18 -19 ] 等.尽管取得了这些成功,但是基于能量函数的蛋白质设计方法准确度仍然较低,在没有多轮实验试错的情况下无法可靠使用,导致蛋白设计实验成功率难以提升[20 ] . ...
Structural resolution of switchable states of a de novo peptide assembly
1
2021
... 依据Anfinsen的折叠热力学假说[4 ] ,蛋白质折叠到最低自由能状态,其3D结构由氨基酸序列决定.然而,在折叠过程中最重要的不是折叠态的绝对能量,而是折叠态与最低的备选态之间的能量差.这种计算不仅涉及到所有可能的氨基酸序列,而且涉及到所有可能的结构,因此多数现有的方法都集中在寻找所需结构的最低能量氨基酸序列这个更容易处理的问题上.目前广泛使用的方法仍然是基于能量函数和启发式采样方法的算法[5 ] .RosettaDesign[6 ] 、FoldX[7 ] 、EvoDesign / EvoEF2[8 ] 等设计方法使用使用蛋白质结构参数化的打分项来量化氨基酸序列和特定三维结构之间的匹配度,其中RosettaDesign是目前使用最为广泛的方法之一.RosettaDesign采用能量函数[9 ] 来捕捉序列-结构关系,对结构中每个残基侧链的氨基酸种类和构象进行采样,并使用蒙特卡洛模拟退火等方法进行优化以获得低能序列和构象.在过去的三十年中,基于能量函数的蛋白计算设计取得了巨大的进展,包括设计新颖的3D折叠[10 ] 、酶[11 ] 和复合物[11 ] ,更包括免疫信号[12 -13 ] 、靶向治疗蛋白[14 -15 ] 、蛋白质开关[16 -17 ] 、自组装蛋白[18 -19 ] 等.尽管取得了这些成功,但是基于能量函数的蛋白质设计方法准确度仍然较低,在没有多轮实验试错的情况下无法可靠使用,导致蛋白设计实验成功率难以提升[20 ] . ...
De novo design of self-assembling helical protein filaments
1
2018
... 依据Anfinsen的折叠热力学假说[4 ] ,蛋白质折叠到最低自由能状态,其3D结构由氨基酸序列决定.然而,在折叠过程中最重要的不是折叠态的绝对能量,而是折叠态与最低的备选态之间的能量差.这种计算不仅涉及到所有可能的氨基酸序列,而且涉及到所有可能的结构,因此多数现有的方法都集中在寻找所需结构的最低能量氨基酸序列这个更容易处理的问题上.目前广泛使用的方法仍然是基于能量函数和启发式采样方法的算法[5 ] .RosettaDesign[6 ] 、FoldX[7 ] 、EvoDesign / EvoEF2[8 ] 等设计方法使用使用蛋白质结构参数化的打分项来量化氨基酸序列和特定三维结构之间的匹配度,其中RosettaDesign是目前使用最为广泛的方法之一.RosettaDesign采用能量函数[9 ] 来捕捉序列-结构关系,对结构中每个残基侧链的氨基酸种类和构象进行采样,并使用蒙特卡洛模拟退火等方法进行优化以获得低能序列和构象.在过去的三十年中,基于能量函数的蛋白计算设计取得了巨大的进展,包括设计新颖的3D折叠[10 ] 、酶[11 ] 和复合物[11 ] ,更包括免疫信号[12 -13 ] 、靶向治疗蛋白[14 -15 ] 、蛋白质开关[16 -17 ] 、自组装蛋白[18 -19 ] 等.尽管取得了这些成功,但是基于能量函数的蛋白质设计方法准确度仍然较低,在没有多轮实验试错的情况下无法可靠使用,导致蛋白设计实验成功率难以提升[20 ] . ...
Design of a hyperstable 60-subunit protein icosahedron
1
2016
... 依据Anfinsen的折叠热力学假说[4 ] ,蛋白质折叠到最低自由能状态,其3D结构由氨基酸序列决定.然而,在折叠过程中最重要的不是折叠态的绝对能量,而是折叠态与最低的备选态之间的能量差.这种计算不仅涉及到所有可能的氨基酸序列,而且涉及到所有可能的结构,因此多数现有的方法都集中在寻找所需结构的最低能量氨基酸序列这个更容易处理的问题上.目前广泛使用的方法仍然是基于能量函数和启发式采样方法的算法[5 ] .RosettaDesign[6 ] 、FoldX[7 ] 、EvoDesign / EvoEF2[8 ] 等设计方法使用使用蛋白质结构参数化的打分项来量化氨基酸序列和特定三维结构之间的匹配度,其中RosettaDesign是目前使用最为广泛的方法之一.RosettaDesign采用能量函数[9 ] 来捕捉序列-结构关系,对结构中每个残基侧链的氨基酸种类和构象进行采样,并使用蒙特卡洛模拟退火等方法进行优化以获得低能序列和构象.在过去的三十年中,基于能量函数的蛋白计算设计取得了巨大的进展,包括设计新颖的3D折叠[10 ] 、酶[11 ] 和复合物[11 ] ,更包括免疫信号[12 -13 ] 、靶向治疗蛋白[14 -15 ] 、蛋白质开关[16 -17 ] 、自组装蛋白[18 -19 ] 等.尽管取得了这些成功,但是基于能量函数的蛋白质设计方法准确度仍然较低,在没有多轮实验试错的情况下无法可靠使用,导致蛋白设计实验成功率难以提升[20 ] . ...
Global analysis of protein folding using massively parallel design, synthesis, and testing
1
2017
... 依据Anfinsen的折叠热力学假说[4 ] ,蛋白质折叠到最低自由能状态,其3D结构由氨基酸序列决定.然而,在折叠过程中最重要的不是折叠态的绝对能量,而是折叠态与最低的备选态之间的能量差.这种计算不仅涉及到所有可能的氨基酸序列,而且涉及到所有可能的结构,因此多数现有的方法都集中在寻找所需结构的最低能量氨基酸序列这个更容易处理的问题上.目前广泛使用的方法仍然是基于能量函数和启发式采样方法的算法[5 ] .RosettaDesign[6 ] 、FoldX[7 ] 、EvoDesign / EvoEF2[8 ] 等设计方法使用使用蛋白质结构参数化的打分项来量化氨基酸序列和特定三维结构之间的匹配度,其中RosettaDesign是目前使用最为广泛的方法之一.RosettaDesign采用能量函数[9 ] 来捕捉序列-结构关系,对结构中每个残基侧链的氨基酸种类和构象进行采样,并使用蒙特卡洛模拟退火等方法进行优化以获得低能序列和构象.在过去的三十年中,基于能量函数的蛋白计算设计取得了巨大的进展,包括设计新颖的3D折叠[10 ] 、酶[11 ] 和复合物[11 ] ,更包括免疫信号[12 -13 ] 、靶向治疗蛋白[14 -15 ] 、蛋白质开关[16 -17 ] 、自组装蛋白[18 -19 ] 等.尽管取得了这些成功,但是基于能量函数的蛋白质设计方法准确度仍然较低,在没有多轮实验试错的情况下无法可靠使用,导致蛋白设计实验成功率难以提升[20 ] . ...
The protein data bank
1
2000
... 以深度学习为代表的人工智能技术,随着算法和算力的发展以及大数据的积累,近期在多个领域取得了重要进展.在生物学和化学领域中,深度神经网络的优势在于可以从蛋白质结构的原子坐标、氨基酸种类、二级结构等简单的输入数据中学习高阶特征.深度学习模型一旦学会了蛋白质特征间的关系,就可以用来为结构生物学和生物分子的设计提供新的见解和指导.海量具备真实性和可用性的数据[21 -24 ] 使得深度学习表现出比经典物理模型或其他机器学习方法更好的性能[25 ] .目前,深度学习已被应用于蛋白质-配体打分[26 -29 ] 、蛋白质-蛋白质相互作用预测[30 -32 ] 、化合物性质预测[33 ] 、分子结构生成[34 -36 ] 等诸多领域[37 ] ,近期更是在蛋白质结构预测上取得了引人注目的进展.以AlphaFold[38 ] 和RoseTTAFold[39 ] 为代表的结构预测算法通过多序列比对(Multiple sequence alignment,MSA)、基于注意力机制的序列分析和蛋白三维结构生成等模块,以端到端的方法大幅提高了蛋白三维结构预测的准确率. ...
SCOPe: Structural Classification of Proteins—Extended, integrating SCOP and ASTRAL data and classification of new structures
0
2014
UniProt: the universal protein knowledgebase
0
2017
Pfam: the protein families database in 2021
1
2021
... 以深度学习为代表的人工智能技术,随着算法和算力的发展以及大数据的积累,近期在多个领域取得了重要进展.在生物学和化学领域中,深度神经网络的优势在于可以从蛋白质结构的原子坐标、氨基酸种类、二级结构等简单的输入数据中学习高阶特征.深度学习模型一旦学会了蛋白质特征间的关系,就可以用来为结构生物学和生物分子的设计提供新的见解和指导.海量具备真实性和可用性的数据[21 -24 ] 使得深度学习表现出比经典物理模型或其他机器学习方法更好的性能[25 ] .目前,深度学习已被应用于蛋白质-配体打分[26 -29 ] 、蛋白质-蛋白质相互作用预测[30 -32 ] 、化合物性质预测[33 ] 、分子结构生成[34 -36 ] 等诸多领域[37 ] ,近期更是在蛋白质结构预测上取得了引人注目的进展.以AlphaFold[38 ] 和RoseTTAFold[39 ] 为代表的结构预测算法通过多序列比对(Multiple sequence alignment,MSA)、基于注意力机制的序列分析和蛋白三维结构生成等模块,以端到端的方法大幅提高了蛋白三维结构预测的准确率. ...
Data-driven computational protein design
1
2021
... 以深度学习为代表的人工智能技术,随着算法和算力的发展以及大数据的积累,近期在多个领域取得了重要进展.在生物学和化学领域中,深度神经网络的优势在于可以从蛋白质结构的原子坐标、氨基酸种类、二级结构等简单的输入数据中学习高阶特征.深度学习模型一旦学会了蛋白质特征间的关系,就可以用来为结构生物学和生物分子的设计提供新的见解和指导.海量具备真实性和可用性的数据[21 -24 ] 使得深度学习表现出比经典物理模型或其他机器学习方法更好的性能[25 ] .目前,深度学习已被应用于蛋白质-配体打分[26 -29 ] 、蛋白质-蛋白质相互作用预测[30 -32 ] 、化合物性质预测[33 ] 、分子结构生成[34 -36 ] 等诸多领域[37 ] ,近期更是在蛋白质结构预测上取得了引人注目的进展.以AlphaFold[38 ] 和RoseTTAFold[39 ] 为代表的结构预测算法通过多序列比对(Multiple sequence alignment,MSA)、基于注意力机制的序列分析和蛋白三维结构生成等模块,以端到端的方法大幅提高了蛋白三维结构预测的准确率. ...
AK-score: Accurate protein-ligand binding affinity prediction using an ensemble of 3D-convolutional neural networks
1
2020
... 以深度学习为代表的人工智能技术,随着算法和算力的发展以及大数据的积累,近期在多个领域取得了重要进展.在生物学和化学领域中,深度神经网络的优势在于可以从蛋白质结构的原子坐标、氨基酸种类、二级结构等简单的输入数据中学习高阶特征.深度学习模型一旦学会了蛋白质特征间的关系,就可以用来为结构生物学和生物分子的设计提供新的见解和指导.海量具备真实性和可用性的数据[21 -24 ] 使得深度学习表现出比经典物理模型或其他机器学习方法更好的性能[25 ] .目前,深度学习已被应用于蛋白质-配体打分[26 -29 ] 、蛋白质-蛋白质相互作用预测[30 -32 ] 、化合物性质预测[33 ] 、分子结构生成[34 -36 ] 等诸多领域[37 ] ,近期更是在蛋白质结构预测上取得了引人注目的进展.以AlphaFold[38 ] 和RoseTTAFold[39 ] 为代表的结构预测算法通过多序列比对(Multiple sequence alignment,MSA)、基于注意力机制的序列分析和蛋白三维结构生成等模块,以端到端的方法大幅提高了蛋白三维结构预测的准确率. ...
InteractionGraphNet: a novel and efficient deep graph representation learning framework for accurate protein-ligand interaction predictions
0
2021
Improved protein-ligand binding affinity prediction with structure-based deep fusion inference
0
2021
K DEEP : Protein-ligand absolute binding affinity prediction via 3D-convolutional neural networks
1
2018
... 以深度学习为代表的人工智能技术,随着算法和算力的发展以及大数据的积累,近期在多个领域取得了重要进展.在生物学和化学领域中,深度神经网络的优势在于可以从蛋白质结构的原子坐标、氨基酸种类、二级结构等简单的输入数据中学习高阶特征.深度学习模型一旦学会了蛋白质特征间的关系,就可以用来为结构生物学和生物分子的设计提供新的见解和指导.海量具备真实性和可用性的数据[21 -24 ] 使得深度学习表现出比经典物理模型或其他机器学习方法更好的性能[25 ] .目前,深度学习已被应用于蛋白质-配体打分[26 -29 ] 、蛋白质-蛋白质相互作用预测[30 -32 ] 、化合物性质预测[33 ] 、分子结构生成[34 -36 ] 等诸多领域[37 ] ,近期更是在蛋白质结构预测上取得了引人注目的进展.以AlphaFold[38 ] 和RoseTTAFold[39 ] 为代表的结构预测算法通过多序列比对(Multiple sequence alignment,MSA)、基于注意力机制的序列分析和蛋白三维结构生成等模块,以端到端的方法大幅提高了蛋白三维结构预测的准确率. ...
D-SCRIPT translates genome to phenome with sequence-based, structure-aware, genome-scale predictions of protein-protein interactions
1
2021
... 以深度学习为代表的人工智能技术,随着算法和算力的发展以及大数据的积累,近期在多个领域取得了重要进展.在生物学和化学领域中,深度神经网络的优势在于可以从蛋白质结构的原子坐标、氨基酸种类、二级结构等简单的输入数据中学习高阶特征.深度学习模型一旦学会了蛋白质特征间的关系,就可以用来为结构生物学和生物分子的设计提供新的见解和指导.海量具备真实性和可用性的数据[21 -24 ] 使得深度学习表现出比经典物理模型或其他机器学习方法更好的性能[25 ] .目前,深度学习已被应用于蛋白质-配体打分[26 -29 ] 、蛋白质-蛋白质相互作用预测[30 -32 ] 、化合物性质预测[33 ] 、分子结构生成[34 -36 ] 等诸多领域[37 ] ,近期更是在蛋白质结构预测上取得了引人注目的进展.以AlphaFold[38 ] 和RoseTTAFold[39 ] 为代表的结构预测算法通过多序列比对(Multiple sequence alignment,MSA)、基于注意力机制的序列分析和蛋白三维结构生成等模块,以端到端的方法大幅提高了蛋白三维结构预测的准确率. ...
Struct2Graph: a graph attention network for structure based predictions of protein-protein interactions
0
2022
RGN: Residue-based graph attention and convolutional network for protein-protein interaction site prediction
1
2022
... 以深度学习为代表的人工智能技术,随着算法和算力的发展以及大数据的积累,近期在多个领域取得了重要进展.在生物学和化学领域中,深度神经网络的优势在于可以从蛋白质结构的原子坐标、氨基酸种类、二级结构等简单的输入数据中学习高阶特征.深度学习模型一旦学会了蛋白质特征间的关系,就可以用来为结构生物学和生物分子的设计提供新的见解和指导.海量具备真实性和可用性的数据[21 -24 ] 使得深度学习表现出比经典物理模型或其他机器学习方法更好的性能[25 ] .目前,深度学习已被应用于蛋白质-配体打分[26 -29 ] 、蛋白质-蛋白质相互作用预测[30 -32 ] 、化合物性质预测[33 ] 、分子结构生成[34 -36 ] 等诸多领域[37 ] ,近期更是在蛋白质结构预测上取得了引人注目的进展.以AlphaFold[38 ] 和RoseTTAFold[39 ] 为代表的结构预测算法通过多序列比对(Multiple sequence alignment,MSA)、基于注意力机制的序列分析和蛋白三维结构生成等模块,以端到端的方法大幅提高了蛋白三维结构预测的准确率. ...
Out-of-the-box deep learning prediction of pharmaceutical properties by broadly learned knowledge-based molecular representations
1
2021
... 以深度学习为代表的人工智能技术,随着算法和算力的发展以及大数据的积累,近期在多个领域取得了重要进展.在生物学和化学领域中,深度神经网络的优势在于可以从蛋白质结构的原子坐标、氨基酸种类、二级结构等简单的输入数据中学习高阶特征.深度学习模型一旦学会了蛋白质特征间的关系,就可以用来为结构生物学和生物分子的设计提供新的见解和指导.海量具备真实性和可用性的数据[21 -24 ] 使得深度学习表现出比经典物理模型或其他机器学习方法更好的性能[25 ] .目前,深度学习已被应用于蛋白质-配体打分[26 -29 ] 、蛋白质-蛋白质相互作用预测[30 -32 ] 、化合物性质预测[33 ] 、分子结构生成[34 -36 ] 等诸多领域[37 ] ,近期更是在蛋白质结构预测上取得了引人注目的进展.以AlphaFold[38 ] 和RoseTTAFold[39 ] 为代表的结构预测算法通过多序列比对(Multiple sequence alignment,MSA)、基于注意力机制的序列分析和蛋白三维结构生成等模块,以端到端的方法大幅提高了蛋白三维结构预测的准确率. ...
Automated de novo molecular design by hybrid machine intelligence and rule-driven chemical synthesis
1
2019
... 以深度学习为代表的人工智能技术,随着算法和算力的发展以及大数据的积累,近期在多个领域取得了重要进展.在生物学和化学领域中,深度神经网络的优势在于可以从蛋白质结构的原子坐标、氨基酸种类、二级结构等简单的输入数据中学习高阶特征.深度学习模型一旦学会了蛋白质特征间的关系,就可以用来为结构生物学和生物分子的设计提供新的见解和指导.海量具备真实性和可用性的数据[21 -24 ] 使得深度学习表现出比经典物理模型或其他机器学习方法更好的性能[25 ] .目前,深度学习已被应用于蛋白质-配体打分[26 -29 ] 、蛋白质-蛋白质相互作用预测[30 -32 ] 、化合物性质预测[33 ] 、分子结构生成[34 -36 ] 等诸多领域[37 ] ,近期更是在蛋白质结构预测上取得了引人注目的进展.以AlphaFold[38 ] 和RoseTTAFold[39 ] 为代表的结构预测算法通过多序列比对(Multiple sequence alignment,MSA)、基于注意力机制的序列分析和蛋白三维结构生成等模块,以端到端的方法大幅提高了蛋白三维结构预测的准确率. ...
MolGAN: An implicit generative model for small molecular graphs
0
2018
Efficient multi-objective molecular optimization in a continuous latent space
1
2019
... 以深度学习为代表的人工智能技术,随着算法和算力的发展以及大数据的积累,近期在多个领域取得了重要进展.在生物学和化学领域中,深度神经网络的优势在于可以从蛋白质结构的原子坐标、氨基酸种类、二级结构等简单的输入数据中学习高阶特征.深度学习模型一旦学会了蛋白质特征间的关系,就可以用来为结构生物学和生物分子的设计提供新的见解和指导.海量具备真实性和可用性的数据[21 -24 ] 使得深度学习表现出比经典物理模型或其他机器学习方法更好的性能[25 ] .目前,深度学习已被应用于蛋白质-配体打分[26 -29 ] 、蛋白质-蛋白质相互作用预测[30 -32 ] 、化合物性质预测[33 ] 、分子结构生成[34 -36 ] 等诸多领域[37 ] ,近期更是在蛋白质结构预测上取得了引人注目的进展.以AlphaFold[38 ] 和RoseTTAFold[39 ] 为代表的结构预测算法通过多序列比对(Multiple sequence alignment,MSA)、基于注意力机制的序列分析和蛋白三维结构生成等模块,以端到端的方法大幅提高了蛋白三维结构预测的准确率. ...
Protein design via deep learning
1
2022
... 以深度学习为代表的人工智能技术,随着算法和算力的发展以及大数据的积累,近期在多个领域取得了重要进展.在生物学和化学领域中,深度神经网络的优势在于可以从蛋白质结构的原子坐标、氨基酸种类、二级结构等简单的输入数据中学习高阶特征.深度学习模型一旦学会了蛋白质特征间的关系,就可以用来为结构生物学和生物分子的设计提供新的见解和指导.海量具备真实性和可用性的数据[21 -24 ] 使得深度学习表现出比经典物理模型或其他机器学习方法更好的性能[25 ] .目前,深度学习已被应用于蛋白质-配体打分[26 -29 ] 、蛋白质-蛋白质相互作用预测[30 -32 ] 、化合物性质预测[33 ] 、分子结构生成[34 -36 ] 等诸多领域[37 ] ,近期更是在蛋白质结构预测上取得了引人注目的进展.以AlphaFold[38 ] 和RoseTTAFold[39 ] 为代表的结构预测算法通过多序列比对(Multiple sequence alignment,MSA)、基于注意力机制的序列分析和蛋白三维结构生成等模块,以端到端的方法大幅提高了蛋白三维结构预测的准确率. ...
Highly accurate protein structure prediction with AlphaFold
2
2021
... 以深度学习为代表的人工智能技术,随着算法和算力的发展以及大数据的积累,近期在多个领域取得了重要进展.在生物学和化学领域中,深度神经网络的优势在于可以从蛋白质结构的原子坐标、氨基酸种类、二级结构等简单的输入数据中学习高阶特征.深度学习模型一旦学会了蛋白质特征间的关系,就可以用来为结构生物学和生物分子的设计提供新的见解和指导.海量具备真实性和可用性的数据[21 -24 ] 使得深度学习表现出比经典物理模型或其他机器学习方法更好的性能[25 ] .目前,深度学习已被应用于蛋白质-配体打分[26 -29 ] 、蛋白质-蛋白质相互作用预测[30 -32 ] 、化合物性质预测[33 ] 、分子结构生成[34 -36 ] 等诸多领域[37 ] ,近期更是在蛋白质结构预测上取得了引人注目的进展.以AlphaFold[38 ] 和RoseTTAFold[39 ] 为代表的结构预测算法通过多序列比对(Multiple sequence alignment,MSA)、基于注意力机制的序列分析和蛋白三维结构生成等模块,以端到端的方法大幅提高了蛋白三维结构预测的准确率. ...
... Anand[106 ] 模型通过定义二级结构和残基接触矩阵约束嵌入到高维空间,再使用IPA模块降维到三维空间中表征蛋白结构.作者使用AlphaFold网络架构中的[38 ] 不变点注意力(Invariant Point Attention, IPA)模块替换transformer中的标准注意力模块保证模型的平移旋转不变性,使用类似于BERT[107 ] 的扩散方法在骨架上生成序列.与其他DDPM模型不同,该模型不使用随机产生的高斯噪声,而是通过随机掩盖部分残基,在[0, 1]中作为t的函数进行线性插值来训练模型;在生成时,模型在t = T时掩蔽所有的残基来进行反向过程,从t = T到t = 0的时间步进行迭代采样.模型还允许人为给定条件信息编码蛋白结构.该模型完全从真实蛋白结构数据中学习,并生成蛋白质拓扑结构的条件约束,以产生全原子骨架构型以及序列和侧链预测.作者用了3个独立训练的模型分别生成蛋白结构、序列和转子,并将模型应用于无序列从头生成、蛋白补全、序列设计、侧链转子重排等任务中,结果表明其具有作为端到端的蛋白质从头设计工具的潜力. ...
Accurate prediction of protein structures and interactions using a three-track neural network
1
2021
... 以深度学习为代表的人工智能技术,随着算法和算力的发展以及大数据的积累,近期在多个领域取得了重要进展.在生物学和化学领域中,深度神经网络的优势在于可以从蛋白质结构的原子坐标、氨基酸种类、二级结构等简单的输入数据中学习高阶特征.深度学习模型一旦学会了蛋白质特征间的关系,就可以用来为结构生物学和生物分子的设计提供新的见解和指导.海量具备真实性和可用性的数据[21 -24 ] 使得深度学习表现出比经典物理模型或其他机器学习方法更好的性能[25 ] .目前,深度学习已被应用于蛋白质-配体打分[26 -29 ] 、蛋白质-蛋白质相互作用预测[30 -32 ] 、化合物性质预测[33 ] 、分子结构生成[34 -36 ] 等诸多领域[37 ] ,近期更是在蛋白质结构预测上取得了引人注目的进展.以AlphaFold[38 ] 和RoseTTAFold[39 ] 为代表的结构预测算法通过多序列比对(Multiple sequence alignment,MSA)、基于注意力机制的序列分析和蛋白三维结构生成等模块,以端到端的方法大幅提高了蛋白三维结构预测的准确率. ...
Protein design automation
1
1996
... 固定骨架蛋白质设计的目标是找到一个最能折叠成目标结构的氨基酸序列,也可以看作是找到一个折叠成目标结构的概率比其他所有序列都大的序列[40 -41 ] . ...
De novo protein design: fully automated sequence selection
1
1997
... 固定骨架蛋白质设计的目标是找到一个最能折叠成目标结构的氨基酸序列,也可以看作是找到一个折叠成目标结构的概率比其他所有序列都大的序列[40 -41 ] . ...
Direct prediction of profiles of sequences compatible with a protein structure by neural networks with fragment-based local and energy-based nonlocal profiles
1
2014
... SPIN使用一个基于片段局部特征和能量非局部轮廓的神经网络来解决基于固定骨架结构的蛋白序列设计问题[42 ] ,其输入特征包括目标蛋白质主链的φ,ψ二面角,通过比较相邻5个残基的结构片段得到局部片段衍生序列图谱[43 ] ,并采用DFIRE统计势[44 ] 计算全局能量.SPIN在500个蛋白质的测试集上平均序列恢复率约为30%. ...
Improving computational protein design by using structure-derived sequence profile
1
2010
... SPIN使用一个基于片段局部特征和能量非局部轮廓的神经网络来解决基于固定骨架结构的蛋白序列设计问题[42 ] ,其输入特征包括目标蛋白质主链的φ,ψ二面角,通过比较相邻5个残基的结构片段得到局部片段衍生序列图谱[43 ] ,并采用DFIRE统计势[44 ] 计算全局能量.SPIN在500个蛋白质的测试集上平均序列恢复率约为30%. ...
Ab initio folding of terminal segments with secondary structures reveals the fine difference between two closely related all-atom statistical energy functions
1
2008
... SPIN使用一个基于片段局部特征和能量非局部轮廓的神经网络来解决基于固定骨架结构的蛋白序列设计问题[42 ] ,其输入特征包括目标蛋白质主链的φ,ψ二面角,通过比较相邻5个残基的结构片段得到局部片段衍生序列图谱[43 ] ,并采用DFIRE统计势[44 ] 计算全局能量.SPIN在500个蛋白质的测试集上平均序列恢复率约为30%. ...
Computational protein design with deep learning neural networks
1
2018
... Qi等人开发了用于蛋白计算设计的神经网络模型,使用目标残基及其相邻残基的距离、主链二面角和二级结构等几何特征,以约3倍于SPIN的训练集对神经网络进行训练,将序列恢复率提高至33%[45 ] .同期,SPIN2[46 ] 使用一个具有三个隐藏层的神经网络,在蛋白骨架特征中添加另外两个骨架二面角θ和ι并改用正弦和余弦表示作为特征输入,将序列恢复率提高至34%. ...
SPIN2: Predicting sequence profiles from protein structures using deep neural networks
1
2018
... Qi等人开发了用于蛋白计算设计的神经网络模型,使用目标残基及其相邻残基的距离、主链二面角和二级结构等几何特征,以约3倍于SPIN的训练集对神经网络进行训练,将序列恢复率提高至33%[45 ] .同期,SPIN2[46 ] 使用一个具有三个隐藏层的神经网络,在蛋白骨架特征中添加另外两个骨架二面角θ和ι并改用正弦和余弦表示作为特征输入,将序列恢复率提高至34%. ...
To improve protein sequence profile prediction through image captioning on pairwise residue distance map
1
2020
... SPIN2仅使用一维结构特征,不足以表征具有复杂三维结构的蛋白质.SPROF[47 ] 则通过两两残基距离的二维距离矩阵来表示蛋白质中残基之间距离(图1 ).SPROF使用双向长短时记忆网络与自注意力二维卷积神经网络来预测蛋白质序列.SPROF方法在独立测试集上取得了39.8 %的序列恢复率,明显高于仅从一维结构特征训练的SPIN2方法取得的34.6 %. ...
ImageNet classification with deep convolutional neural networks
1
2017
... 卷积神经网络(Convolutional Neural Network, CNN)[48 ] 是最成功的神经网络架构之一,主要包括卷积和池化两种基本操作.在蛋白质设计中,卷积层用于对蛋白质残基间距离图或蛋白质在三维空间网格中的密度距离分布进行变换并提取特征,更深的卷积网络能从输入特征中迭代提取更复杂的特征.池化层通过连续降采样的方式逐渐降低数据的空间尺寸,以减少网络中的参数数量,使得计算资源耗费变少,也有效控制过拟合.另外卷积使得模型能够处理大小可变的输入数据. ...
ProDCoNN: Protein design using a convolutional neural network
1
2020
... ProDCoNN[49 ] , Anand等人发展的方法[50 ] 和DenseCPD[51 ] 均使用三维卷积网络从目标残基周围的三维结构环境特征来预测残基类型(图2 ).模型以残基周围的原子密度和原子类型网格作为输入,使用DenseNet[52 ] 等多层卷积网络对密度分布数据进行学习,捕获不同尺度下的结构信息.网络中的卷积层提取蛋白质共价键信息、键角、二面角和二级结构的特征图,池化层精简特征图数量,最后输出目标残基为20种天然氨基酸的概率大小.其中, ProDCoNN和Anand模型分别在相同的T500和TS50上达到约40%的序列恢复率,DenseCPD则达到51%. ...
Protein sequence design with a learned potential
1
2022
... ProDCoNN[49 ] , Anand等人发展的方法[50 ] 和DenseCPD[51 ] 均使用三维卷积网络从目标残基周围的三维结构环境特征来预测残基类型(图2 ).模型以残基周围的原子密度和原子类型网格作为输入,使用DenseNet[52 ] 等多层卷积网络对密度分布数据进行学习,捕获不同尺度下的结构信息.网络中的卷积层提取蛋白质共价键信息、键角、二面角和二级结构的特征图,池化层精简特征图数量,最后输出目标残基为20种天然氨基酸的概率大小.其中, ProDCoNN和Anand模型分别在相同的T500和TS50上达到约40%的序列恢复率,DenseCPD则达到51%. ...
DenseCPD: Improving the accuracy of neural-network-based computational protein sequence design with DenseNet
1
2020
... ProDCoNN[49 ] , Anand等人发展的方法[50 ] 和DenseCPD[51 ] 均使用三维卷积网络从目标残基周围的三维结构环境特征来预测残基类型(图2 ).模型以残基周围的原子密度和原子类型网格作为输入,使用DenseNet[52 ] 等多层卷积网络对密度分布数据进行学习,捕获不同尺度下的结构信息.网络中的卷积层提取蛋白质共价键信息、键角、二面角和二级结构的特征图,池化层精简特征图数量,最后输出目标残基为20种天然氨基酸的概率大小.其中, ProDCoNN和Anand模型分别在相同的T500和TS50上达到约40%的序列恢复率,DenseCPD则达到51%. ...
Densely connected convolutional networks
1
2017
... ProDCoNN[49 ] , Anand等人发展的方法[50 ] 和DenseCPD[51 ] 均使用三维卷积网络从目标残基周围的三维结构环境特征来预测残基类型(图2 ).模型以残基周围的原子密度和原子类型网格作为输入,使用DenseNet[52 ] 等多层卷积网络对密度分布数据进行学习,捕获不同尺度下的结构信息.网络中的卷积层提取蛋白质共价键信息、键角、二面角和二级结构的特征图,池化层精简特征图数量,最后输出目标残基为20种天然氨基酸的概率大小.其中, ProDCoNN和Anand模型分别在相同的T500和TS50上达到约40%的序列恢复率,DenseCPD则达到51%. ...
Discovery of novel gain-of-function mutations guided by structure-based deep learning
1
2020
... MutCompute[53 ] 使用残基原子(C,H,O,N,S)坐标、部分电荷(partialcharge)和溶剂可及表面积(SASA,solvent-accessible surface area)作为结构特征输入3D-CNN网络.MutCompute以蛋白质中心目标残基的Cα为中心,掩蔽20 Å立方体内的所有肽原子,构造为该残基的局部化学微环境(microenvironment)样本,以这种方式从19300个蛋白质结构中构造170万个微环境作为训练集.训练后的模型能够识别稳定的突变,根据残基局部化学微环境预测蛋白质中不稳定的位点.Lu[54 ] 使用MutCompute模型设计了一种聚对苯二甲酸乙二醇酯(PET)水解酶,指导野生型水解酶PETase组合N233K / R224Q / S121E和骨架的D186H / R280A五个位点的突变,得到的突变体FAST-PETase具有优异的催化活性和热稳定性.FAST-PETase在30至50摄氏度和一系列pH水平之间显示出优越的PET水解活性,适用于至少51种未经处理的PET降解,工业上可广泛用于塑料的回收与循环. ...
Machine learning-aided engineering of hydrolases for PET depolymerization
1
2022
... MutCompute[53 ] 使用残基原子(C,H,O,N,S)坐标、部分电荷(partialcharge)和溶剂可及表面积(SASA,solvent-accessible surface area)作为结构特征输入3D-CNN网络.MutCompute以蛋白质中心目标残基的Cα为中心,掩蔽20 Å立方体内的所有肽原子,构造为该残基的局部化学微环境(microenvironment)样本,以这种方式从19300个蛋白质结构中构造170万个微环境作为训练集.训练后的模型能够识别稳定的突变,根据残基局部化学微环境预测蛋白质中不稳定的位点.Lu[54 ] 使用MutCompute模型设计了一种聚对苯二甲酸乙二醇酯(PET)水解酶,指导野生型水解酶PETase组合N233K / R224Q / S121E和骨架的D186H / R280A五个位点的突变,得到的突变体FAST-PETase具有优异的催化活性和热稳定性.FAST-PETase在30至50摄氏度和一系列pH水平之间显示出优越的PET水解活性,适用于至少51种未经处理的PET降解,工业上可广泛用于塑料的回收与循环. ...
Protein sequence design by conformational landscape optimization
2
2021
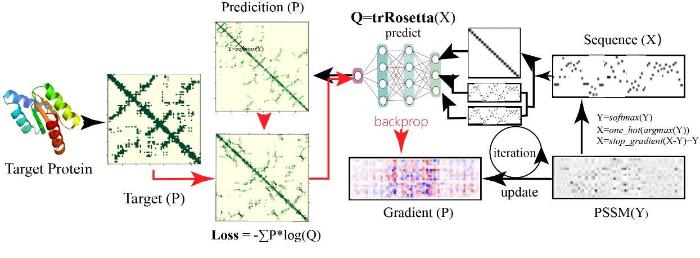
... TrDesign[55 ] 使用基于卷积神经网络的结构预测模型trRosetta进行反向序列设计.首先将随机氨基酸序列输入到蛋白质结构预测模型trRosetta[56 ] 中,输出残基之间距离、角度和二面角的分布(图3 ).其次计算预测分布与目标蛋白结构分布之间的差异,使用梯度反向传播来更新氨基酸序列,重复该过程直到收敛.TrDesign通过trRosetta遍历全局构象势能面,和RosettaDesign单点能量计算方法相比,能够多方面捕获序列折叠势能,保证设计蛋白质的可折叠性和稳定性.高分辨率的Rosetta模型用于创建目标结构的深度能量极小值,而低分辨率的trRosetta模型用于减少在能量极小值点备选序列的数量.将两种方法结合,能够在遍历势能面的同时减少的候选序列的数量.然而使用trRosetta进行反向序列设计所需要反复运行trRosetta模型,计算效率不高并且容易陷入势能面上次优解. ...
... Family-wide hallucination集成了无限制幻想设计[80 , 82 ] 与Rosetta序列设计方法[55 ] ,对环(loop)和可变区域(variable regions)的序列和结构进行从头设计,并对核心区域的结构进行序列优化.该方法从2000个天然NTF2s序列出发,在序列空间中进行蒙特卡洛搜索,每一步都进行一次序列变化,并使用trRosetta进行结构预测.模型的损失函数由两部分构成:结构保守区域基于与NTF2 - like蛋白实验结构的输入残基距离和方向分布的一致性进行评估;而可变区域基于网络预测与背景分布之间的KL散度计算的预测残基间几何结构的置信度进行评估.氢键网络也被纳入设计的结构中,以增加结构特异性.实验数据显示Family-wide hallucination生成的1615个骨架在原生结构的空间内采样更多,并且比原生骨架或非深度学习能量优化生成的骨架具有更强的序列结构关系. ...
Improved protein structure prediction using predicted interresidue orientations
1
2020
... TrDesign[55 ] 使用基于卷积神经网络的结构预测模型trRosetta进行反向序列设计.首先将随机氨基酸序列输入到蛋白质结构预测模型trRosetta[56 ] 中,输出残基之间距离、角度和二面角的分布(图3 ).其次计算预测分布与目标蛋白结构分布之间的差异,使用梯度反向传播来更新氨基酸序列,重复该过程直到收敛.TrDesign通过trRosetta遍历全局构象势能面,和RosettaDesign单点能量计算方法相比,能够多方面捕获序列折叠势能,保证设计蛋白质的可折叠性和稳定性.高分辨率的Rosetta模型用于创建目标结构的深度能量极小值,而低分辨率的trRosetta模型用于减少在能量极小值点备选序列的数量.将两种方法结合,能够在遍历势能面的同时减少的候选序列的数量.然而使用trRosetta进行反向序列设计所需要反复运行trRosetta模型,计算效率不高并且容易陷入势能面上次优解. ...
Protein docking model evaluation by graph neural networks
1
2021
... 图神经网络(Graph neural network, GNN)运行在图这种非欧氏数据结构上,已被广泛应用于知识图谱、社交网络、药物发现和蛋白质生物信息学等领域[57 -58 ] .蛋白质结构可用图进行编码,残基信息编码在节点特征中,空间中相邻残基之间的关系可编码为边特征. ...
Semi-supervised classification with graph convolutional networks
1
2016
... 图神经网络(Graph neural network, GNN)运行在图这种非欧氏数据结构上,已被广泛应用于知识图谱、社交网络、药物发现和蛋白质生物信息学等领域[57 -58 ] .蛋白质结构可用图进行编码,残基信息编码在节点特征中,空间中相邻残基之间的关系可编码为边特征. ...
Generative models for graph-based protein design
1
2019
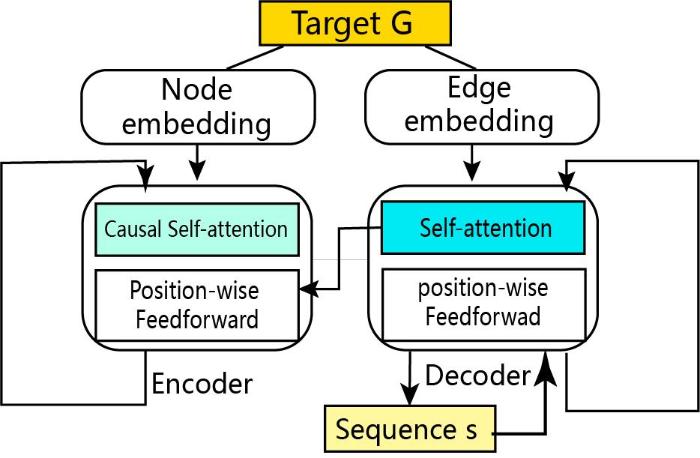
... GraphTrans[59 ] 使用图G = { V , E } V = { v 1 , v 2 … . v N } { e i j } i≠ j 捕捉它们之间的关系(图4 ).模型通过三维结构的自回归解码Transformer[60 ] 以捕获序列残基之间稀疏的成对依赖关系信息.GraphTrans模型可以有效地捕获序列和结构之间的高阶依赖关系,序列恢复率在Ollikainen 40测试集上达到39.2%,高于RosettaDesign的33.1%;在CATH测试集上残基困惑度(per-residue perplexities)为6.85,精度比以往基于神经网络(LSTM:17.13,SPIN2:12.61)的模型显著提高. ...
Attention is all You need
2
2017
... GraphTrans[59 ] 使用图G = { V , E } V = { v 1 , v 2 … . v N } { e i j } i≠ j 捕捉它们之间的关系(图4 ).模型通过三维结构的自回归解码Transformer[60 ] 以捕获序列残基之间稀疏的成对依赖关系信息.GraphTrans模型可以有效地捕获序列和结构之间的高阶依赖关系,序列恢复率在Ollikainen 40测试集上达到39.2%,高于RosettaDesign的33.1%;在CATH测试集上残基困惑度(per-residue perplexities)为6.85,精度比以往基于神经网络(LSTM:17.13,SPIN2:12.61)的模型显著提高. ...
... 随着transformer模型[60 ] 在自然语言处理领域大放异彩,越来越多的研究者将transformer架构应用到蛋白质序列生成领域,由此产生了许多基于transformer的序列生成模型.2020年Madami等人提出了ProGen模型[114 ] .ProGen是一种条件transformer语言模型.该模型使用带有一系列蛋白性质标签的氨基酸序列进行训练,实现可控生成.ProGen生成的蛋白质在能量上与天然蛋白质相近,具有理想的生物功能.由 Elnaggar等[115 ] 提出的ProtTrans模型,使用四种不同的语言模型(两种自回归语言模型Tranformer-XL, XLNet以及两种自编码模型Bert, Albert)在蛋白质数据集上进行预训练,从序列中学习提取有用的特征,而后引入下游监督任务,以实现单个残基和单个蛋白性质的预测.这些模型原则上具有序列生成能力.2021年Gligorijević等[116 ] 提出了一种序列去噪自编码器,该模型与一个功能预测器相结合,可以从大量未标记的蛋白质数据中学习蛋白质序列的多样性,而功能预测器可对序列采样的方向进行指导.在测试阶段,研究者进一步探究了模型在设计带有金属结合位点的序列以及重新设计功能增强的角质酶的能力. ...
Fast and flexible protein design using deep graph neural networks
1
2020
... 一个给定的蛋白质结构,对应于单一的距离矩阵,可以由许多不同的满足距离矩阵约束的同源序列折叠而成.ProteinSolver[61 ] 是一个预训练的图卷积神经网络,将使用氨基酸序列填充特定目标结构表述为一个约束满足问题( Constraint satisfaction problem),其目标是在兼顾长程和短程的约束的同时,为链中的残基分配氨基酸标签,使得残基之间的作用力是有利的.训练好的ProteinSolver网络能够以很高的准确度快速生成数千个匹配特定蛋白质拓扑结构的序列. ...
Learning from protein structure with geometric vector perceptrons
2
2020
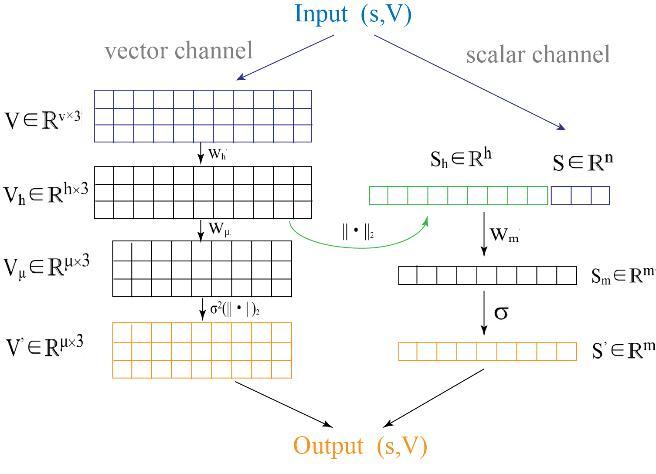
... 为同时将蛋白质残基几何结构和关系特征的纳入统一网络架构, Jing等提出使用几何向量感知器(Geometric vector perceptron,GVP)[62 ] (图5 )来代替多层感知器(multi-layer perceptron, MLPs).给定一个标量和向量输入特征( s, V)的元组,GVPs 将残基原子三维坐标转化为残基距离特征,并将其与标量特征组合,输出一个更新的元组( s′, V′).GVP模型在标量特征进行转换之前,会将其与转换后向量特征的范数进行拼接,这允许模型从输入向量中提取旋转不变信息,以便图中节点的信息传播.GVP-GNN[62 ] 使用GVP层来增强GNN对于几何结构特征的感知,并能够在欧氏向量特征上执行和表达,在蛋白质结构的质量评估和序列设计方面具有独特的优势. ...
... [62 ]使用GVP层来增强GNN对于几何结构特征的感知,并能够在欧氏向量特征上执行和表达,在蛋白质结构的质量评估和序列设计方面具有独特的优势. ...
Protein sequence sampling and prediction from structural data
1
2021
... Orellana[63 ] 对上述GVP的结构提出了改进,使用图卷积神经网络(Graph convolutional neural network, GCN)同时对节点和结构信息进行端到端的学习.模型添加每个氨基酸骨架中所有原子之间的归一化距离作为节点特征;将每个氨基酸的Cα 与其k个最近邻氨基酸的Cα 之间的标准化距离(K值邻近,k=35)作为边特征,然后将节点和边特征嵌入空间进行编码,并将其引入到GCN模型中,输出为序列中每个位置的氨基酸种类,可用于指导基于能量函数的蛋白设计方法.该模型的序列恢复率从以往模型的40.2 %提高到44.7 %. ...
Neural network-derived Potts models for structure-based protein design using backbone atomic coordinates and tertiary motifs
1
2023
... TERMinator[64 ] 使用三级motifs(TERM)捕获序列-结构关系[65 ] ,融合了残基原子坐标信息作为特征.TERMinator提取目标蛋白中与TERM结构匹配的信息来构建节点和边,嵌入空间编码后输入图神经网络中,输出序列空间中拟合了能量函数的Potts模型.GNN Potts模型编码器接受TERM数据并提取特征,使用使用马尔科夫链蒙特卡洛(Markov Chain Monte Carlo,MCMC)模拟退火算法生成最优序列,输出位置氨基酸标签.作者还进行消融了实验,完整的TERMinator模型(恢复率41.73%)性能强于消融TERM信息输入的模型(恢复率40.29%),表明联合使用TERM和空间坐标作为特征有利于蛋白质设计. ...
Tertiary structural propensities reveal fundamental sequence/structure relationships
1
2015
... TERMinator[64 ] 使用三级motifs(TERM)捕获序列-结构关系[65 ] ,融合了残基原子坐标信息作为特征.TERMinator提取目标蛋白中与TERM结构匹配的信息来构建节点和边,嵌入空间编码后输入图神经网络中,输出序列空间中拟合了能量函数的Potts模型.GNN Potts模型编码器接受TERM数据并提取特征,使用使用马尔科夫链蒙特卡洛(Markov Chain Monte Carlo,MCMC)模拟退火算法生成最优序列,输出位置氨基酸标签.作者还进行消融了实验,完整的TERMinator模型(恢复率41.73%)性能强于消融TERM信息输入的模型(恢复率40.29%),表明联合使用TERM和空间坐标作为特征有利于蛋白质设计. ...
Learning inverse folding from millions of predicted structures
1
2022
... ESM-IF1[66 ] 使用GVP来学习向量特征的等变转换和标量特征的不变变换.该工作尝试以下三种架构:(1)GVP-GNN;(2) 更宽和更深的GVP-GNN-large;(3)由GVP-GNN结构编码器和Transformer组成的混合模型.ESM-IF1使用AlphaFold2预测的1200万个结构,将训练数据增加了近3个数据级,克服了实验数据的限制,最终在CATH 4.3测试集上进行评估并根据残基困惑度(Perplexity,越低越好)和序列恢复率进行比较.GVP-GNN-large和GVP-Transformer模型均在序列恢复率上比简单GVP-GNN提高约9%,达到与DenseCPD相当的51%,且困惑度由6降低至4.在突变效应的zero-shot多项预测测试中(包括复合物稳定性、结合亲合力和插入效应),ESM-IF1均取得优异的性能表现. ...
A Deep SE(3)-Equivariant Model for Learning Inverse Protein Folding
1
202
... McPartion[67 ] 引入了一种深度 SE(3)-等变图 transformer 架构,直接对从蛋白质主链结构衍生的特征进行操作,实现了同时预测每个残基的氨基酸类型和侧链构象.局部感知图(Locality Aware Graph)transformer利用蛋白质主链的几何形状来优化单个残基和残基对的特征表示,并将注意力限制在空间上相邻的残基对上.该模块的输出和蛋白质主链坐标一起被传递到张量融合网络(Tensor fusion network,TFN)[68 ] 输出一个标量和残基位置,然后由TFN-transformer为每个输入残基产生侧链构象和氨基酸类型.作者评估了五种不同的残基掩蔽方法并分别进行了损失函数、网络架构和模型超参数的消融实验,发现从损失函数中移除侧链坐标均方根偏差 (Root Mean Squared Deviation,RMSD)和预测的侧链原子之间的成对距离两个特征显著降低了测试蛋白上的天然序列恢复率.除此之外,移除模型中的 TFN-Transformer 层对恢复率的影响最大.与几种现有的序列设计方法对比而言,该模型在四个测试集上展现了更高的序列恢复率. ...
Tensor fusion network for multimodal sentiment analysis
1
2017
... McPartion[67 ] 引入了一种深度 SE(3)-等变图 transformer 架构,直接对从蛋白质主链结构衍生的特征进行操作,实现了同时预测每个残基的氨基酸类型和侧链构象.局部感知图(Locality Aware Graph)transformer利用蛋白质主链的几何形状来优化单个残基和残基对的特征表示,并将注意力限制在空间上相邻的残基对上.该模块的输出和蛋白质主链坐标一起被传递到张量融合网络(Tensor fusion network,TFN)[68 ] 输出一个标量和残基位置,然后由TFN-transformer为每个输入残基产生侧链构象和氨基酸类型.作者评估了五种不同的残基掩蔽方法并分别进行了损失函数、网络架构和模型超参数的消融实验,发现从损失函数中移除侧链坐标均方根偏差 (Root Mean Squared Deviation,RMSD)和预测的侧链原子之间的成对距离两个特征显著降低了测试蛋白上的天然序列恢复率.除此之外,移除模型中的 TFN-Transformer 层对恢复率的影响最大.与几种现有的序列设计方法对比而言,该模型在四个测试集上展现了更高的序列恢复率. ...
Increasing the efficiency and accuracy of the ABACUS protein sequence design method
2
2020
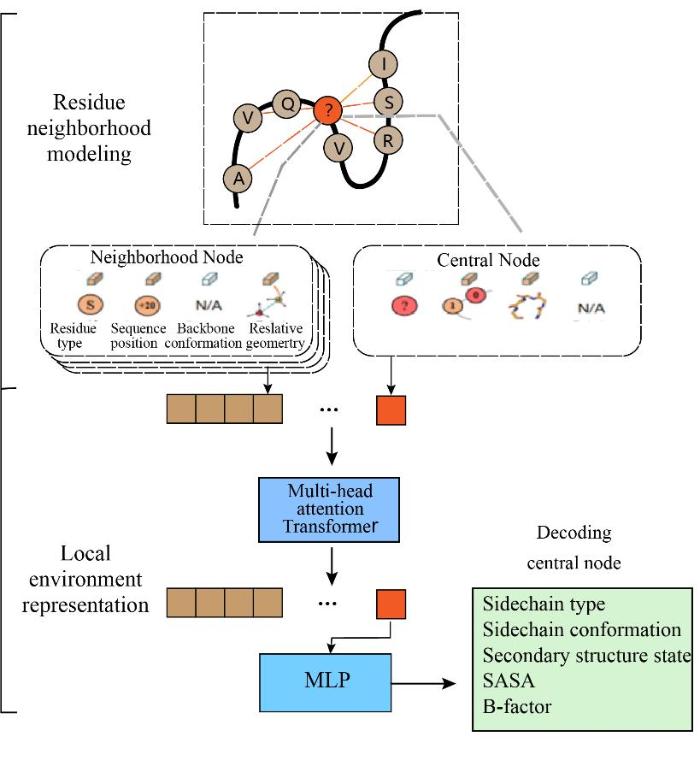
... ABACUS-R[69 -71 ] 使用一个多任务学习的编码器-解码器网络,根据固定骨架上局部环境预测中心位置的残基类型(图6 ).网络的输入是目标残基与最邻近k个残基联合形成的局部特征,包含空间层面的相对位置与取向信息、序列层面的相对位置信息以及邻近残基的残基类型.ABACUS-R模型不需要显示地模拟侧链,从而避免优化结构的过程.模型学习到给定结构下侧链类型的能量打分函数,通过在目标骨架上残基的迭代,逐轮降低随机残基数目,使得设计结果逐渐收敛,产生自洽的整体序列.ABACUS-R在单个残基平均序列恢复率达到53%,多个湿实验结果(包括X射线晶体学解析的晶体结构)表明,ABACUS-R在设计精度和成功率方面都优于基于能量函数的从头序列设计方法. ...
... 在模板未知条件下,使用神经网络形式的能量项模型SCUBA 驱动的随机动力学(Stochastic dynamics)和模拟退火算法(Simulated annealing)来生成可设计的新蛋白质主链骨架,再使用前文中提到的ABACUS2[69 ] 对主链骨架序列进行序列优化和骨架松弛[10 ] 设计的迭代,从而完成对蛋白质的可变骨架从头设计任务.在9种用SCUBA设计的高精度骨架蛋白结构中,其中有4种具有新颖的非天然结构.对于这一结果充分展示了SCUBA在蛋白设计中的实用性,特别是在设计功能蛋白时,能量函数驱动的骨架采样和优化可以很容易地进行定制,以促进对结构空间的广泛探索.另外,SCUBA+ ABACUS2[87 ] 策略所设计的蛋白质具有高于天然蛋白质骨架的热稳定性,设计成功率为~ 42 % ( 38个经实验验证的蛋白质中有16个成功折叠,14个H2E4蛋白质和4个H4蛋白质),设计的骨架与实验获得的结构一致,具有原子精度,同时设计的H2E4和H4蛋白与具有相似结构的已知天然蛋白质具有低序列同一性(平均同一性14%). ...
Rotamer-free protein sequence design based on deep learning and self-consistency
0
2022
Protein design with a comprehensive statistical energy function and boosted by experimental selection for foldability
1
2014
... ABACUS-R[69 -71 ] 使用一个多任务学习的编码器-解码器网络,根据固定骨架上局部环境预测中心位置的残基类型(图6 ).网络的输入是目标残基与最邻近k个残基联合形成的局部特征,包含空间层面的相对位置与取向信息、序列层面的相对位置信息以及邻近残基的残基类型.ABACUS-R模型不需要显示地模拟侧链,从而避免优化结构的过程.模型学习到给定结构下侧链类型的能量打分函数,通过在目标骨架上残基的迭代,逐轮降低随机残基数目,使得设计结果逐渐收敛,产生自洽的整体序列.ABACUS-R在单个残基平均序列恢复率达到53%,多个湿实验结果(包括X射线晶体学解析的晶体结构)表明,ABACUS-R在设计精度和成功率方面都优于基于能量函数的从头序列设计方法. ...
State-of-the-art estimation of protein model accuracy using AlphaFold
1
2022
... Roney[72 ] 认为Alphafold从蛋白质的共进化数据中学习了一个高精度的能量函数,可以在不使用任何共进化数据的情况下,确定蛋白质3D结构和序列之间的关系,从而用于蛋白质设计问题中.该流程类似于TrDesign,将目标蛋白骨架结构提供给AlphaFold作为模板,最小化目标结构和预测结构之间的差异,并优化关于输入序列的复合置信度评分(Composite Confidence Score).该设计方法达到约30%的序列恢复率. ...
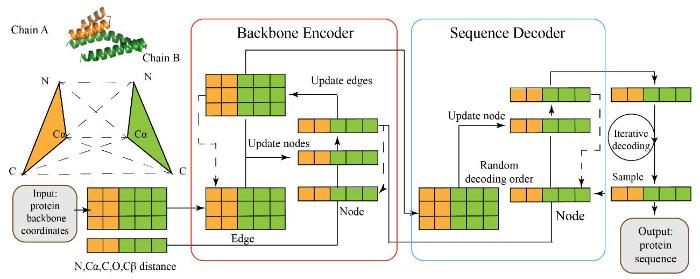
Robust deep learning-based protein sequence design using ProteinMPNN
2
2022
... ProteinMPNN[73 ] 参考GraphTrans,使用具有 3 个编码器和 3 个解码器层以及每层宽度为128 的消息传递网络(Message passing network, MPNN).作者认为相较于残基主链二面角和旋转走向,残基N、Cα、C、O 和Cβ原子之间的距离提供了更好的归纳偏置来捕获残基之间的相互作用.将上述特征输入MPNN网络(图7 ),使模型预测序列恢复从 41.2%增加到 49.0%. ...
... 其二,目前对于蛋白设计模型的性能评估大多为天然序列恢复率和预测结构与原结构之间的差异,然而这两个指标仅能够衡量设计序列或结构与原蛋白的全局相似程度,并不能很好量化设计蛋白的物理化学性质.Dauparas[73 ] 在ProteinMPNN文章中也指出天然序列恢复率对结构分辨率敏感,并且与局部残基距离误差相关性不高(Rpearson ~0.5),并不是一个能够很好评价蛋白序列预测模型性能的指标.单个关键残基预测的错误对整体天然序列恢复率影响不大,但对序列折叠能力是毁灭性的.未来的方向可能是引入更多的评价指标,局部指标包括二级结构恢复率、溶剂可及表面、设计序列中无序残基比例等[120 ] .设计结构的全局评估可以使用结构预测模型折叠的结构并计算与目标结构的差异;长时间分子动力学模拟能够衡量序列折叠后结构的稳定性、展现结合蛋白与靶点之间的相互作用构象.将深度学习方法与传统的基于能量函数的蛋白质设计方法联用或前后相接,将深度学习模型生成的大量候选序列或结构输入基于物理化学的能量函数模型中进行验证和筛选,挑选出最优序列进行实验验证.充分发挥深度学习模型的高通量序列生成能力和物理化学模型对于蛋白的可表达性、可溶性以及聚集效应等物理化学性质的把握能力. ...
Accurate and efficient protein sequence design through learning concise local environment of residues
1
2023
... 如果一个设计氨基酸序列的每个残基都与其局部环境很好地吻合,那么它就有望折叠成一个与目标结构相似的结构,ProDESIGN-LE[74 ] 便采用该思路.ProDESIGN-LE以每个邻近残基的残基类型和相对于中心残基的3×3变换矩阵R 和三维平移向量t 来表示中心残基的局部环境,将特征输入一个3层的transformer来学习残基对其局部环境的依赖性,并输出其嵌入图,后进一步使用全连接层将嵌入图转化为20种氨基酸类型的分布.训练好的transformer模型在目标结构的序列上迭代地选择合适的残基,并相应地更新相邻残基的局部环境,最终获得所有残基都与自身局部环境匹配良好的设计序列.ProDESIGN - LE模型在计算指标评估和实验验证上均取得不错的结果,在设计的5个CATⅢ蛋白中,有3个具有良好的溶解性. ...
Structure-informed language models are protein designers
2
2023
... 固定骨架序列设计模型在CATH 4.2测试集上的序列恢复率和困惑度[75 ] ...
... Perplexity (↓)
GraphTrans 35.82 6.63 StructGNN[76 ] 37.1 6.49 GVP-GNN-large 39.20 6.17 GVP-GNN-Transformer 38.30 6.44 GVP-GNN-Transformer+AF2 51.60 4.01 ProteinMPNN 45.96 4.61 ProDesign 50.22 4.69 PiFold[77 ] 50.22 4.62 LM-DESIGN[75 ] (PiFold) 55.65 4.52 表2 固定骨架序列设计模型在TS50 &TS500测试集上的序列恢复率和困惑度[77 ] ...
Generative models for graph-based protein design
1
2019
... Perplexity (↓)
GraphTrans 35.82 6.63 StructGNN[76 ] 37.1 6.49 GVP-GNN-large 39.20 6.17 GVP-GNN-Transformer 38.30 6.44 GVP-GNN-Transformer+AF2 51.60 4.01 ProteinMPNN 45.96 4.61 ProDesign 50.22 4.69 PiFold[77 ] 50.22 4.62 LM-DESIGN[75 ] (PiFold) 55.65 4.52 表2 固定骨架序列设计模型在TS50 &TS500测试集上的序列恢复率和困惑度[77 ] ...
ProDesign: Toward effective and efficient protein design
2
2022
... Perplexity (↓)
GraphTrans 35.82 6.63 StructGNN[76 ] 37.1 6.49 GVP-GNN-large 39.20 6.17 GVP-GNN-Transformer 38.30 6.44 GVP-GNN-Transformer+AF2 51.60 4.01 ProteinMPNN 45.96 4.61 ProDesign 50.22 4.69 PiFold[77 ] 50.22 4.62 LM-DESIGN[75 ] (PiFold) 55.65 4.52 表2 固定骨架序列设计模型在TS50 &TS500测试集上的序列恢复率和困惑度[77 ] ...
... 固定骨架序列设计模型在TS50 &TS500测试集上的序列恢复率和困惑度[77 ] ...
Generative De Novo Protein Design with Global Context
1
2022
... GNN
StructGNN 43.89 5.40 45.69 4.98 GraphTrans 42.20 5.60 44.66 5.16 GVP-GNN 44.14 4.71 49.14 4.20 GCA[78 ] 47.02 5.09 47.74 4.72 ADesign[79 ] 48.36 5.25 49.23 4.93 ProteinMPNN 54.43 3.93 58.08 3.53 PiFold 58.72 3.86 60.42 3.44 LM-DESIGN(PiFold) 57.89 3.50 67.78 3.19 2 可变骨架的序列设计 与固定骨架设计问题不同,在可变骨架设计问题中,蛋白质确切的骨架结构通常都是未知的,因此在设计过程中需要同时考虑优化序列和结构. ...
AlphaDesign: a graph protein design method and benchmark on AlphaFoldDB
1
2022
... GNN
StructGNN 43.89 5.40 45.69 4.98 GraphTrans 42.20 5.60 44.66 5.16 GVP-GNN 44.14 4.71 49.14 4.20 GCA[78 ] 47.02 5.09 47.74 4.72 ADesign[79 ] 48.36 5.25 49.23 4.93 ProteinMPNN 54.43 3.93 58.08 3.53 PiFold 58.72 3.86 60.42 3.44 LM-DESIGN(PiFold) 57.89 3.50 67.78 3.19 2 可变骨架的序列设计 与固定骨架设计问题不同,在可变骨架设计问题中,蛋白质确切的骨架结构通常都是未知的,因此在设计过程中需要同时考虑优化序列和结构. ...
De novo protein design by deep network hallucination
3
2021
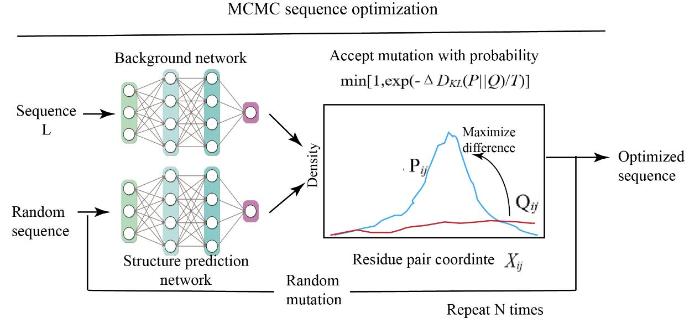
... 前文提到trRosetta能够快速预测一个蛋白质序列的空间约束, Ivan[80 ] 重新训练了一个背景网络,将输入trRosetta的序列在自身的输出结构上不断迭代,使预测结构的空间约束逐渐具有清晰的分布,这种方法被称为幻想(Hallucination)设计.首先将一个随机序列转换为折叠蛋白序列的编码,同时输入随机噪音得到背景的空间约束.使用马尔科夫链蒙特卡洛(Markov Chain Monte Carlo,MCMC )算法对序列进行随机突变,再将其输入trRosetta模型中逐轮预测空间约束,以Kullback-Leibler(KL)散度对序列约束和背景约束的分布差异进行优化,使得到的空间约束逐渐逼近真实蛋白质,并借此折叠蛋白3D结构(图8 ). ...
... Family-wide hallucination集成了无限制幻想设计[80 , 82 ] 与Rosetta序列设计方法[55 ] ,对环(loop)和可变区域(variable regions)的序列和结构进行从头设计,并对核心区域的结构进行序列优化.该方法从2000个天然NTF2s序列出发,在序列空间中进行蒙特卡洛搜索,每一步都进行一次序列变化,并使用trRosetta进行结构预测.模型的损失函数由两部分构成:结构保守区域基于与NTF2 - like蛋白实验结构的输入残基距离和方向分布的一致性进行评估;而可变区域基于网络预测与背景分布之间的KL散度计算的预测残基间几何结构的置信度进行评估.氢键网络也被纳入设计的结构中,以增加结构特异性.实验数据显示Family-wide hallucination生成的1615个骨架在原生结构的空间内采样更多,并且比原生骨架或非深度学习能量优化生成的骨架具有更强的序列结构关系. ...
... 此外基于VAE的模型还有Guo 等[96 ] 提出的DECO-VAE模型.在该模型中,训练数据集中的3D结构首先表示为二维接触图,而后经由图神经网络提取节点和边特征,输入编码器,解码器的输出以既定的方法还原为蛋白质3D结构. Harteveld等[97 ] 提出的GENESIS模型通过优化蛋白质拓扑晶格模型在距离和角度特征图中的2D表示来去噪蛋白质拓扑晶格模型草图.GENESIS结合trRosetta[80 ] 设计框架,为不同的蛋白质折叠生成了大量的不同序列. ...
Design of proteins presenting discontinuous functional sites using deep learning
1
2020
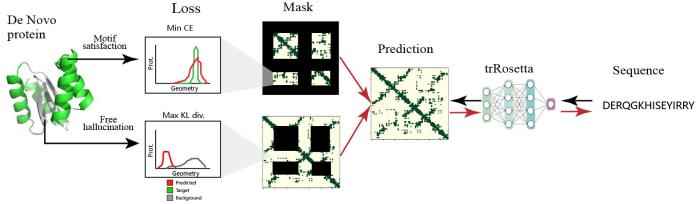
... TrDesign-motif[81 ] 将trRosetta和hallucination有机结合起来用以蛋白质结合motif的设计.对于活性位点,初始输入骨架的2D特征作为目标分布,让motif功能部分预测序列与原结构尽可能地相似;而在自由幻想部分,将随机噪音的2D特征分布作为背景,让生成的序列尽可能远离其分布.使用混合的损失函数来优化结构和序列,创造出一个携带功能motif片段的新蛋白结构. ...
Scaffolding protein functional sites using deep learning
5
2022
... RFDesign使用Constrained hallucination[82 ] 对幻想算法进行约束,优化序列,在保证预测结构的功能基序(motif)与目标结构接近的同时,自由幻想生成其非功能位点 (图9 ).Inpainting[82 ] 进行蛋白结构补全(即RFjoint2[82 ] ),将trRosetta换成RoseTTAFold框架,并尝试不同的结构掩蔽方式训练一个蛋白结构和序列模型,从功能位点出发填充非功能区的序列和结构,创建一个可行的蛋白质主链.Inpainting可以同时进行结构和序列生成,不依赖于trRosetta或反向传播的更新,可以通过输入主链走向来提高性能. ...
... [82 ]进行蛋白结构补全(即RFjoint2[82 ] ),将trRosetta换成RoseTTAFold框架,并尝试不同的结构掩蔽方式训练一个蛋白结构和序列模型,从功能位点出发填充非功能区的序列和结构,创建一个可行的蛋白质主链.Inpainting可以同时进行结构和序列生成,不依赖于trRosetta或反向传播的更新,可以通过输入主链走向来提高性能. ...
... [82 ]),将trRosetta换成RoseTTAFold框架,并尝试不同的结构掩蔽方式训练一个蛋白结构和序列模型,从功能位点出发填充非功能区的序列和结构,创建一个可行的蛋白质主链.Inpainting可以同时进行结构和序列生成,不依赖于trRosetta或反向传播的更新,可以通过输入主链走向来提高性能. ...
... 研究人员使用以上三种幻想方法设计了金属蛋白、酶活性位点和蛋白结合蛋白等,并都进行了计算机模拟和实验测试相结合的验证[82 ] .模型中的inpainting和hallucinate模块能够实现大肠杆菌铁蛋白(E. coli bacterioferritin)双铁结合位点的重新构建,在设计的96个铁蛋白结构中有76个可溶性表达,8个具有金属结合的特征光谱位移,3个具有与AlphaFold折叠结构一致的二级结构(圆二色光谱鉴定),并且能够稳定地与金属络合.幻想设计能够产生碳酸酐酶Ⅱ上三个Zn2+ 配位组氨酸和环上苏氨酸组成的基序,并正确放置Zn2+ 配位:幻想模型还构建了参与甾体激素生物合成的D5-3-酮甾体异构酶(KSI)的催化侧链,两种酶的活性位点与天然晶体结构几乎完全匹配.文章中还展示了幻想设计通过固定靶点蛋白和结合蛋白部分位点,修复缺失位点(inpainting)或自由幻想(hallucinate)全新的骨架结构来设计蛋白质结合蛋白的过程.其中设计的结合蛋白pdl1_inp_1与PD-L1结合能力(Kd=326 nM)相较于野生型PD-1(Kd=3.9 mM)增强;设计的TrkA在配体结合时呈现与天然结构相同的二聚化现象; 多种设计的Mdm2癌基因结合蛋白与抑癌蛋白p53的天然N端螺旋结合紧密. ...
... Family-wide hallucination集成了无限制幻想设计[80 , 82 ] 与Rosetta序列设计方法[55 ] ,对环(loop)和可变区域(variable regions)的序列和结构进行从头设计,并对核心区域的结构进行序列优化.该方法从2000个天然NTF2s序列出发,在序列空间中进行蒙特卡洛搜索,每一步都进行一次序列变化,并使用trRosetta进行结构预测.模型的损失函数由两部分构成:结构保守区域基于与NTF2 - like蛋白实验结构的输入残基距离和方向分布的一致性进行评估;而可变区域基于网络预测与背景分布之间的KL散度计算的预测残基间几何结构的置信度进行评估.氢键网络也被纳入设计的结构中,以增加结构特异性.实验数据显示Family-wide hallucination生成的1615个骨架在原生结构的空间内采样更多,并且比原生骨架或非深度学习能量优化生成的骨架具有更强的序列结构关系. ...
AutoFoldFinder: an automated adaptive optimization toolkit for de novo protein fold design
1
... Zhang[83 ] 基于上文提到的hallucinate方法,提出一种从头设计蛋白质折叠的自动自适应优化工具包AutoFoldFinder,通过序列优化的方式产生具有新蛋白元件排列方式的氨基酸序列与结构,使用同余系数图对齐(congruence coefficient map alignment,CM-align)替换hallucinate方法中的KL散度,无需对整个接触图的全局比较,能够更精细地反映接触图在局部二级结构上的特征差异.AutoFoldFinder通过序列优化将生成一千条蛋白质序列中低相似度序列比例从22%提升至30.9%,加入CM-Align方法后,超过50%的结构与已知结构有显著差异. ...
De novo design of luciferases using deep learning
1
2023
... 最近Baker等[84 ] 发布了首个使用深度学习工具从头设计荧光酶结构的工作.研究人员选择合成荧光素酶底物二苯基特拉嗪(diphenylterrazine,DTZ)作为目标酶的作用底物,作者首先构建了DTZ阴离子构象系综,随后围绕每个构象,使用RIFGen方法[85 -86 ] 枚举了与DTZ相互作用的氨基酸侧链旋转异构体相互作用场( RIFs ),最后使用RIFDock将每个DTZ构象和RIF在约4000个天然蛋白骨架的中心腔中进行对接,以最大化蛋白- DTZ相互作用.此方法发现与DTZ结构互补的结合口袋中大多为核转运因子2 ( nuclear transport factor 2,NTF2 )家族蛋白,将对接获得的骨架和口袋使用Family-wide hallucination方法进行优化设计. ...
De novo design of a fluorescence-activating β-barrel
1
2018
... 最近Baker等[84 ] 发布了首个使用深度学习工具从头设计荧光酶结构的工作.研究人员选择合成荧光素酶底物二苯基特拉嗪(diphenylterrazine,DTZ)作为目标酶的作用底物,作者首先构建了DTZ阴离子构象系综,随后围绕每个构象,使用RIFGen方法[85 -86 ] 枚举了与DTZ相互作用的氨基酸侧链旋转异构体相互作用场( RIFs ),最后使用RIFDock将每个DTZ构象和RIF在约4000个天然蛋白骨架的中心腔中进行对接,以最大化蛋白- DTZ相互作用.此方法发现与DTZ结构互补的结合口袋中大多为核转运因子2 ( nuclear transport factor 2,NTF2 )家族蛋白,将对接获得的骨架和口袋使用Family-wide hallucination方法进行优化设计. ...
Design of protein-binding proteins from the target structure alone
1
2022
... 最近Baker等[84 ] 发布了首个使用深度学习工具从头设计荧光酶结构的工作.研究人员选择合成荧光素酶底物二苯基特拉嗪(diphenylterrazine,DTZ)作为目标酶的作用底物,作者首先构建了DTZ阴离子构象系综,随后围绕每个构象,使用RIFGen方法[85 -86 ] 枚举了与DTZ相互作用的氨基酸侧链旋转异构体相互作用场( RIFs ),最后使用RIFDock将每个DTZ构象和RIF在约4000个天然蛋白骨架的中心腔中进行对接,以最大化蛋白- DTZ相互作用.此方法发现与DTZ结构互补的结合口袋中大多为核转运因子2 ( nuclear transport factor 2,NTF2 )家族蛋白,将对接获得的骨架和口袋使用Family-wide hallucination方法进行优化设计. ...
A backbone-centred energy function of neural networks for protein design
2
2022
... 可变骨架的蛋白质设计可以分解成骨架结构的生成和固定骨架设计两个独立的子任务.中国科学技术大学刘海燕组提出了一种全新的,使用神经网络形式能量项的统计模型——SCUBA[87 ] ,使基于连续采样和优化主链中心能量面来设计新主链的方法成为可能.SCUBA模型将主链的可设计性分解为几个关键因素的作用,包括局部构象倾向性、肽主链氢键几何构象以及手性附着和紧密排列的侧链所需的骨架空间.研究者使用统计能量项来表示各种相互作用,用一种名为邻接计数神经网络(Neighbor counting-neural network, NC-NN)的通用方法训练.NC-NN包含两步过程,首先通过基于核的密度估计(即邻接计数)从原始结构数据估计统计能量值,然后训练神经网络(三层全连接感知机)表示势.得到的统计能量项,除了可以提供易于计算的函数值和导数用于结构采样和优化外,还可以高保真地表示复杂的、高维且高度相关的真实结构数据分布. ...
... 在模板未知条件下,使用神经网络形式的能量项模型SCUBA 驱动的随机动力学(Stochastic dynamics)和模拟退火算法(Simulated annealing)来生成可设计的新蛋白质主链骨架,再使用前文中提到的ABACUS2[69 ] 对主链骨架序列进行序列优化和骨架松弛[10 ] 设计的迭代,从而完成对蛋白质的可变骨架从头设计任务.在9种用SCUBA设计的高精度骨架蛋白结构中,其中有4种具有新颖的非天然结构.对于这一结果充分展示了SCUBA在蛋白设计中的实用性,特别是在设计功能蛋白时,能量函数驱动的骨架采样和优化可以很容易地进行定制,以促进对结构空间的广泛探索.另外,SCUBA+ ABACUS2[87 ] 策略所设计的蛋白质具有高于天然蛋白质骨架的热稳定性,设计成功率为~ 42 % ( 38个经实验验证的蛋白质中有16个成功折叠,14个H2E4蛋白质和4个H4蛋白质),设计的骨架与实验获得的结构一致,具有原子精度,同时设计的H2E4和H4蛋白与具有相似结构的已知天然蛋白质具有低序列同一性(平均同一性14%). ...
De novo protein design by an energy function based on series expansion in distance and orientation dependence
1
2021
... Liang等[88 ] 随后发展了一个基于级数展开的能量函数模型OSCAR-Design.在四个独立的阶段中优化目标函数E total =E side + E bb + E ref 的各项参数,最大化原结构和其他旋转异构体之间的能量差;最小化天然环结构中选择环诱饵之间的RMSD,最大化氨基酸组成与天然序列的相似性;惩罚埋藏的非氢键极性原子.作者使用Monte Carlo模拟退火算法对OSCAR-Design进行测试.OSCAR-Design在侧链和loop预测任务中与OSCAR[89 -90 ] 和LEAP[91 ] 一样准确.在从头设计任务中,OSCAR-Design在测试集达到38 ~43 %天然序列恢复率,在75 %的情况下成功保留了亲疏水性残基,在氨基酸组成的整体相似性达到90 %. ...
Fast and accurate prediction of protein side-chain conformations
1
2011
... Liang等[88 ] 随后发展了一个基于级数展开的能量函数模型OSCAR-Design.在四个独立的阶段中优化目标函数E total =E side + E bb + E ref 的各项参数,最大化原结构和其他旋转异构体之间的能量差;最小化天然环结构中选择环诱饵之间的RMSD,最大化氨基酸组成与天然序列的相似性;惩罚埋藏的非氢键极性原子.作者使用Monte Carlo模拟退火算法对OSCAR-Design进行测试.OSCAR-Design在侧链和loop预测任务中与OSCAR[89 -90 ] 和LEAP[91 ] 一样准确.在从头设计任务中,OSCAR-Design在测试集达到38 ~43 %天然序列恢复率,在75 %的情况下成功保留了亲疏水性残基,在氨基酸组成的整体相似性达到90 %. ...
Protein side chain modeling with orientation-dependent atomic force fields derived by series expansions
1
2011
... Liang等[88 ] 随后发展了一个基于级数展开的能量函数模型OSCAR-Design.在四个独立的阶段中优化目标函数E total =E side + E bb + E ref 的各项参数,最大化原结构和其他旋转异构体之间的能量差;最小化天然环结构中选择环诱饵之间的RMSD,最大化氨基酸组成与天然序列的相似性;惩罚埋藏的非氢键极性原子.作者使用Monte Carlo模拟退火算法对OSCAR-Design进行测试.OSCAR-Design在侧链和loop预测任务中与OSCAR[89 -90 ] 和LEAP[91 ] 一样准确.在从头设计任务中,OSCAR-Design在测试集达到38 ~43 %天然序列恢复率,在75 %的情况下成功保留了亲疏水性残基,在氨基酸组成的整体相似性达到90 %. ...
LEAP: highly accurate prediction of protein loop conformations by integrating coarse-grained sampling and optimized energy scores with all-atom refinement of backbone and side chains
1
2014
... Liang等[88 ] 随后发展了一个基于级数展开的能量函数模型OSCAR-Design.在四个独立的阶段中优化目标函数E total =E side + E bb + E ref 的各项参数,最大化原结构和其他旋转异构体之间的能量差;最小化天然环结构中选择环诱饵之间的RMSD,最大化氨基酸组成与天然序列的相似性;惩罚埋藏的非氢键极性原子.作者使用Monte Carlo模拟退火算法对OSCAR-Design进行测试.OSCAR-Design在侧链和loop预测任务中与OSCAR[89 -90 ] 和LEAP[91 ] 一样准确.在从头设计任务中,OSCAR-Design在测试集达到38 ~43 %天然序列恢复率,在75 %的情况下成功保留了亲疏水性残基,在氨基酸组成的整体相似性达到90 %. ...
Generative Modeling for Protein Structures
1
3
... Huang等[92 ] 提出了一种基于生成对抗网络(Generative adversarial network, GAN)的生成模型,策略具体细节如图11 所示.蛋白质的结构使用蛋白质主链上成对Cα 之间的距离(以埃为单位)来表示.GAN模型中的生成器通过输入一个正态分布随机变量z~N (0, I ),输出一个成对距离图,判别器判断生成器输出的结果是真实的(数据样本)或是虚假的(生成器输出),而后生成器对生成的结果不断迭代优化用以欺骗判别器,整个模型最终输出得到合理的成对距离图.得到的距离图随后通过交替方向乘子法(Alternating direction multiplier method,ADMM)折叠成3D结构从而得到Cα 的坐标,最后使用一个快速追踪脚本将Cα 原子的坐标匹配到一个合理的蛋白质骨架.研究者将此方案应用于补全蛋白质结构中缺失残基的任务,同时还扩展生成建模程序来解决端到端的结构恢复问题,并减少当前模型在精细局部结构中出错的问题.在后续研究中,Huang等人进一步优化了他们的方案[93 ] ,通过所有主链原子之间的成对距离来表示蛋白质结构,并提出了一种以可微分的方式直接恢复和细化相应主链坐标的方法(图10 ).具体来说,在GAN生成骨架原子距离矩阵之后,采用卷积神经网络,通过自编码器损失从成对距离矩阵中恢复蛋白质骨架坐标.相较于ADMM恢复方法,这种新提出的方案是一种快速、完全可微分的方法,即生成的3D骨架坐标的错误可以反向传播到生成器网络. ...
Fully differentiable full-atom protein backbone generation
1
2019
... Huang等[92 ] 提出了一种基于生成对抗网络(Generative adversarial network, GAN)的生成模型,策略具体细节如图11 所示.蛋白质的结构使用蛋白质主链上成对Cα 之间的距离(以埃为单位)来表示.GAN模型中的生成器通过输入一个正态分布随机变量z~N (0, I ),输出一个成对距离图,判别器判断生成器输出的结果是真实的(数据样本)或是虚假的(生成器输出),而后生成器对生成的结果不断迭代优化用以欺骗判别器,整个模型最终输出得到合理的成对距离图.得到的距离图随后通过交替方向乘子法(Alternating direction multiplier method,ADMM)折叠成3D结构从而得到Cα 的坐标,最后使用一个快速追踪脚本将Cα 原子的坐标匹配到一个合理的蛋白质骨架.研究者将此方案应用于补全蛋白质结构中缺失残基的任务,同时还扩展生成建模程序来解决端到端的结构恢复问题,并减少当前模型在精细局部结构中出错的问题.在后续研究中,Huang等人进一步优化了他们的方案[93 ] ,通过所有主链原子之间的成对距离来表示蛋白质结构,并提出了一种以可微分的方式直接恢复和细化相应主链坐标的方法(图10 ).具体来说,在GAN生成骨架原子距离矩阵之后,采用卷积神经网络,通过自编码器损失从成对距离矩阵中恢复蛋白质骨架坐标.相较于ADMM恢复方法,这种新提出的方案是一种快速、完全可微分的方法,即生成的3D骨架坐标的错误可以反向传播到生成器网络. ...
Ig-VAE: Generative modeling of protein structure by direct 3D coordinate generation
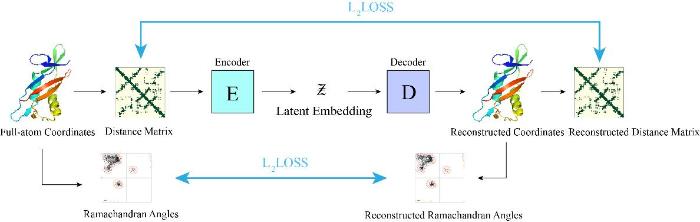
3
2022
... 以上提到的GAN方法在结构生成领域表现出了较好的性能,但也存在一定的弊端,例如生成的距离约束不能保证是欧氏有效的,因此不能恢复完全满足生成的约束的3D坐标[94 ] .2020年Huang等[94 ] 提出了一种构建蛋白质骨架的新方法—Ig-VAE,使用变分自编码器(Variational autoencoder,VAE)直接生成免疫球蛋白的三维坐标.模型的架构如图12 所示.首先通过输入蛋白的原子坐标计算出主链残基二面角和距离矩阵,其次将距离矩阵输入编码器压缩特征得到低维的潜在空间表征,潜在空间表征传递给解码器,解码器直接生成蛋白3D空间中的坐标(图12 ).通过重构出的坐标重新计算主链残基二面角和距离矩阵,角度和距离矩阵的误差都通过3D坐标反向传播进网络中.训练完成后,Ig-VAE在结构嵌入及重构、隐空间插值以及生成能力方面表现良好,是一种构建单结构域抗体的有效工具. ...
... [94 ]提出了一种构建蛋白质骨架的新方法—Ig-VAE,使用变分自编码器(Variational autoencoder,VAE)直接生成免疫球蛋白的三维坐标.模型的架构如图12 所示.首先通过输入蛋白的原子坐标计算出主链残基二面角和距离矩阵,其次将距离矩阵输入编码器压缩特征得到低维的潜在空间表征,潜在空间表征传递给解码器,解码器直接生成蛋白3D空间中的坐标(图12 ).通过重构出的坐标重新计算主链残基二面角和距离矩阵,角度和距离矩阵的误差都通过3D坐标反向传播进网络中.训练完成后,Ig-VAE在结构嵌入及重构、隐空间插值以及生成能力方面表现良好,是一种构建单结构域抗体的有效工具. ...
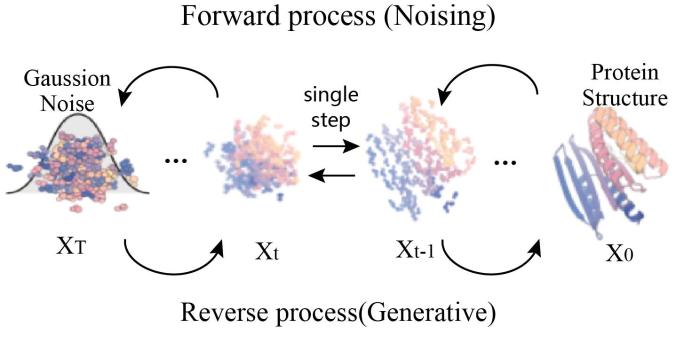
... 现有的蛋白质3D结构生成方法仅限于在高度约束的环境中生成蛋白的拓扑结构[94 ] .去噪扩散概率模型(Denoising diffusion probabilistic models, DDPMs)是一类从复杂数据分布中采样的生成模型.DDPMs定义了一个正向扩散过程,将数据扰动为噪声,学习反向过程中每一步的噪声为何,再逐步从数据分布中将随机高斯噪声去噪最终产生样本.近年来被训练用来以重建被不同数量的噪声破坏的数据(例如图像或文本).DDPMs应用于蛋白设计领域则是将加噪后的蛋白质结构多步迭代后还原为真实结构用以训练;使用训练好的模型对输入随机的高斯噪音逐步“去噪”来生成折叠性质完好的蛋白结构,实现蛋白设计或结构生成. ...
End-to-End deep structure generative model for protein design
1
2022
... 2022年许锦波组提出了一种直接在三维坐标空间中对蛋白质结构进行建模的、基于VAE的模型[95 ] ,相比于先前提出的直接坐标生成模型[3 ] 其应用仅限于固定长度的蛋白质,新提出的模型通过提取蛋白质几何形状的不变表示(Invariant representations),并使用局部对齐的坐标损失函数直接在坐标空间上执行梯度优化,解决了输入和输出空间中的旋转和平移等方差,因此可以直接、灵活地对三维结构进行建模. ...
Generating tertiary protein structures via interpretable graph variational autoencoders
1
2021
... 此外基于VAE的模型还有Guo 等[96 ] 提出的DECO-VAE模型.在该模型中,训练数据集中的3D结构首先表示为二维接触图,而后经由图神经网络提取节点和边特征,输入编码器,解码器的输出以既定的方法还原为蛋白质3D结构. Harteveld等[97 ] 提出的GENESIS模型通过优化蛋白质拓扑晶格模型在距离和角度特征图中的2D表示来去噪蛋白质拓扑晶格模型草图.GENESIS结合trRosetta[80 ] 设计框架,为不同的蛋白质折叠生成了大量的不同序列. ...
Deep sharpening of topological features for de novo protein design
1
2022
... 此外基于VAE的模型还有Guo 等[96 ] 提出的DECO-VAE模型.在该模型中,训练数据集中的3D结构首先表示为二维接触图,而后经由图神经网络提取节点和边特征,输入编码器,解码器的输出以既定的方法还原为蛋白质3D结构. Harteveld等[97 ] 提出的GENESIS模型通过优化蛋白质拓扑晶格模型在距离和角度特征图中的2D表示来去噪蛋白质拓扑晶格模型草图.GENESIS结合trRosetta[80 ] 设计框架,为不同的蛋白质折叠生成了大量的不同序列. ...
Denoising diffusion probabilistic models
1
2020
... DDPMs模型[98 -99 ] 输入的随机性使得去噪轨迹和输出的结构具备高度多样性,模型不需要起始的三维拓扑结构信息,但可以通过提供额外初始结构信息或施加外部约束条件,引导结构生成过程中每个步骤的迭代,直至特定的设计目标(图13 ). ...
Deep unsupervised learning using nonequilibrium thermodynamics
1
2015
... DDPMs模型[98 -99 ] 输入的随机性使得去噪轨迹和输出的结构具备高度多样性,模型不需要起始的三维拓扑结构信息,但可以通过提供额外初始结构信息或施加外部约束条件,引导结构生成过程中每个步骤的迭代,直至特定的设计目标(图13 ). ...
Broadly applicable and accurate protein design by integrating structure prediction networks and diffusion generative models
2
2022
... DDPMs模型
[98 -99 ] 输入的随机性使得去噪轨迹和输出的结构具备高度多样性,模型不需要起始的三维拓扑结构信息,但可以通过提供额外初始结构信息或施加外部约束条件,引导结构生成过程中每个步骤的迭代,直至特定的设计目标(
图13 ).
图 13 蛋白质结构生成扩散模型的原理示意图<sup>[<xref ref-type="bibr" rid="R100">100</xref>]</sup> Schematic diagram of diffusion model for protein structure generation Fig. 13 ![]()
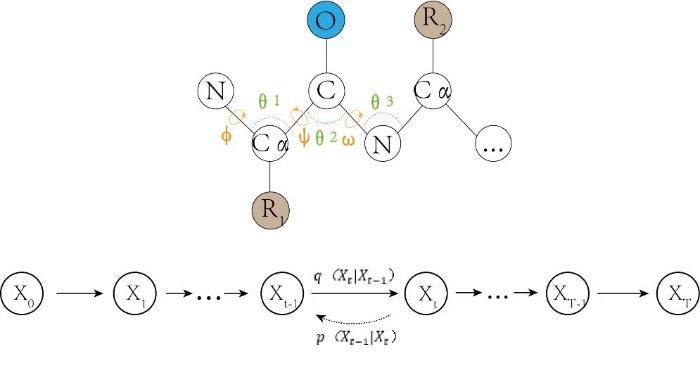
Trippe等[101 ] 开发了ProtDiff(一种蛋白骨架扩散概率模型)以及SMCDiff(一种以模体为条件的骨架生成方法).ProtDiff模型采用分子 E(3) 等变扩散模型用于蛋白质结构生成 .SMCDiff是一种基于顺序蒙特卡洛的模体-骨架问题解决模型,将无条件训练的扩散概率模型用于条件采样.模体-骨架生成整体框架包含两个步骤,首先训练 ProtDiff 来学习蛋白质骨架上的分布,然后使用 SMCDiff 和 ProtDiff 来修补给定模体.评估结果表明,该框架能够生成多样化的超过20个残基的支架,计算时间在几分钟或更短的数量级.2022年Wu等[102 ] 提出了FoldingDiff,一种使用Transformer作为主干训练的去噪扩散概率模型(图14 ).FoldingDiff的训练流程如下图所示.对于蛋白质的3D结构,研究者们使用氨基酸残基间的角度(ψ、ω、φ、θ1 、θ2 、θ3 )来表示,其中三个角为二面角,另外三个角为键角.训练天然蛋白骨架X0 开始,通过正向过程向其中迭代添加高斯噪声,直到Xt时刻角度无法辨识.反向过程中,研究者们采用了一个双向的transformer架构,在正向过程中得到的实例上学习反向去噪过程.经过训练得到的扩散模型可以生成高质量的、多样化的、在生物学上合理的蛋白质结构.生成的结构可带有手性,同时表现出高度的可设计性. ...
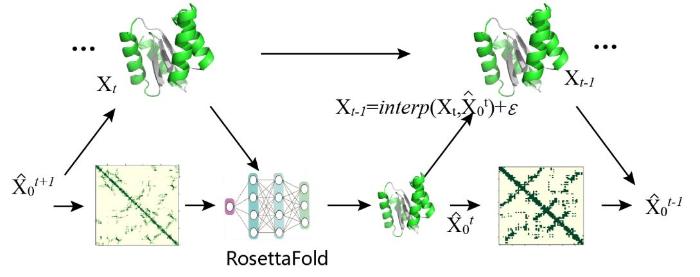
... Baker组随后推出基于RoseTTAFold(RF)的扩散模型RFdiffusion[100 ] . 将扩散模型建模为预训练后微调的RoseTTAFold模型 (图16 ).在使用RoseTTAFold进行经典结构预测时,模型的结构输入来自同源模板结构,每个模板结构都有相关的每个残基的"置信度"值.在RFdiffusion中,结构输入来自于部分(去)噪声的结构,置信度特征被重新参数化以表示当前的去噪时间步,模型在该时间步的条件上进行结构预测,然后计算当前输入结构到预测的最终结构的噪声插值,生成去噪的结构并输入到下一个时间步.RFdiffusion有着RF的序列信息通道,类似于前文中提到的RFjoint,能够在扩散生成时逐渐地恢复被遮蔽的序列,通过输入部分遮蔽的序列和完整结构模板来预测未知位置的氨基酸分布,实现部分序列设计.为了生成用于训练或推断的加噪蛋白质结构,作者用N - Cα - C骨架对残基编码并进行正向扩散.对于平移,用3D高斯噪声对残基Cα坐标进行局部扰动;对于旋转,使用等变的SO ( 3 )-transformer [108 ] 在旋转矩阵上模拟布朗运动生成噪声[109 ] ,使得模型具有全局的旋转不变性和高维的表征能力.在后续无条件约束策略设计和限制拓扑结构设计两种策略下,RFdiffusion设计了包括蛋白质单体、蛋白质-肽复合物、对称寡聚体、酶和金属结合蛋白等多种类型的蛋白,证明了RFdiffusion的在蛋白设计任务中的有效性和通用性. ...
Diffusion probabilistic modeling of protein backbones in 3D for the motif-scaffolding problem
1
2022
... Trippe等[101 ] 开发了ProtDiff(一种蛋白骨架扩散概率模型)以及SMCDiff(一种以模体为条件的骨架生成方法).ProtDiff模型采用分子 E(3) 等变扩散模型用于蛋白质结构生成 .SMCDiff是一种基于顺序蒙特卡洛的模体-骨架问题解决模型,将无条件训练的扩散概率模型用于条件采样.模体-骨架生成整体框架包含两个步骤,首先训练 ProtDiff 来学习蛋白质骨架上的分布,然后使用 SMCDiff 和 ProtDiff 来修补给定模体.评估结果表明,该框架能够生成多样化的超过20个残基的支架,计算时间在几分钟或更短的数量级.2022年Wu等[102 ] 提出了FoldingDiff,一种使用Transformer作为主干训练的去噪扩散概率模型(图14 ).FoldingDiff的训练流程如下图所示.对于蛋白质的3D结构,研究者们使用氨基酸残基间的角度(ψ、ω、φ、θ1 、θ2 、θ3 )来表示,其中三个角为二面角,另外三个角为键角.训练天然蛋白骨架X0 开始,通过正向过程向其中迭代添加高斯噪声,直到Xt时刻角度无法辨识.反向过程中,研究者们采用了一个双向的transformer架构,在正向过程中得到的实例上学习反向去噪过程.经过训练得到的扩散模型可以生成高质量的、多样化的、在生物学上合理的蛋白质结构.生成的结构可带有手性,同时表现出高度的可设计性. ...
Protein structure generation via folding diffusion
1
2022
... Trippe等[101 ] 开发了ProtDiff(一种蛋白骨架扩散概率模型)以及SMCDiff(一种以模体为条件的骨架生成方法).ProtDiff模型采用分子 E(3) 等变扩散模型用于蛋白质结构生成 .SMCDiff是一种基于顺序蒙特卡洛的模体-骨架问题解决模型,将无条件训练的扩散概率模型用于条件采样.模体-骨架生成整体框架包含两个步骤,首先训练 ProtDiff 来学习蛋白质骨架上的分布,然后使用 SMCDiff 和 ProtDiff 来修补给定模体.评估结果表明,该框架能够生成多样化的超过20个残基的支架,计算时间在几分钟或更短的数量级.2022年Wu等[102 ] 提出了FoldingDiff,一种使用Transformer作为主干训练的去噪扩散概率模型(图14 ).FoldingDiff的训练流程如下图所示.对于蛋白质的3D结构,研究者们使用氨基酸残基间的角度(ψ、ω、φ、θ1 、θ2 、θ3 )来表示,其中三个角为二面角,另外三个角为键角.训练天然蛋白骨架X0 开始,通过正向过程向其中迭代添加高斯噪声,直到Xt时刻角度无法辨识.反向过程中,研究者们采用了一个双向的transformer架构,在正向过程中得到的实例上学习反向去噪过程.经过训练得到的扩散模型可以生成高质量的、多样化的、在生物学上合理的蛋白质结构.生成的结构可带有手性,同时表现出高度的可设计性. ...
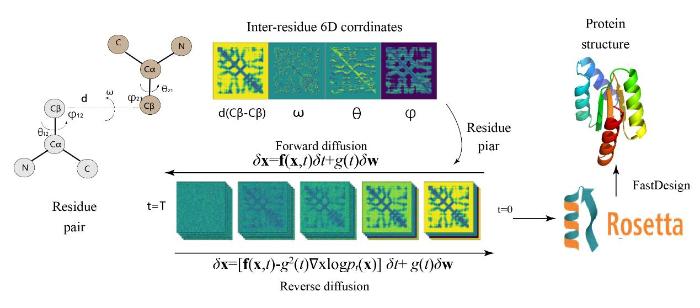
ProteinSGM: Score-based generative modeling for de novo protein design
1
... ProteinSGM[103 ] 模型可以从头产生真实的蛋白质,并且可以将输入的蛋白骨架和功能位点修复为预定义长度的完整蛋白结构.ProteinSGM将两个残基之间的6D坐标特征作为输入特征,将其转化为2D的蛋白质残基接触矩阵(图15 ).扩散模型在2D接触矩阵上逐渐添加噪音并迭代进行学习正向扩散的进程,训练完成的模型再对噪声反向逐步去噪,从噪声中生成真实的残基接触矩阵样本,后转化为蛋白质6D坐标.使用模型的输出残基约束指导Rosetta Design[104 ] 和Relax生成与6D坐标约束相对应的蛋白质结构.因为连续时间扩散模型的采样需要大量正向传播的得分网络来求解反向梯度,而RosettaDesign依赖于昂贵的蒙特卡洛(MCMC)算法来遍历结构势能面找到局部最小值对应的低能量结构,因此模型在高通量设计任务中选择外接结构预测算法(如AlphaFold2等)来减小计算量. ...
ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules
1
2011
... ProteinSGM[103 ] 模型可以从头产生真实的蛋白质,并且可以将输入的蛋白骨架和功能位点修复为预定义长度的完整蛋白结构.ProteinSGM将两个残基之间的6D坐标特征作为输入特征,将其转化为2D的蛋白质残基接触矩阵(图15 ).扩散模型在2D接触矩阵上逐渐添加噪音并迭代进行学习正向扩散的进程,训练完成的模型再对噪声反向逐步去噪,从噪声中生成真实的残基接触矩阵样本,后转化为蛋白质6D坐标.使用模型的输出残基约束指导Rosetta Design[104 ] 和Relax生成与6D坐标约束相对应的蛋白质结构.因为连续时间扩散模型的采样需要大量正向传播的得分网络来求解反向梯度,而RosettaDesign依赖于昂贵的蒙特卡洛(MCMC)算法来遍历结构势能面找到局部最小值对应的低能量结构,因此模型在高通量设计任务中选择外接结构预测算法(如AlphaFold2等)来减小计算量. ...
Illuminating protein space with a programmable generative model
1
2022
... Ingraham等[105 ] 提出的Chroma模型,能够直接对新的蛋白质结构和序列进行采样,并调节生成过程,使其达到所需的特性和功能,同时实现完整蛋白复合物的3D结构和序列的联合建模且计算效率十分可观.模型可以在不同线索下实现条件采样,而无需重新训练.Chroma 实现了一种可编程蛋白质设计的新模式,这种模式为生成特定和量身定制的蛋白质提供了可行性. ...
Protein structure and sequence generation with equivariant denoising diffusion probabilistic models
1
2022
... Anand[106 ] 模型通过定义二级结构和残基接触矩阵约束嵌入到高维空间,再使用IPA模块降维到三维空间中表征蛋白结构.作者使用AlphaFold网络架构中的[38 ] 不变点注意力(Invariant Point Attention, IPA)模块替换transformer中的标准注意力模块保证模型的平移旋转不变性,使用类似于BERT[107 ] 的扩散方法在骨架上生成序列.与其他DDPM模型不同,该模型不使用随机产生的高斯噪声,而是通过随机掩盖部分残基,在[0, 1]中作为t的函数进行线性插值来训练模型;在生成时,模型在t = T时掩蔽所有的残基来进行反向过程,从t = T到t = 0的时间步进行迭代采样.模型还允许人为给定条件信息编码蛋白结构.该模型完全从真实蛋白结构数据中学习,并生成蛋白质拓扑结构的条件约束,以产生全原子骨架构型以及序列和侧链预测.作者用了3个独立训练的模型分别生成蛋白结构、序列和转子,并将模型应用于无序列从头生成、蛋白补全、序列设计、侧链转子重排等任务中,结果表明其具有作为端到端的蛋白质从头设计工具的潜力. ...
BERT: Pre-training of deep bidirectional transformers for language understanding
1
2018
... Anand[106 ] 模型通过定义二级结构和残基接触矩阵约束嵌入到高维空间,再使用IPA模块降维到三维空间中表征蛋白结构.作者使用AlphaFold网络架构中的[38 ] 不变点注意力(Invariant Point Attention, IPA)模块替换transformer中的标准注意力模块保证模型的平移旋转不变性,使用类似于BERT[107 ] 的扩散方法在骨架上生成序列.与其他DDPM模型不同,该模型不使用随机产生的高斯噪声,而是通过随机掩盖部分残基,在[0, 1]中作为t的函数进行线性插值来训练模型;在生成时,模型在t = T时掩蔽所有的残基来进行反向过程,从t = T到t = 0的时间步进行迭代采样.模型还允许人为给定条件信息编码蛋白结构.该模型完全从真实蛋白结构数据中学习,并生成蛋白质拓扑结构的条件约束,以产生全原子骨架构型以及序列和侧链预测.作者用了3个独立训练的模型分别生成蛋白结构、序列和转子,并将模型应用于无序列从头生成、蛋白补全、序列设计、侧链转子重排等任务中,结果表明其具有作为端到端的蛋白质从头设计工具的潜力. ...
Riemannian score-based generative modelling
1
2022
... Baker组随后推出基于RoseTTAFold(RF)的扩散模型RFdiffusion[100 ] . 将扩散模型建模为预训练后微调的RoseTTAFold模型 (图16 ).在使用RoseTTAFold进行经典结构预测时,模型的结构输入来自同源模板结构,每个模板结构都有相关的每个残基的"置信度"值.在RFdiffusion中,结构输入来自于部分(去)噪声的结构,置信度特征被重新参数化以表示当前的去噪时间步,模型在该时间步的条件上进行结构预测,然后计算当前输入结构到预测的最终结构的噪声插值,生成去噪的结构并输入到下一个时间步.RFdiffusion有着RF的序列信息通道,类似于前文中提到的RFjoint,能够在扩散生成时逐渐地恢复被遮蔽的序列,通过输入部分遮蔽的序列和完整结构模板来预测未知位置的氨基酸分布,实现部分序列设计.为了生成用于训练或推断的加噪蛋白质结构,作者用N - Cα - C骨架对残基编码并进行正向扩散.对于平移,用3D高斯噪声对残基Cα坐标进行局部扰动;对于旋转,使用等变的SO ( 3 )-transformer [108 ] 在旋转矩阵上模拟布朗运动生成噪声[109 ] ,使得模型具有全局的旋转不变性和高维的表征能力.在后续无条件约束策略设计和限制拓扑结构设计两种策略下,RFdiffusion设计了包括蛋白质单体、蛋白质-肽复合物、对称寡聚体、酶和金属结合蛋白等多种类型的蛋白,证明了RFdiffusion的在蛋白设计任务中的有效性和通用性. ...
Denoising diffusion probabilistic models on SO(3) for rotational alignment
1
2022
... Baker组随后推出基于RoseTTAFold(RF)的扩散模型RFdiffusion[100 ] . 将扩散模型建模为预训练后微调的RoseTTAFold模型 (图16 ).在使用RoseTTAFold进行经典结构预测时,模型的结构输入来自同源模板结构,每个模板结构都有相关的每个残基的"置信度"值.在RFdiffusion中,结构输入来自于部分(去)噪声的结构,置信度特征被重新参数化以表示当前的去噪时间步,模型在该时间步的条件上进行结构预测,然后计算当前输入结构到预测的最终结构的噪声插值,生成去噪的结构并输入到下一个时间步.RFdiffusion有着RF的序列信息通道,类似于前文中提到的RFjoint,能够在扩散生成时逐渐地恢复被遮蔽的序列,通过输入部分遮蔽的序列和完整结构模板来预测未知位置的氨基酸分布,实现部分序列设计.为了生成用于训练或推断的加噪蛋白质结构,作者用N - Cα - C骨架对残基编码并进行正向扩散.对于平移,用3D高斯噪声对残基Cα坐标进行局部扰动;对于旋转,使用等变的SO ( 3 )-transformer [108 ] 在旋转矩阵上模拟布朗运动生成噪声[109 ] ,使得模型具有全局的旋转不变性和高维的表征能力.在后续无条件约束策略设计和限制拓扑结构设计两种策略下,RFdiffusion设计了包括蛋白质单体、蛋白质-肽复合物、对称寡聚体、酶和金属结合蛋白等多种类型的蛋白,证明了RFdiffusion的在蛋白设计任务中的有效性和通用性. ...
De novo protein backbone generation based on diffusion with structured priors and adversarial training
1
2022
... 2022年刘海燕组提出的SCUBA-D[110 ] ,可以从包含不同类型或数量噪声的原始骨架中生成高质量的骨架.整个模型包含三个主要部分:一个低分辨率去噪模块,用于从初始骨架结构生成先验骨架结构;一个语言模型辅助的结构扩散模块,用于生成高分辨率的输出结构以及一个判别器网络,用于辅助训练去噪扩散模块.在此框架中,初始结构可以是完全随机的也可以带有若干约束,低分辨率去噪模块经过训练可以处理不同类型的初始结构.对不同的初始结构,该模块的目标是生成一个经过优化的粗糙的骨架结构,并保留所有初始结构中包含的拓扑信息.而后语言模型辅助的结构扩散模块获取低分辨率去噪模块的输出先验骨架结构,使用一系列去噪步骤对其进行细化,最终得到高分辨率的输出结构,其中使用氨基酸序列语言模型(ESM1b模型[111 ] )辅助结构扩散过程.为了保证生成结构的高物理可信度,在架构中还使用了两个GAN风格的判别器,在训练中提供额外的损失.而后研究者将结构预测用于在生成骨架上设计的序列,来评估模型生成骨架的质量.结果表明,模型可以始终生成高质量的骨架结构,具有十分广阔的应用前景. ...
Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences
1
2021
... 2022年刘海燕组提出的SCUBA-D[110 ] ,可以从包含不同类型或数量噪声的原始骨架中生成高质量的骨架.整个模型包含三个主要部分:一个低分辨率去噪模块,用于从初始骨架结构生成先验骨架结构;一个语言模型辅助的结构扩散模块,用于生成高分辨率的输出结构以及一个判别器网络,用于辅助训练去噪扩散模块.在此框架中,初始结构可以是完全随机的也可以带有若干约束,低分辨率去噪模块经过训练可以处理不同类型的初始结构.对不同的初始结构,该模块的目标是生成一个经过优化的粗糙的骨架结构,并保留所有初始结构中包含的拓扑信息.而后语言模型辅助的结构扩散模块获取低分辨率去噪模块的输出先验骨架结构,使用一系列去噪步骤对其进行细化,最终得到高分辨率的输出结构,其中使用氨基酸序列语言模型(ESM1b模型[111 ] )辅助结构扩散过程.为了保证生成结构的高物理可信度,在架构中还使用了两个GAN风格的判别器,在训练中提供额外的损失.而后研究者将结构预测用于在生成骨架上设计的序列,来评估模型生成骨架的质量.结果表明,模型可以始终生成高质量的骨架结构,具有十分广阔的应用前景. ...
Antigen-specific antibody design and optimization with diffusion-based generative models for protein structures
1
2022
... 目前,扩散模型在抗体设计中的应用已有报道的工作.2022年Luo等[112 ] 提出了DiffAb模型,该模型基于扩散概率模型以及等变神经网络对抗原抗体互补决定区(complementarity-determining regions)进行联合建模,可以生成针对特定抗原结构的抗体.研究者们同时对蛋白序列、坐标以及每个氨基酸的方向都进行了建模,使得模型可以实现原子分辨率级别的抗体设计且对旋转和平移等变.模型训练完成后,研究者将模型应用于序列结构协同设计、基于主链的抗体序列设计以及抗体优化任务中,结果表明模型在三个任务上均有出色的表现. ...
Expanding functional protein sequence spaces using generative adversarial networks
1
2021
... 蛋白质序列生成模型的发展主要受到自然语言处理领域出色模型的启发.Repecka等[113 ] 提出了一种基于生成对抗网络的蛋白质序列生成模型—ProteinGAN(图17 ).ProteinGAN模型使用生成对抗网络架构,训练数据为苹果脱氢酶家族的16706个蛋白序列.模型输入长为128的随机向量(均值为0,方差为0.5),由生成器生成蛋白质序列并将其呈递给判别器.在与自然蛋白质序列比较后,判别器对得到的序列进行打分,判断其为自然序列或是生成序列.生成器学习生成与自然序列近似的氨基酸序列用以欺骗判别器.经过2.5M步训练之后,98%的生成序列包含苹果脱氢酶的全部主要结构域,同时序列聚类中的不同氨基酸序列之间相似度不超过10%,这表明模型已极大程度上探索了苹果脱氢酶家族的序列空间. ...
ProGen: Language modeling for protein generation
1
2020
... 随着transformer模型[60 ] 在自然语言处理领域大放异彩,越来越多的研究者将transformer架构应用到蛋白质序列生成领域,由此产生了许多基于transformer的序列生成模型.2020年Madami等人提出了ProGen模型[114 ] .ProGen是一种条件transformer语言模型.该模型使用带有一系列蛋白性质标签的氨基酸序列进行训练,实现可控生成.ProGen生成的蛋白质在能量上与天然蛋白质相近,具有理想的生物功能.由 Elnaggar等[115 ] 提出的ProtTrans模型,使用四种不同的语言模型(两种自回归语言模型Tranformer-XL, XLNet以及两种自编码模型Bert, Albert)在蛋白质数据集上进行预训练,从序列中学习提取有用的特征,而后引入下游监督任务,以实现单个残基和单个蛋白性质的预测.这些模型原则上具有序列生成能力.2021年Gligorijević等[116 ] 提出了一种序列去噪自编码器,该模型与一个功能预测器相结合,可以从大量未标记的蛋白质数据中学习蛋白质序列的多样性,而功能预测器可对序列采样的方向进行指导.在测试阶段,研究者进一步探究了模型在设计带有金属结合位点的序列以及重新设计功能增强的角质酶的能力. ...
ProtTrans: towards cracking the language of lifes code through self-supervised deep learning and high performance computing
1
2021
... 随着transformer模型[60 ] 在自然语言处理领域大放异彩,越来越多的研究者将transformer架构应用到蛋白质序列生成领域,由此产生了许多基于transformer的序列生成模型.2020年Madami等人提出了ProGen模型[114 ] .ProGen是一种条件transformer语言模型.该模型使用带有一系列蛋白性质标签的氨基酸序列进行训练,实现可控生成.ProGen生成的蛋白质在能量上与天然蛋白质相近,具有理想的生物功能.由 Elnaggar等[115 ] 提出的ProtTrans模型,使用四种不同的语言模型(两种自回归语言模型Tranformer-XL, XLNet以及两种自编码模型Bert, Albert)在蛋白质数据集上进行预训练,从序列中学习提取有用的特征,而后引入下游监督任务,以实现单个残基和单个蛋白性质的预测.这些模型原则上具有序列生成能力.2021年Gligorijević等[116 ] 提出了一种序列去噪自编码器,该模型与一个功能预测器相结合,可以从大量未标记的蛋白质数据中学习蛋白质序列的多样性,而功能预测器可对序列采样的方向进行指导.在测试阶段,研究者进一步探究了模型在设计带有金属结合位点的序列以及重新设计功能增强的角质酶的能力. ...
Function-guided protein design by deep manifold sampling
1
2021
... 随着transformer模型[60 ] 在自然语言处理领域大放异彩,越来越多的研究者将transformer架构应用到蛋白质序列生成领域,由此产生了许多基于transformer的序列生成模型.2020年Madami等人提出了ProGen模型[114 ] .ProGen是一种条件transformer语言模型.该模型使用带有一系列蛋白性质标签的氨基酸序列进行训练,实现可控生成.ProGen生成的蛋白质在能量上与天然蛋白质相近,具有理想的生物功能.由 Elnaggar等[115 ] 提出的ProtTrans模型,使用四种不同的语言模型(两种自回归语言模型Tranformer-XL, XLNet以及两种自编码模型Bert, Albert)在蛋白质数据集上进行预训练,从序列中学习提取有用的特征,而后引入下游监督任务,以实现单个残基和单个蛋白性质的预测.这些模型原则上具有序列生成能力.2021年Gligorijević等[116 ] 提出了一种序列去噪自编码器,该模型与一个功能预测器相结合,可以从大量未标记的蛋白质数据中学习蛋白质序列的多样性,而功能预测器可对序列采样的方向进行指导.在测试阶段,研究者进一步探究了模型在设计带有金属结合位点的序列以及重新设计功能增强的角质酶的能力. ...
Design in the DARK: Learning deep generative models for de novo protein design
1
2022
... 2022年Moffat等[117 ] 提出了DARK架构,用于在不断迭代扩展的合成蛋白质序列上有效地训练生成模型,该模型使用了标准的transformer 解码器架构,可生成具有不同有序结构的新序列.随后,Ferruz等人提出了ProtGPT2模型[118 ] ,该模型是一个自回归transformer模型,拥有7.38亿参数.模型的训练在Uniref-50数据集上进行.训练完成后生成的序列显示出与自然序列相似的预测稳定性与动态特性,同时在进化上与当前的蛋白质序列空间相距甚远.Hesslow等[119 ] 提出RITA模型是一个拥有12亿的参数的自回归生成模型.该模型在UniRef-100数据集超过2.8亿个蛋白质序列上进行训练.研究者们探究了模型大小对自回归模型性能的影响,结果表明随着模型规模的增大,模型的表现有了显著的提升.而后Nijkamp等[118 ] 提出的ProGen2自回归transformer模型具有更大的规模,模型参数最多可达64亿,模型的训练在从基因组、宏基因组和免疫库数据库中提取的超过 10 亿种蛋白质的不同序列组成的数据集上进行.为了评估ProGen2生成序列的能力,研究者选择在以下三种情境对模型进行评估,即预训练后一般序列的生成、微调后的可以折叠成特殊结构的序列生成,以及在抗体序列数据集上进行预训练后的抗体序列生成.结果表明,截至ProGen2模型的提出,ProGen2在生成合理序列方面的表现为当前最佳. ...
ProGen2: exploring the boundaries of protein language models
2
2022
... 2022年Moffat等[117 ] 提出了DARK架构,用于在不断迭代扩展的合成蛋白质序列上有效地训练生成模型,该模型使用了标准的transformer 解码器架构,可生成具有不同有序结构的新序列.随后,Ferruz等人提出了ProtGPT2模型[118 ] ,该模型是一个自回归transformer模型,拥有7.38亿参数.模型的训练在Uniref-50数据集上进行.训练完成后生成的序列显示出与自然序列相似的预测稳定性与动态特性,同时在进化上与当前的蛋白质序列空间相距甚远.Hesslow等[119 ] 提出RITA模型是一个拥有12亿的参数的自回归生成模型.该模型在UniRef-100数据集超过2.8亿个蛋白质序列上进行训练.研究者们探究了模型大小对自回归模型性能的影响,结果表明随着模型规模的增大,模型的表现有了显著的提升.而后Nijkamp等[118 ] 提出的ProGen2自回归transformer模型具有更大的规模,模型参数最多可达64亿,模型的训练在从基因组、宏基因组和免疫库数据库中提取的超过 10 亿种蛋白质的不同序列组成的数据集上进行.为了评估ProGen2生成序列的能力,研究者选择在以下三种情境对模型进行评估,即预训练后一般序列的生成、微调后的可以折叠成特殊结构的序列生成,以及在抗体序列数据集上进行预训练后的抗体序列生成.结果表明,截至ProGen2模型的提出,ProGen2在生成合理序列方面的表现为当前最佳. ...
... [118 ]提出的ProGen2自回归transformer模型具有更大的规模,模型参数最多可达64亿,模型的训练在从基因组、宏基因组和免疫库数据库中提取的超过 10 亿种蛋白质的不同序列组成的数据集上进行.为了评估ProGen2生成序列的能力,研究者选择在以下三种情境对模型进行评估,即预训练后一般序列的生成、微调后的可以折叠成特殊结构的序列生成,以及在抗体序列数据集上进行预训练后的抗体序列生成.结果表明,截至ProGen2模型的提出,ProGen2在生成合理序列方面的表现为当前最佳. ...
RITA: a study on scaling up generative protein sequence models
1
2022
... 2022年Moffat等[117 ] 提出了DARK架构,用于在不断迭代扩展的合成蛋白质序列上有效地训练生成模型,该模型使用了标准的transformer 解码器架构,可生成具有不同有序结构的新序列.随后,Ferruz等人提出了ProtGPT2模型[118 ] ,该模型是一个自回归transformer模型,拥有7.38亿参数.模型的训练在Uniref-50数据集上进行.训练完成后生成的序列显示出与自然序列相似的预测稳定性与动态特性,同时在进化上与当前的蛋白质序列空间相距甚远.Hesslow等[119 ] 提出RITA模型是一个拥有12亿的参数的自回归生成模型.该模型在UniRef-100数据集超过2.8亿个蛋白质序列上进行训练.研究者们探究了模型大小对自回归模型性能的影响,结果表明随着模型规模的增大,模型的表现有了显著的提升.而后Nijkamp等[118 ] 提出的ProGen2自回归transformer模型具有更大的规模,模型参数最多可达64亿,模型的训练在从基因组、宏基因组和免疫库数据库中提取的超过 10 亿种蛋白质的不同序列组成的数据集上进行.为了评估ProGen2生成序列的能力,研究者选择在以下三种情境对模型进行评估,即预训练后一般序列的生成、微调后的可以折叠成特殊结构的序列生成,以及在抗体序列数据集上进行预训练后的抗体序列生成.结果表明,截至ProGen2模型的提出,ProGen2在生成合理序列方面的表现为当前最佳. ...
Energy functions in de novo protein design: current challenges and future prospects
1
2013
... 其二,目前对于蛋白设计模型的性能评估大多为天然序列恢复率和预测结构与原结构之间的差异,然而这两个指标仅能够衡量设计序列或结构与原蛋白的全局相似程度,并不能很好量化设计蛋白的物理化学性质.Dauparas[73 ] 在ProteinMPNN文章中也指出天然序列恢复率对结构分辨率敏感,并且与局部残基距离误差相关性不高(Rpearson ~0.5),并不是一个能够很好评价蛋白序列预测模型性能的指标.单个关键残基预测的错误对整体天然序列恢复率影响不大,但对序列折叠能力是毁灭性的.未来的方向可能是引入更多的评价指标,局部指标包括二级结构恢复率、溶剂可及表面、设计序列中无序残基比例等[120 ] .设计结构的全局评估可以使用结构预测模型折叠的结构并计算与目标结构的差异;长时间分子动力学模拟能够衡量序列折叠后结构的稳定性、展现结合蛋白与靶点之间的相互作用构象.将深度学习方法与传统的基于能量函数的蛋白质设计方法联用或前后相接,将深度学习模型生成的大量候选序列或结构输入基于物理化学的能量函数模型中进行验证和筛选,挑选出最优序列进行实验验证.充分发挥深度学习模型的高通量序列生成能力和物理化学模型对于蛋白的可表达性、可溶性以及聚集效应等物理化学性质的把握能力. ...










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}