Hybrid schemes based on quantum mechanics/molecular mechanics simulations goals to success, problems, and perspectives
1
2011
... 酶一般是功能性的蛋白质,在各种生物反应中作为生物催化剂参与,作为生物细胞发挥功能不可或缺的部分.经过漫长的岁月进化,天然酶为了适应自然环境而拥有了特定的功能[1-2].一般适宜在温和环境下且具有特定作用.由于酶的高效特定作用,且无污染的特性,酶非常受工业生产研究人员的青睐.例如用于酿酒的酵母菌、用于降解塑料的酶等等,都是酶分子应用在工业领域中的经典例子.但实际生产工业过程中,发现在工业环境中直接应用天然酶并没有达到满意的效果.错误的折叠,出现副产物,功能不适宜等缺陷对酶在工业行业的应用发出了挑战[3]. ...
Machine learning in enzyme engineering
2
2020
... 酶一般是功能性的蛋白质,在各种生物反应中作为生物催化剂参与,作为生物细胞发挥功能不可或缺的部分.经过漫长的岁月进化,天然酶为了适应自然环境而拥有了特定的功能[1-2].一般适宜在温和环境下且具有特定作用.由于酶的高效特定作用,且无污染的特性,酶非常受工业生产研究人员的青睐.例如用于酿酒的酵母菌、用于降解塑料的酶等等,都是酶分子应用在工业领域中的经典例子.但实际生产工业过程中,发现在工业环境中直接应用天然酶并没有达到满意的效果.错误的折叠,出现副产物,功能不适宜等缺陷对酶在工业行业的应用发出了挑战[3]. ...
... 人工智能在酶的设计改造过程中的应用,有助于对酶序列,功能以及结构空间的快速探索.对于酶的计算机智能辅助设计,通常集中于酶的热稳定性,耐酸碱性,催化活性,底物特异性以及酶的从头设计等方面[2].前面几种的设计着重于对酶的功能空间的探索,提高酶的某种已有功能特性,且不影响其原有的其他功能特性.而酶的从头设计则侧重于设计一种新酶,其目标功能可能只是具有8个β“片段桶”(barrel)这样的形状要求,或者是这个“桶”从结构上更为松散的这种功能性要求,也或者是β片段的排列方式这种结构上的要求.这意味着酶的设计要从结构和功能上达到统一. ...
Current advances in design and engineering strategies of industrial enzymes
1
2021
... 酶一般是功能性的蛋白质,在各种生物反应中作为生物催化剂参与,作为生物细胞发挥功能不可或缺的部分.经过漫长的岁月进化,天然酶为了适应自然环境而拥有了特定的功能[1-2].一般适宜在温和环境下且具有特定作用.由于酶的高效特定作用,且无污染的特性,酶非常受工业生产研究人员的青睐.例如用于酿酒的酵母菌、用于降解塑料的酶等等,都是酶分子应用在工业领域中的经典例子.但实际生产工业过程中,发现在工业环境中直接应用天然酶并没有达到满意的效果.错误的折叠,出现副产物,功能不适宜等缺陷对酶在工业行业的应用发出了挑战[3]. ...
Molecular engineering of industrial enzymes: recent advances and future prospects
1
2014
... 要想解决这一问题,酶必须进行改造或者设计新酶来满足特定的工业环境或者功能需求.那么,认识酶的结构与功能的关系是非常重要的[4].传统的酶改造过程涉及到修改酶的基因,使其在细胞中被成功表达纯化[5].然后对得到的突变体进行试验验证是否提高性能.这期间的时间,人力成本是巨大的,而且成功率是非常低的.随着人工智能技术的发展,利用计算方法辅助指导酶的改造或者设计开始成为主流[6-10].计算算法的快速实现,极大的降低了遍历穷举整个可能计算空间的搜索,同时利用优化算法很容易寻找到可行解.例如中科院微生物研究所吴边课题组使用多种计算工具,根据塑料降解酶的序列从保守性,结构能量值等角度筛选可能存在的突变位点,获得了塑料降解酶PETase的突变体DuraPETase[11].该突变体的熔融温度提高了35℃,温和温度下对塑料降解酶的降解能力提升了23%.根据特定的改造或者设计目标,智能计算方法一般是基于酶的序列或者结构挖掘和酶功能之间的映射关系,并希望借此能了解酶的各种作用机制,比如催化作用,特异性结合能力等. ...
Biocatalysis engineering: the big picture
1
2017
... 要想解决这一问题,酶必须进行改造或者设计新酶来满足特定的工业环境或者功能需求.那么,认识酶的结构与功能的关系是非常重要的[4].传统的酶改造过程涉及到修改酶的基因,使其在细胞中被成功表达纯化[5].然后对得到的突变体进行试验验证是否提高性能.这期间的时间,人力成本是巨大的,而且成功率是非常低的.随着人工智能技术的发展,利用计算方法辅助指导酶的改造或者设计开始成为主流[6-10].计算算法的快速实现,极大的降低了遍历穷举整个可能计算空间的搜索,同时利用优化算法很容易寻找到可行解.例如中科院微生物研究所吴边课题组使用多种计算工具,根据塑料降解酶的序列从保守性,结构能量值等角度筛选可能存在的突变位点,获得了塑料降解酶PETase的突变体DuraPETase[11].该突变体的熔融温度提高了35℃,温和温度下对塑料降解酶的降解能力提升了23%.根据特定的改造或者设计目标,智能计算方法一般是基于酶的序列或者结构挖掘和酶功能之间的映射关系,并希望借此能了解酶的各种作用机制,比如催化作用,特异性结合能力等. ...
Can machine learning revolutionize directed evolution of selective enzymes?
1
2019
... 要想解决这一问题,酶必须进行改造或者设计新酶来满足特定的工业环境或者功能需求.那么,认识酶的结构与功能的关系是非常重要的[4].传统的酶改造过程涉及到修改酶的基因,使其在细胞中被成功表达纯化[5].然后对得到的突变体进行试验验证是否提高性能.这期间的时间,人力成本是巨大的,而且成功率是非常低的.随着人工智能技术的发展,利用计算方法辅助指导酶的改造或者设计开始成为主流[6-10].计算算法的快速实现,极大的降低了遍历穷举整个可能计算空间的搜索,同时利用优化算法很容易寻找到可行解.例如中科院微生物研究所吴边课题组使用多种计算工具,根据塑料降解酶的序列从保守性,结构能量值等角度筛选可能存在的突变位点,获得了塑料降解酶PETase的突变体DuraPETase[11].该突变体的熔融温度提高了35℃,温和温度下对塑料降解酶的降解能力提升了23%.根据特定的改造或者设计目标,智能计算方法一般是基于酶的序列或者结构挖掘和酶功能之间的映射关系,并希望借此能了解酶的各种作用机制,比如催化作用,特异性结合能力等. ...
De novo computational design of retro-aldol enzymes
0
2008
Kemp elimination catalysts by computational enzyme design
0
2008
Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction
0
2010
Machine-learning-guided directed evolution for protein engineering
1
2019
... 要想解决这一问题,酶必须进行改造或者设计新酶来满足特定的工业环境或者功能需求.那么,认识酶的结构与功能的关系是非常重要的[4].传统的酶改造过程涉及到修改酶的基因,使其在细胞中被成功表达纯化[5].然后对得到的突变体进行试验验证是否提高性能.这期间的时间,人力成本是巨大的,而且成功率是非常低的.随着人工智能技术的发展,利用计算方法辅助指导酶的改造或者设计开始成为主流[6-10].计算算法的快速实现,极大的降低了遍历穷举整个可能计算空间的搜索,同时利用优化算法很容易寻找到可行解.例如中科院微生物研究所吴边课题组使用多种计算工具,根据塑料降解酶的序列从保守性,结构能量值等角度筛选可能存在的突变位点,获得了塑料降解酶PETase的突变体DuraPETase[11].该突变体的熔融温度提高了35℃,温和温度下对塑料降解酶的降解能力提升了23%.根据特定的改造或者设计目标,智能计算方法一般是基于酶的序列或者结构挖掘和酶功能之间的映射关系,并希望借此能了解酶的各种作用机制,比如催化作用,特异性结合能力等. ...
GRAPE, a greedy accumulated strategy for computational protein engineering
2
2021
... 要想解决这一问题,酶必须进行改造或者设计新酶来满足特定的工业环境或者功能需求.那么,认识酶的结构与功能的关系是非常重要的[4].传统的酶改造过程涉及到修改酶的基因,使其在细胞中被成功表达纯化[5].然后对得到的突变体进行试验验证是否提高性能.这期间的时间,人力成本是巨大的,而且成功率是非常低的.随着人工智能技术的发展,利用计算方法辅助指导酶的改造或者设计开始成为主流[6-10].计算算法的快速实现,极大的降低了遍历穷举整个可能计算空间的搜索,同时利用优化算法很容易寻找到可行解.例如中科院微生物研究所吴边课题组使用多种计算工具,根据塑料降解酶的序列从保守性,结构能量值等角度筛选可能存在的突变位点,获得了塑料降解酶PETase的突变体DuraPETase[11].该突变体的熔融温度提高了35℃,温和温度下对塑料降解酶的降解能力提升了23%.根据特定的改造或者设计目标,智能计算方法一般是基于酶的序列或者结构挖掘和酶功能之间的映射关系,并希望借此能了解酶的各种作用机制,比如催化作用,特异性结合能力等. ...
... 利用酶的序列以及功能性指标数据对,构建模型,然后利用模型指导酶分子改造.其构建的模型输入一般是基于序列或者结构提取的描述符,输出则是蛋白质适应性的预测目标,一般对应于要改造的具体功能性指标.一旦模型建立,即可通过预测大量突变序列的性能快速筛选不理想的突变体.以Frances H. Arnold团队发表在PNAS上的一个工作为例[25].该工作主要是改造一氧化氮双加氧酶 (NOD) 立体选择性,并选择多个机器学习模型去构建NOD的立体选择性催化模型,包括但不仅限于K最近邻、线性模型、决策树、随机森林,将76% (S)-ee初始突变体提升至93% (S)-ee及反转至79% (R)-ee.中科院微生物研究所吴边团队提出一种新型蛋白质稳定性计算设计策略GRAPE[11].该策略对传统筛选突变体策略进行补充,并通过系统聚类分析对得到的单点有益突变进行聚类,同时结合贪婪算法进行网络迭代叠加,大幅度规避了以往遇到的累积突变所带来的负协同相互作用.设计出的突变体DuraPETase可在中等温度下有效降解塑料,为酶的设计的计算策略提供了非常重要的方向.当特定类型的酶数据比较小的时候,可以借助在大量通用酶类数据上的预训练模型来学习氨基酸对之间的互作用关系或者邻居结构环境信息,指导后续的酶改造任务.这种方法的好处是可以根据特定任务在具体的数据集上对预训练模型进行微调,以适应于不同的小数据集的下游任务.2021年提出的Low-N模型充分利用了UniRep中大量的蛋白质序列,通过无监督语言预训练任务提取了蛋白质的一般功能特征,然后通过特定家族序列上微调学习到了该家族的特异性特征[26].这样学习到的蛋白质表示后,仅需要少量的序列和目标功能的数据,就可以训练一个简单且有效的监督模型.将该模型应用到实际中,最少仅需24个avGFP突变体的数据集,就设计出了新的荧光蛋白,可以与高保真且高通量的蛋白质工程产物sfGFP相媲美.Low-N以较少的数量实现了蛋白质序列到功能模式的转变.类似工作还有[27]中提到的SEMA. ...
De novo protein fold design through sequence-independent fragment assembly simulations
1
2023
... 那么,对于设计或者改造后的新酶,是否可以按照实验要求折叠成给定的构象,实现要设计的功能?这个能力一般称之为“可折叠性”[12-13].实验验证是将新酶序列在大肠杆菌中纯化表达,同时测定是否具有给定的功能.但是,现在很多工作随机生成新酶,可以产生大量要求的序列.这些序列如果都通过实验室测定其是否合理,并不符合通过计算手段降低实验成本的初衷.迄今为止,尽管从头酶设计有了诸多成果,但大多都表现出低效率.有研究表明错误折叠是大多数酶设计工作失败的原因.如果在设计或者改造的过程中,考虑加入结构约束的话,则在很大程度上可以提高新酶的“可折叠性”.最近五年来,基于人工智能与数据驱动技术的蛋白质结构预测取得了一系列的突破性进展[14-16].例如,AlphaFold2[14]预测了人类蛋白组的98.5%蛋白结构,极大丰富了蛋白结构数据并促进对人类生命机制的研究.实际上,蛋白质结构预测实际上可以称之为“逆式”的蛋白质设计.那么,在蛋白质设计领域,蛋白质结构预测这些相对成熟化的工具,是否能从结构约束角度促进酶的改造设计工具更快速且精确化促进酶的“可折叠性”研究呢? ...
Assessing and enhancing foldability in designed proteins
3
2022
... 那么,对于设计或者改造后的新酶,是否可以按照实验要求折叠成给定的构象,实现要设计的功能?这个能力一般称之为“可折叠性”[12-13].实验验证是将新酶序列在大肠杆菌中纯化表达,同时测定是否具有给定的功能.但是,现在很多工作随机生成新酶,可以产生大量要求的序列.这些序列如果都通过实验室测定其是否合理,并不符合通过计算手段降低实验成本的初衷.迄今为止,尽管从头酶设计有了诸多成果,但大多都表现出低效率.有研究表明错误折叠是大多数酶设计工作失败的原因.如果在设计或者改造的过程中,考虑加入结构约束的话,则在很大程度上可以提高新酶的“可折叠性”.最近五年来,基于人工智能与数据驱动技术的蛋白质结构预测取得了一系列的突破性进展[14-16].例如,AlphaFold2[14]预测了人类蛋白组的98.5%蛋白结构,极大丰富了蛋白结构数据并促进对人类生命机制的研究.实际上,蛋白质结构预测实际上可以称之为“逆式”的蛋白质设计.那么,在蛋白质设计领域,蛋白质结构预测这些相对成熟化的工具,是否能从结构约束角度促进酶的改造设计工具更快速且精确化促进酶的“可折叠性”研究呢? ...
... Sarel Jacob Fleishman课题组提出,现有功能蛋白设计方面由于错误折叠等导致的失败使得可靠的高效酶从头设计目标仍然遥不可及,因此设计了一种改善设计蛋白中不是很合理的位置方法[13].该方法首先利用Rosetta进行单点突变扫描,筛选有超过5种以上降低自然状态能量突变的位置标记为“次优”位置.然后应用FuncLib集中在这些低效率酶的“次优”位置上设计突变,将催化效率提高了330倍.最后利用AlphaFold2预测的pLDDT得分和计算的RMSD标记了可能错误折叠的区域,合理规避或者重新设计不合理区域,大大提高了其催化效率[13].这种思路类似于在小节1.1中讨论的根据残基在当前结构环境中的“不合理”值,判断是否要在此位点突变.该工作指出,AlphaFold2分析可以提供有关新设计的骨架结构可能的准确性和可折叠性的关键信息,指示可能错误折叠的区域,并评估旨在减轻错误折叠的突变. ...
... [13].这种思路类似于在小节1.1中讨论的根据残基在当前结构环境中的“不合理”值,判断是否要在此位点突变.该工作指出,AlphaFold2分析可以提供有关新设计的骨架结构可能的准确性和可折叠性的关键信息,指示可能错误折叠的区域,并评估旨在减轻错误折叠的突变. ...
Highly accurate protein structure prediction for the human proteome
5
2021
... 那么,对于设计或者改造后的新酶,是否可以按照实验要求折叠成给定的构象,实现要设计的功能?这个能力一般称之为“可折叠性”[12-13].实验验证是将新酶序列在大肠杆菌中纯化表达,同时测定是否具有给定的功能.但是,现在很多工作随机生成新酶,可以产生大量要求的序列.这些序列如果都通过实验室测定其是否合理,并不符合通过计算手段降低实验成本的初衷.迄今为止,尽管从头酶设计有了诸多成果,但大多都表现出低效率.有研究表明错误折叠是大多数酶设计工作失败的原因.如果在设计或者改造的过程中,考虑加入结构约束的话,则在很大程度上可以提高新酶的“可折叠性”.最近五年来,基于人工智能与数据驱动技术的蛋白质结构预测取得了一系列的突破性进展[14-16].例如,AlphaFold2[14]预测了人类蛋白组的98.5%蛋白结构,极大丰富了蛋白结构数据并促进对人类生命机制的研究.实际上,蛋白质结构预测实际上可以称之为“逆式”的蛋白质设计.那么,在蛋白质设计领域,蛋白质结构预测这些相对成熟化的工具,是否能从结构约束角度促进酶的改造设计工具更快速且精确化促进酶的“可折叠性”研究呢? ...
... [14]预测了人类蛋白组的98.5%蛋白结构,极大丰富了蛋白结构数据并促进对人类生命机制的研究.实际上,蛋白质结构预测实际上可以称之为“逆式”的蛋白质设计.那么,在蛋白质设计领域,蛋白质结构预测这些相对成熟化的工具,是否能从结构约束角度促进酶的改造设计工具更快速且精确化促进酶的“可折叠性”研究呢? ...
... 2019年Mohammed AlQuraishi提出RGN方法首次尝试使用深度学习算法端到端的从蛋白质序列直接预测最终的3D坐标[79],而不是通过前面介绍的“两步式”方法.其主要思想是将每个残基作为一个可微基元,然后从两个方向:N端到C端,C端到N端,预测在已有的所有残基的局部结构下,当前残基加入后的空间结构,从而将整个蛋白质残基序列串联起来,得到最终蛋白质结构.这个过程中,考虑了当前残基与相邻残基之间的相互作用关系,并实现了“多个尺寸”的蛋白质表示学习.实验证明相比CASP11,CASP12上排名第一的Server组来说,该方法在对于具有新折叠的自由建模中,表现优异.但是该方法输入是蛋白质序列独热 (one-hot) 编码以及位置保守性特异矩阵 (position-specific scoring matrices,PSSM) ,然后通过LSTM去实现序列的编码框架,预测出每个残基的扭转角参数.PSSM相比前面提到的MSA中提取的共进化信息,并不包含残基对间的相互作用,只着重单个残基在单个位置上的进化保守性.因此,导致该方法:1)依赖PSSM矩阵的特征准确性;2)忽略残基对间的相互作用(MSA中共进化信息不是线性的,成本高,且不适合RGN的循环方法).而之后在CASP14比赛上,DeepMind提出AlphaFold2[14],完全抛弃了AlphaFold1传统的“两步式”思路,通过图推理的方式直接实现了“端到端”(end-to-end) 的蛋白质结构预测方法,转变了结合人工智能研究蛋白质结构研究新范式.因此,由该方法引发的“AI蛋白质折叠”被《MIT Technology Review》评为“全球十大突破性技术”.AlphaFold2主要由神经网络EvoFormer和结构模块两部分组成.EvoFormer中序列信息和从MSA中抽取的进化特征之间进行信息交换,直接推理出在空间和进化关系中残基对的配对表征.结构模块则用于将得到的特征转化为三维坐标结构.AlphaFold2的优势在于信息流之间的注意力机制,包括从MSA中学习到配对特征表示与序列上每个残基的特征表示之间的相互信息交流(基于注意力机制),通过几何空间约束形成的具有共残基的相互作用残基对之间的信息交流(三角注意力机制).得到更新后的配对残基特征以及单残基特征后,通过结构模块不断迭代更新坐标系预测当前残基和相邻残基之间肽键的角度和距离偏移,最终得到整个蛋白质的全局笛卡尔系坐标.平均自由建模精度(GDT打分)达到80以上,而在CASAP13(AlphaFold出现)之前,这一个值最高是40左右. ...
... 在众多优秀的蛋白质结构预测工具中,不得不提AlphaFold2[14].AlphaFold2实现了对人类蛋白组58%的准确性预测(pLDDT高于70,可信),36%的结构位置预测高可信.其与欧洲生物信息研究所(EMBL-EBI)合作建立的平台AlphaFold DB(AlphaFold 蛋白质结构数据库,AlphaFold Protein Structure Database (ebi.ac.uk)),涵盖了几乎98.5% 的所有人类蛋白.因此,本文以AlphaFold2为代表,探索如何借助蛋白质结构预测工具增加酶设计改造的准确性.其他结构预测工具,可以根据具体研究的数据或者任务不同,替代AlphaFold2的结构预测工作. ...
... 本文从头梳理了酶改造设计在利用人工智能技术方面的一系列工作,指出现有工作中错误折叠甚至无法折叠导致失败,以及设计大量序列需要人工实验验证的成本的问题.同时基于现有蛋白质结构预测工具的高效快速预测性,可以作为结构“分析器”,突变“筛选器”,折叠“监督器”在设计过程中帮助提高酶的“可折叠性”.正因为考虑“可折叠”能力,设计的新酶的质量相比传统大量序列中质量较高,帮助后续的实验验证降低成本的同时又提高了成功率.值得注意的是,这里面结构预测工具与酶设计工具共同采用,结构预测工具本身只是作为辅助任务.我们在讨论结构预测工具应用的时候,是以AlphaFold2[14]为代表展开介绍的. ...
Improved protein structure prediction using potentials from deep learning
1
2020
... 除了上面提到的接触约束,CASP13上DeepMind提出的AlphaFold1,则将这一约束扩展到了残基间的距离约束.然后将离散化的距离预测值通过采样插值转化成可微的残基距离分布函数,进而通过直接优化该函数求解距离和角度的最优解,从而确定最终的蛋白质三维结构[15].AlphaFold1的成功不仅仅是预测精度的显著提高,更是作为一种信号:深度神经网络可以有效识别蛋白质序列中的信号以及共进化信息的模式,并将其转化到高精度的距离分布上.考虑到三维空间的特性,trRosetta相比AlphaFold1还引入了5个角度的预测值来表示残基间的相对方向进一步加强了残基间的约束,并且精度提高了6.5%[16].David T. Jones 组提出的DMPfold,预测的是相对残基间的距离,主链氢键以及扭转角[75].当学习到这些约束后,类似于RaptorX,输入到crystallography and NMR system (CNS) [76]中作为约束指导蛋白质从头折叠.在2022年的CASP15上,张阳组郑伟博士在已有的I-TASSER基础上提出的D-I-TASSER算法[77],将AttentionPotential以及DeepPotential[78]两个深度学习算法预测出的高准确度的氢键 (hydrogen-bond) 网络,接触图以及距离图等约束加入到I-TASSER 中采用的力场能量项中,然后通过蒙特卡洛模拟进行迭代的片段组装装配最终的蛋白质结构构象,位列蛋白质单体单结构域比赛第一名. ...
Improved protein structure prediction using predicted interresidue orientations
3
2020
... 那么,对于设计或者改造后的新酶,是否可以按照实验要求折叠成给定的构象,实现要设计的功能?这个能力一般称之为“可折叠性”[12-13].实验验证是将新酶序列在大肠杆菌中纯化表达,同时测定是否具有给定的功能.但是,现在很多工作随机生成新酶,可以产生大量要求的序列.这些序列如果都通过实验室测定其是否合理,并不符合通过计算手段降低实验成本的初衷.迄今为止,尽管从头酶设计有了诸多成果,但大多都表现出低效率.有研究表明错误折叠是大多数酶设计工作失败的原因.如果在设计或者改造的过程中,考虑加入结构约束的话,则在很大程度上可以提高新酶的“可折叠性”.最近五年来,基于人工智能与数据驱动技术的蛋白质结构预测取得了一系列的突破性进展[14-16].例如,AlphaFold2[14]预测了人类蛋白组的98.5%蛋白结构,极大丰富了蛋白结构数据并促进对人类生命机制的研究.实际上,蛋白质结构预测实际上可以称之为“逆式”的蛋白质设计.那么,在蛋白质设计领域,蛋白质结构预测这些相对成熟化的工具,是否能从结构约束角度促进酶的改造设计工具更快速且精确化促进酶的“可折叠性”研究呢? ...
... 除了上面提到的接触约束,CASP13上DeepMind提出的AlphaFold1,则将这一约束扩展到了残基间的距离约束.然后将离散化的距离预测值通过采样插值转化成可微的残基距离分布函数,进而通过直接优化该函数求解距离和角度的最优解,从而确定最终的蛋白质三维结构[15].AlphaFold1的成功不仅仅是预测精度的显著提高,更是作为一种信号:深度神经网络可以有效识别蛋白质序列中的信号以及共进化信息的模式,并将其转化到高精度的距离分布上.考虑到三维空间的特性,trRosetta相比AlphaFold1还引入了5个角度的预测值来表示残基间的相对方向进一步加强了残基间的约束,并且精度提高了6.5%[16].David T. Jones 组提出的DMPfold,预测的是相对残基间的距离,主链氢键以及扭转角[75].当学习到这些约束后,类似于RaptorX,输入到crystallography and NMR system (CNS) [76]中作为约束指导蛋白质从头折叠.在2022年的CASP15上,张阳组郑伟博士在已有的I-TASSER基础上提出的D-I-TASSER算法[77],将AttentionPotential以及DeepPotential[78]两个深度学习算法预测出的高准确度的氢键 (hydrogen-bond) 网络,接触图以及距离图等约束加入到I-TASSER 中采用的力场能量项中,然后通过蒙特卡洛模拟进行迭代的片段组装装配最终的蛋白质结构构象,位列蛋白质单体单结构域比赛第一名. ...
... 第一部分中我们提到,对于酶的改造和设计这两个应用场景,设计新酶的折叠能力是至关重要的.不论是在给定结构还是在给定功能约束下,设计的新酶如果不能正常折叠或者折叠后偏离预设结构,则减弱甚至丧失给定的功能.因此在设计过程中结合设计后新酶的折叠状态,相比不考虑再去实验验证筛选(几千几万条),在时间和实验成本上都占有优势.然而,折叠后的构象,实际上就是蛋白质结构预测的目标.结合第二部分中对蛋白质结构预测工具的梳理,可以看到蛋白质结构预测在人工智能强大的拟合能力帮助下最近几年来获得了突破性的进展.许多蛋白质结构预测工具由于预测的高效快速被广泛应用,例如:trRosetta[16],RoseTTAFold[84]等.那么,从设计酶的“可折叠性”出发,探索将蛋白质结构预测工具与现有的酶设计改造方法相结合,将会是一条有效的酶智能设计改造思路,有助于探索更为广阔的蛋白质序列空间. ...
AAindex: Amino Acid index database
1
2000
... 利用人工智能解决问题是在已有的数据挖掘内部隐藏的看不见的模式,即序列、结构与功能之间的内在的关系映射.第一步则需要合理的将酶的描述特征提取并表示成机器识别的模式,一般分为以下几类:基于序列的,基于结构的,基于嵌入的.基于序列的,包含一些常见的one-hot编码,物理化学特性编码(疏水性,电荷等),进化保守性, AA-index[17],zScales[18]等.基于结构的,包含一些基于统计的残基对间的接触势,相邻结构域的类型及物理化学性质,骨架扭转角度,键长,距离活性位点的远近等[19].而基于嵌入的,是指的模型通过在大量蛋白质家族序列或者结构上进行类似于“完形填空”的训练过程中,学习到序列/结构邻居的上下文信息.在此过程中,模型学习氨基酸的有意义的中间表示,并提炼出每个氨基酸位置周围的重要结构环境.比如:ProtVec[20],ESM-1V[21],TAPE[22],dMaSIF[23]等.接下来需要构建合适的模型预测或者生成目标.这部分的差异,可参考综述 [24].接下来根据目标从酶的智能改造和设计两部分展开. ...
New chemical descriptors relevant for the design of biologically active peptides. A multivariate characterization of 87 amino acids
1
1998
... 利用人工智能解决问题是在已有的数据挖掘内部隐藏的看不见的模式,即序列、结构与功能之间的内在的关系映射.第一步则需要合理的将酶的描述特征提取并表示成机器识别的模式,一般分为以下几类:基于序列的,基于结构的,基于嵌入的.基于序列的,包含一些常见的one-hot编码,物理化学特性编码(疏水性,电荷等),进化保守性, AA-index[17],zScales[18]等.基于结构的,包含一些基于统计的残基对间的接触势,相邻结构域的类型及物理化学性质,骨架扭转角度,键长,距离活性位点的远近等[19].而基于嵌入的,是指的模型通过在大量蛋白质家族序列或者结构上进行类似于“完形填空”的训练过程中,学习到序列/结构邻居的上下文信息.在此过程中,模型学习氨基酸的有意义的中间表示,并提炼出每个氨基酸位置周围的重要结构环境.比如:ProtVec[20],ESM-1V[21],TAPE[22],dMaSIF[23]等.接下来需要构建合适的模型预测或者生成目标.这部分的差异,可参考综述 [24].接下来根据目标从酶的智能改造和设计两部分展开. ...
Learning the local landscape of protein structures with convolutional neural networks
2
2021
... 利用人工智能解决问题是在已有的数据挖掘内部隐藏的看不见的模式,即序列、结构与功能之间的内在的关系映射.第一步则需要合理的将酶的描述特征提取并表示成机器识别的模式,一般分为以下几类:基于序列的,基于结构的,基于嵌入的.基于序列的,包含一些常见的one-hot编码,物理化学特性编码(疏水性,电荷等),进化保守性, AA-index[17],zScales[18]等.基于结构的,包含一些基于统计的残基对间的接触势,相邻结构域的类型及物理化学性质,骨架扭转角度,键长,距离活性位点的远近等[19].而基于嵌入的,是指的模型通过在大量蛋白质家族序列或者结构上进行类似于“完形填空”的训练过程中,学习到序列/结构邻居的上下文信息.在此过程中,模型学习氨基酸的有意义的中间表示,并提炼出每个氨基酸位置周围的重要结构环境.比如:ProtVec[20],ESM-1V[21],TAPE[22],dMaSIF[23]等.接下来需要构建合适的模型预测或者生成目标.这部分的差异,可参考综述 [24].接下来根据目标从酶的智能改造和设计两部分展开. ...
... 除此之外,随着日益丰富的结构数据与逐渐成熟的深度网络学习能力,从酶的结构数据集中直接挖掘结构与功能之间的关系也成为可能.2022年,得克萨斯大学奥斯汀分校 McKetta 化学工程系教授Hal S. Alper结合人工智能技术和酶工程,改造出一系列塑料降解酶的变体,相关工作发表在Nature上[28].其中最优秀的突变体FAST-PETase优于现有的PET降解酶的变体的降解效率,且能在更广泛环境中具有较好的活性,证明了在工业规模上酶塑料回收的可行途径.该方法首先筛选有效突变位点的方法是利用一个深度学习算法MutCompute[19]来有效过滤筛选突变位点 .MutCompute通过一个3D的自监督的卷积网络模型,对每一个残基构造一个局部微环境,统计该环境中C, H, O, N, S原子出现的次数,电荷,溶剂可达面积来编码该局部环境,最后预测每个残基的序列类型(分类问题).根据该残基一个已有突变体上的预测概率值与在野生型中的概率差异值大小,衡量出残基在野生型结构中的“不匹配度” (disfavoured) ,进而筛选出这种得分较大的突变位点,结合以往文献中报道的有效突变位点以及活性口袋位点,指导后续进一步筛选有效组合突变.该方法捕获了由结构决定的功能模式的指导转化,筛选条件是该残基在给定的蛋白质折叠环境中是否适配的能力.相比单纯使用序列的模型,考虑残基在结构环境中是否适配或从已有结构数据中学习这种规律,约束了改造酶的合理性并且增加了可能的改造位点方案.类似的工作还被应用在TEM-1 β-内酰胺酶和白色念珠菌磷化异构酶 (CaPMI) 中[29]. ...
Continuous distributed representation of biological sequences for deep proteomics and genomics
1
2015
... 利用人工智能解决问题是在已有的数据挖掘内部隐藏的看不见的模式,即序列、结构与功能之间的内在的关系映射.第一步则需要合理的将酶的描述特征提取并表示成机器识别的模式,一般分为以下几类:基于序列的,基于结构的,基于嵌入的.基于序列的,包含一些常见的one-hot编码,物理化学特性编码(疏水性,电荷等),进化保守性, AA-index[17],zScales[18]等.基于结构的,包含一些基于统计的残基对间的接触势,相邻结构域的类型及物理化学性质,骨架扭转角度,键长,距离活性位点的远近等[19].而基于嵌入的,是指的模型通过在大量蛋白质家族序列或者结构上进行类似于“完形填空”的训练过程中,学习到序列/结构邻居的上下文信息.在此过程中,模型学习氨基酸的有意义的中间表示,并提炼出每个氨基酸位置周围的重要结构环境.比如:ProtVec[20],ESM-1V[21],TAPE[22],dMaSIF[23]等.接下来需要构建合适的模型预测或者生成目标.这部分的差异,可参考综述 [24].接下来根据目标从酶的智能改造和设计两部分展开. ...
Language models enable zero-shot prediction of the effects of mutations on protein function
2
2021
... 利用人工智能解决问题是在已有的数据挖掘内部隐藏的看不见的模式,即序列、结构与功能之间的内在的关系映射.第一步则需要合理的将酶的描述特征提取并表示成机器识别的模式,一般分为以下几类:基于序列的,基于结构的,基于嵌入的.基于序列的,包含一些常见的one-hot编码,物理化学特性编码(疏水性,电荷等),进化保守性, AA-index[17],zScales[18]等.基于结构的,包含一些基于统计的残基对间的接触势,相邻结构域的类型及物理化学性质,骨架扭转角度,键长,距离活性位点的远近等[19].而基于嵌入的,是指的模型通过在大量蛋白质家族序列或者结构上进行类似于“完形填空”的训练过程中,学习到序列/结构邻居的上下文信息.在此过程中,模型学习氨基酸的有意义的中间表示,并提炼出每个氨基酸位置周围的重要结构环境.比如:ProtVec[20],ESM-1V[21],TAPE[22],dMaSIF[23]等.接下来需要构建合适的模型预测或者生成目标.这部分的差异,可参考综述 [24].接下来根据目标从酶的智能改造和设计两部分展开. ...
... 第二个难点是对于深度学习模型来说,从海量数据中挖掘模式是合适的.但是现有的状况是酶的相关数据量小,没有统一的标准格式,冗余的.当然这也与特定学科有关系.很多研究工作利用迁移学习来解决数据量小的问题,比如DeepET在大的蛋白质序列-最佳生长温度 (OGT) 数据集上训练模型,然后迁移到预测酶的最佳催化温度和蛋白质的熔融温度[98].或者利用自然语言处理 (NLP) 中广泛使用的大规模语言预训练模型学习序列的表示,然后小数据集上微调,进行一些功能预测[21,26]. ...
Evaluating protein transfer learning with TAPE
1
2019
... 利用人工智能解决问题是在已有的数据挖掘内部隐藏的看不见的模式,即序列、结构与功能之间的内在的关系映射.第一步则需要合理的将酶的描述特征提取并表示成机器识别的模式,一般分为以下几类:基于序列的,基于结构的,基于嵌入的.基于序列的,包含一些常见的one-hot编码,物理化学特性编码(疏水性,电荷等),进化保守性, AA-index[17],zScales[18]等.基于结构的,包含一些基于统计的残基对间的接触势,相邻结构域的类型及物理化学性质,骨架扭转角度,键长,距离活性位点的远近等[19].而基于嵌入的,是指的模型通过在大量蛋白质家族序列或者结构上进行类似于“完形填空”的训练过程中,学习到序列/结构邻居的上下文信息.在此过程中,模型学习氨基酸的有意义的中间表示,并提炼出每个氨基酸位置周围的重要结构环境.比如:ProtVec[20],ESM-1V[21],TAPE[22],dMaSIF[23]等.接下来需要构建合适的模型预测或者生成目标.这部分的差异,可参考综述 [24].接下来根据目标从酶的智能改造和设计两部分展开. ...
Fast end-to-end learning on protein surfaces
1
2021
... 利用人工智能解决问题是在已有的数据挖掘内部隐藏的看不见的模式,即序列、结构与功能之间的内在的关系映射.第一步则需要合理的将酶的描述特征提取并表示成机器识别的模式,一般分为以下几类:基于序列的,基于结构的,基于嵌入的.基于序列的,包含一些常见的one-hot编码,物理化学特性编码(疏水性,电荷等),进化保守性, AA-index[17],zScales[18]等.基于结构的,包含一些基于统计的残基对间的接触势,相邻结构域的类型及物理化学性质,骨架扭转角度,键长,距离活性位点的远近等[19].而基于嵌入的,是指的模型通过在大量蛋白质家族序列或者结构上进行类似于“完形填空”的训练过程中,学习到序列/结构邻居的上下文信息.在此过程中,模型学习氨基酸的有意义的中间表示,并提炼出每个氨基酸位置周围的重要结构环境.比如:ProtVec[20],ESM-1V[21],TAPE[22],dMaSIF[23]等.接下来需要构建合适的模型预测或者生成目标.这部分的差异,可参考综述 [24].接下来根据目标从酶的智能改造和设计两部分展开. ...
Data-driven enzyme engineering to identify function-enhancing enzymes
1
2023
... 利用人工智能解决问题是在已有的数据挖掘内部隐藏的看不见的模式,即序列、结构与功能之间的内在的关系映射.第一步则需要合理的将酶的描述特征提取并表示成机器识别的模式,一般分为以下几类:基于序列的,基于结构的,基于嵌入的.基于序列的,包含一些常见的one-hot编码,物理化学特性编码(疏水性,电荷等),进化保守性, AA-index[17],zScales[18]等.基于结构的,包含一些基于统计的残基对间的接触势,相邻结构域的类型及物理化学性质,骨架扭转角度,键长,距离活性位点的远近等[19].而基于嵌入的,是指的模型通过在大量蛋白质家族序列或者结构上进行类似于“完形填空”的训练过程中,学习到序列/结构邻居的上下文信息.在此过程中,模型学习氨基酸的有意义的中间表示,并提炼出每个氨基酸位置周围的重要结构环境.比如:ProtVec[20],ESM-1V[21],TAPE[22],dMaSIF[23]等.接下来需要构建合适的模型预测或者生成目标.这部分的差异,可参考综述 [24].接下来根据目标从酶的智能改造和设计两部分展开. ...
Machine learning-assisted directed protein evolution with combinatorial libraries
1
2019
... 利用酶的序列以及功能性指标数据对,构建模型,然后利用模型指导酶分子改造.其构建的模型输入一般是基于序列或者结构提取的描述符,输出则是蛋白质适应性的预测目标,一般对应于要改造的具体功能性指标.一旦模型建立,即可通过预测大量突变序列的性能快速筛选不理想的突变体.以Frances H. Arnold团队发表在PNAS上的一个工作为例[25].该工作主要是改造一氧化氮双加氧酶 (NOD) 立体选择性,并选择多个机器学习模型去构建NOD的立体选择性催化模型,包括但不仅限于K最近邻、线性模型、决策树、随机森林,将76% (S)-ee初始突变体提升至93% (S)-ee及反转至79% (R)-ee.中科院微生物研究所吴边团队提出一种新型蛋白质稳定性计算设计策略GRAPE[11].该策略对传统筛选突变体策略进行补充,并通过系统聚类分析对得到的单点有益突变进行聚类,同时结合贪婪算法进行网络迭代叠加,大幅度规避了以往遇到的累积突变所带来的负协同相互作用.设计出的突变体DuraPETase可在中等温度下有效降解塑料,为酶的设计的计算策略提供了非常重要的方向.当特定类型的酶数据比较小的时候,可以借助在大量通用酶类数据上的预训练模型来学习氨基酸对之间的互作用关系或者邻居结构环境信息,指导后续的酶改造任务.这种方法的好处是可以根据特定任务在具体的数据集上对预训练模型进行微调,以适应于不同的小数据集的下游任务.2021年提出的Low-N模型充分利用了UniRep中大量的蛋白质序列,通过无监督语言预训练任务提取了蛋白质的一般功能特征,然后通过特定家族序列上微调学习到了该家族的特异性特征[26].这样学习到的蛋白质表示后,仅需要少量的序列和目标功能的数据,就可以训练一个简单且有效的监督模型.将该模型应用到实际中,最少仅需24个avGFP突变体的数据集,就设计出了新的荧光蛋白,可以与高保真且高通量的蛋白质工程产物sfGFP相媲美.Low-N以较少的数量实现了蛋白质序列到功能模式的转变.类似工作还有[27]中提到的SEMA. ...
Low-N protein engineering with data-efficient deep learning
2
2021
... 利用酶的序列以及功能性指标数据对,构建模型,然后利用模型指导酶分子改造.其构建的模型输入一般是基于序列或者结构提取的描述符,输出则是蛋白质适应性的预测目标,一般对应于要改造的具体功能性指标.一旦模型建立,即可通过预测大量突变序列的性能快速筛选不理想的突变体.以Frances H. Arnold团队发表在PNAS上的一个工作为例[25].该工作主要是改造一氧化氮双加氧酶 (NOD) 立体选择性,并选择多个机器学习模型去构建NOD的立体选择性催化模型,包括但不仅限于K最近邻、线性模型、决策树、随机森林,将76% (S)-ee初始突变体提升至93% (S)-ee及反转至79% (R)-ee.中科院微生物研究所吴边团队提出一种新型蛋白质稳定性计算设计策略GRAPE[11].该策略对传统筛选突变体策略进行补充,并通过系统聚类分析对得到的单点有益突变进行聚类,同时结合贪婪算法进行网络迭代叠加,大幅度规避了以往遇到的累积突变所带来的负协同相互作用.设计出的突变体DuraPETase可在中等温度下有效降解塑料,为酶的设计的计算策略提供了非常重要的方向.当特定类型的酶数据比较小的时候,可以借助在大量通用酶类数据上的预训练模型来学习氨基酸对之间的互作用关系或者邻居结构环境信息,指导后续的酶改造任务.这种方法的好处是可以根据特定任务在具体的数据集上对预训练模型进行微调,以适应于不同的小数据集的下游任务.2021年提出的Low-N模型充分利用了UniRep中大量的蛋白质序列,通过无监督语言预训练任务提取了蛋白质的一般功能特征,然后通过特定家族序列上微调学习到了该家族的特异性特征[26].这样学习到的蛋白质表示后,仅需要少量的序列和目标功能的数据,就可以训练一个简单且有效的监督模型.将该模型应用到实际中,最少仅需24个avGFP突变体的数据集,就设计出了新的荧光蛋白,可以与高保真且高通量的蛋白质工程产物sfGFP相媲美.Low-N以较少的数量实现了蛋白质序列到功能模式的转变.类似工作还有[27]中提到的SEMA. ...
... 第二个难点是对于深度学习模型来说,从海量数据中挖掘模式是合适的.但是现有的状况是酶的相关数据量小,没有统一的标准格式,冗余的.当然这也与特定学科有关系.很多研究工作利用迁移学习来解决数据量小的问题,比如DeepET在大的蛋白质序列-最佳生长温度 (OGT) 数据集上训练模型,然后迁移到预测酶的最佳催化温度和蛋白质的熔融温度[98].或者利用自然语言处理 (NLP) 中广泛使用的大规模语言预训练模型学习序列的表示,然后小数据集上微调,进行一些功能预测[21,26]. ...
SEMA: Antigen B-cell conformational epitope prediction using deep transfer learning
1
2022
... 利用酶的序列以及功能性指标数据对,构建模型,然后利用模型指导酶分子改造.其构建的模型输入一般是基于序列或者结构提取的描述符,输出则是蛋白质适应性的预测目标,一般对应于要改造的具体功能性指标.一旦模型建立,即可通过预测大量突变序列的性能快速筛选不理想的突变体.以Frances H. Arnold团队发表在PNAS上的一个工作为例[25].该工作主要是改造一氧化氮双加氧酶 (NOD) 立体选择性,并选择多个机器学习模型去构建NOD的立体选择性催化模型,包括但不仅限于K最近邻、线性模型、决策树、随机森林,将76% (S)-ee初始突变体提升至93% (S)-ee及反转至79% (R)-ee.中科院微生物研究所吴边团队提出一种新型蛋白质稳定性计算设计策略GRAPE[11].该策略对传统筛选突变体策略进行补充,并通过系统聚类分析对得到的单点有益突变进行聚类,同时结合贪婪算法进行网络迭代叠加,大幅度规避了以往遇到的累积突变所带来的负协同相互作用.设计出的突变体DuraPETase可在中等温度下有效降解塑料,为酶的设计的计算策略提供了非常重要的方向.当特定类型的酶数据比较小的时候,可以借助在大量通用酶类数据上的预训练模型来学习氨基酸对之间的互作用关系或者邻居结构环境信息,指导后续的酶改造任务.这种方法的好处是可以根据特定任务在具体的数据集上对预训练模型进行微调,以适应于不同的小数据集的下游任务.2021年提出的Low-N模型充分利用了UniRep中大量的蛋白质序列,通过无监督语言预训练任务提取了蛋白质的一般功能特征,然后通过特定家族序列上微调学习到了该家族的特异性特征[26].这样学习到的蛋白质表示后,仅需要少量的序列和目标功能的数据,就可以训练一个简单且有效的监督模型.将该模型应用到实际中,最少仅需24个avGFP突变体的数据集,就设计出了新的荧光蛋白,可以与高保真且高通量的蛋白质工程产物sfGFP相媲美.Low-N以较少的数量实现了蛋白质序列到功能模式的转变.类似工作还有[27]中提到的SEMA. ...
Machine learning-aided engineering of hydrolases for PET depolymerization
1
2022
... 除此之外,随着日益丰富的结构数据与逐渐成熟的深度网络学习能力,从酶的结构数据集中直接挖掘结构与功能之间的关系也成为可能.2022年,得克萨斯大学奥斯汀分校 McKetta 化学工程系教授Hal S. Alper结合人工智能技术和酶工程,改造出一系列塑料降解酶的变体,相关工作发表在Nature上[28].其中最优秀的突变体FAST-PETase优于现有的PET降解酶的变体的降解效率,且能在更广泛环境中具有较好的活性,证明了在工业规模上酶塑料回收的可行途径.该方法首先筛选有效突变位点的方法是利用一个深度学习算法MutCompute[19]来有效过滤筛选突变位点 .MutCompute通过一个3D的自监督的卷积网络模型,对每一个残基构造一个局部微环境,统计该环境中C, H, O, N, S原子出现的次数,电荷,溶剂可达面积来编码该局部环境,最后预测每个残基的序列类型(分类问题).根据该残基一个已有突变体上的预测概率值与在野生型中的概率差异值大小,衡量出残基在野生型结构中的“不匹配度” (disfavoured) ,进而筛选出这种得分较大的突变位点,结合以往文献中报道的有效突变位点以及活性口袋位点,指导后续进一步筛选有效组合突变.该方法捕获了由结构决定的功能模式的指导转化,筛选条件是该残基在给定的蛋白质折叠环境中是否适配的能力.相比单纯使用序列的模型,考虑残基在结构环境中是否适配或从已有结构数据中学习这种规律,约束了改造酶的合理性并且增加了可能的改造位点方案.类似的工作还被应用在TEM-1 β-内酰胺酶和白色念珠菌磷化异构酶 (CaPMI) 中[29]. ...
Discovery of novel gain-of-function mutations guided by structure-based deep learning
1
2020
... 除此之外,随着日益丰富的结构数据与逐渐成熟的深度网络学习能力,从酶的结构数据集中直接挖掘结构与功能之间的关系也成为可能.2022年,得克萨斯大学奥斯汀分校 McKetta 化学工程系教授Hal S. Alper结合人工智能技术和酶工程,改造出一系列塑料降解酶的变体,相关工作发表在Nature上[28].其中最优秀的突变体FAST-PETase优于现有的PET降解酶的变体的降解效率,且能在更广泛环境中具有较好的活性,证明了在工业规模上酶塑料回收的可行途径.该方法首先筛选有效突变位点的方法是利用一个深度学习算法MutCompute[19]来有效过滤筛选突变位点 .MutCompute通过一个3D的自监督的卷积网络模型,对每一个残基构造一个局部微环境,统计该环境中C, H, O, N, S原子出现的次数,电荷,溶剂可达面积来编码该局部环境,最后预测每个残基的序列类型(分类问题).根据该残基一个已有突变体上的预测概率值与在野生型中的概率差异值大小,衡量出残基在野生型结构中的“不匹配度” (disfavoured) ,进而筛选出这种得分较大的突变位点,结合以往文献中报道的有效突变位点以及活性口袋位点,指导后续进一步筛选有效组合突变.该方法捕获了由结构决定的功能模式的指导转化,筛选条件是该残基在给定的蛋白质折叠环境中是否适配的能力.相比单纯使用序列的模型,考虑残基在结构环境中是否适配或从已有结构数据中学习这种规律,约束了改造酶的合理性并且增加了可能的改造位点方案.类似的工作还被应用在TEM-1 β-内酰胺酶和白色念珠菌磷化异构酶 (CaPMI) 中[29]. ...
Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences
1
2021
... 实际上在酶改造过程中,序列和结构信息并不是互相割裂的.Connor W. Coley组提出一种将结构约束在序列表示上,就是一种有效的思路.相比仅用ESM-1b[30]提取蛋白质序列的平均池化模式得到的序列特征,融入离酶活中心远近的结构性差异构建的池化策略,则在增强酶的嵌入性表达的同时还提高了酶活性预测任务的模型性能[31].丰富的酶结构信息,是非常重要且有效的(参见上面加入结构约束之后几个工作的性能提升).随着AlphaFold2等高精度有效的蛋白质结构预测方法的提出,如何结合预测出来的海量结构数据扩展对酶的功能改造,是具有研究价值的. ...
Predicting novel substrates for enzymes with minimal experimental effort with active learning
1
2017
... 实际上在酶改造过程中,序列和结构信息并不是互相割裂的.Connor W. Coley组提出一种将结构约束在序列表示上,就是一种有效的思路.相比仅用ESM-1b[30]提取蛋白质序列的平均池化模式得到的序列特征,融入离酶活中心远近的结构性差异构建的池化策略,则在增强酶的嵌入性表达的同时还提高了酶活性预测任务的模型性能[31].丰富的酶结构信息,是非常重要且有效的(参见上面加入结构约束之后几个工作的性能提升).随着AlphaFold2等高精度有效的蛋白质结构预测方法的提出,如何结合预测出来的海量结构数据扩展对酶的功能改造,是具有研究价值的. ...
A backbone-centred energy function of neural networks for protein design
2
2022
... Summary of protein design tools
Table 1| 方法名称/作者 | 类型 | 模型框架 | 输入 | 输出 | 训练集 | 应用 | 特点 | 网页/GitHub |
|---|
| SCUBA[32] | 骨架设计 | NC-NN | 二级结构motifs | 骨架 | PDB | 两层α/β蛋白;四螺旋束蛋白;EXTD | 突破之前方法仅限于已有模式的限制,基于核密度估计构造神经网络形式的能量函数 | https://doi.org/10.5281/zenodo.4533424 |
| Namrata Anand[33-34] | 骨架设计 | DCGAN | - | 距离图 | distance maps | 补齐完整的结构 | Cα原子之间的相对距离作为约束并优化 | - |
| Mire Zloh[35] | 序列生成 | LSTM | - | 序列 | CAMP+DBAASP+DRAMP+YADAMP | - | 设计对大肠杆菌具有潜在抗菌活性的短肽,并通过结构和表面性能与典型的AMP结构进行比较 | - |
| Gisbert Schneider[36] | 序列生成 | RNN | - | 序列 | ADAM/APD/DADP | 设计具有抗菌功能的肽 | 设计出的肽相比随机生成的肽具有抗菌活性的较高 | https://github.com/alexarnimueller/LSTM_peptides |
| ProteinGAN[37] | 序列生成 | GAN | - | 序列 | MDH 序列 | MDH酶 | 设计与苹果酸脱氢酶同样功能的酶,可同时出现100多个位点 | https://github.com/Biomatter-Designs/ProteinGAN |
| Mostafa Karimi[38] | 序列生成,给定折叠方式 | gcWGAN | - | 序列 | SCOPe v. 2.07 | - | 设计了一个从序列到折叠的预测器作为“oracle”,监督序列折叠成给定的折叠类型 | https://github.com/Shen-Lab/gcWGAN |
| ProteinMPNN[39] | 序列设计,结构约束 | 结构编码-序列解码的自回归模型 | 3D结构 | 序列 | CATH 4.2 | 单体、环状低聚物、蛋白质纳米颗粒 | 从结构中学习残基类型,将原子配对距离势融入到边的特征表示中,使序列恢复率直接提高约7.8% | https://github.com/dauparas/ProteinMPNN |
| ABACUS-R[40] | 序列设计,结构约束 | 结构编码-序列解码 | 3D结构 | 序列 | CATH 4.2 | PDB ID: 1r26, 1cy5 and 1ubq 3个骨架结构 | 从结构中学习残基类型,多任务学习 | https://github.com/liuyf020419/ABACUS-R |
| Transformer |
| David T. Jones[41] | 序列设计,结构约束 | 贪婪的半随机游走,逐步突变起始序列进行迭代的端到端设计 | 序列 | 序列 | - | Top7;Peak6; Foldit1; Ferredog-Diesel | 利用AlphaFold2预测生成序列的结构以及pLDDT打分,判断突变位点以及用距离图约束结构符合给定结构;对于最初始的序列,通过生成模型以及AlphaFold2结构约束产生初始序列 | |
| AlphaDesign[42] | 序列设计,结构约束 | 基于进化的遗传算法迭代生成序列 | 随机序列 | 序列 | - | 设计稳定的单体,二聚体直到六聚体 | 利用AlphaFold2预测的结构与要设计的骨架结构的差异来调整序列的优化 | - |
| trDesign[43] | 序列设计,结构约束 | trRosetta | 随机序列 | 序列 | - | - | 二维距离直方图的损失来更新梯度,更新被表示为PSSM的序列,可以理解为“折叠”的逆问题 | https://github.com/gjoni/trDesign |
| Hallucination[44] | 序列设计,结构约束,不固定骨架结构 | trRosetta | 随机序列 | 序列/结构 | PDB训练背景分布概率 | 设计2000条新的幻觉序列,聚类后129条表达后,62个蛋白可溶,高稳定 | 随机出发设计一条序列,通过最大化与随机背景序列的结构差异,约束该序列具有一个典型的2维结构特性 | https://github.com/gjoni/trDesign |
| Constrained hallucination2[45] | 序列设计,结构约束 | RoseTTAFold | 序列/结构 | 序列/结构 | RoseTTAFold训练集 | 免疫原;金属结合;新酶;特定结合的蛋白 | 设计具有给定motif的序列,通过神经网络不断迭代推理以及反向传播来设计序列 | https://github.com/RosettaCommons/RFDesign |
| RFjoint[45] | 序列设计,结构约束 | 训练RoseTTAFold | 序列/结构 | 序列/结构 | 微调,其中25%: PDB (2020-02-17);75%:AF2 预测结构 | 添加同时恢复序列和结构信息的损失,直接训练全新的模型 |
| PiFold[46] | 序列设计 | GNN | 3D结构 | 序列 | CATH | 序列恢复率:51.66%( CATH4.2),58.72%( TS50),60.42%( TS500) | 设计了新的残基特征器,PiGNN层学习多尺度(节点,边,全局)的残基相互作用信息 | https://github.com/A4Bio/PiFold |
| ProDESIGN-LE[47] | 序列设计 | Transformer+MLP | 3D结构 | 序列 | PDB40 | 设计CAT III酶新序列,3/5可表达且可溶;GFP | 通过Transformer学习当前残基在局部结构环境中的依赖性,使设计序列中的残基类型适配于当前的局部环境 | http://81.70.37.223/; https://github.com/bigict/ProDESIGN-LE |
1.2.1 主链结构设计主链结构设计,指的是在给定目标结构的拓扑下(例如:二级结构基本单元的组成以及顺序,相对位置等),设计出符合这种堆叠方式的蛋白质结构.这里介绍一个非常典型且由突破性的工作,SCUBA[32]. 该工作由中国科学技术大学刘海燕和陈泉团队提出,是一个具有高自主可设计性的主链设计算法,且并不依赖侧链类型.该算法在结构数据中基于核密度估计构造神经网络形式的能量函数来捕获高阶相关关系,可在不确定序列(即设计的能量函数不依赖于侧链,充分考虑柔性)的情况下,连续广泛搜索主链结构空间,突破之前方法仅限于已有模式的限制.再辅以该团队提出的给定主链设计序列的能量统计模型ABACUS[48],形成了一套全新的蛋白质自主设计新路线. ...
... 主链结构设计,指的是在给定目标结构的拓扑下(例如:二级结构基本单元的组成以及顺序,相对位置等),设计出符合这种堆叠方式的蛋白质结构.这里介绍一个非常典型且由突破性的工作,SCUBA[32]. 该工作由中国科学技术大学刘海燕和陈泉团队提出,是一个具有高自主可设计性的主链设计算法,且并不依赖侧链类型.该算法在结构数据中基于核密度估计构造神经网络形式的能量函数来捕获高阶相关关系,可在不确定序列(即设计的能量函数不依赖于侧链,充分考虑柔性)的情况下,连续广泛搜索主链结构空间,突破之前方法仅限于已有模式的限制.再辅以该团队提出的给定主链设计序列的能量统计模型ABACUS[48],形成了一套全新的蛋白质自主设计新路线. ...
Generative modeling for protein structures
2
2018
... Summary of protein design tools
Table 1| 方法名称/作者 | 类型 | 模型框架 | 输入 | 输出 | 训练集 | 应用 | 特点 | 网页/GitHub |
|---|
| SCUBA[32] | 骨架设计 | NC-NN | 二级结构motifs | 骨架 | PDB | 两层α/β蛋白;四螺旋束蛋白;EXTD | 突破之前方法仅限于已有模式的限制,基于核密度估计构造神经网络形式的能量函数 | https://doi.org/10.5281/zenodo.4533424 |
| Namrata Anand[33-34] | 骨架设计 | DCGAN | - | 距离图 | distance maps | 补齐完整的结构 | Cα原子之间的相对距离作为约束并优化 | - |
| Mire Zloh[35] | 序列生成 | LSTM | - | 序列 | CAMP+DBAASP+DRAMP+YADAMP | - | 设计对大肠杆菌具有潜在抗菌活性的短肽,并通过结构和表面性能与典型的AMP结构进行比较 | - |
| Gisbert Schneider[36] | 序列生成 | RNN | - | 序列 | ADAM/APD/DADP | 设计具有抗菌功能的肽 | 设计出的肽相比随机生成的肽具有抗菌活性的较高 | https://github.com/alexarnimueller/LSTM_peptides |
| ProteinGAN[37] | 序列生成 | GAN | - | 序列 | MDH 序列 | MDH酶 | 设计与苹果酸脱氢酶同样功能的酶,可同时出现100多个位点 | https://github.com/Biomatter-Designs/ProteinGAN |
| Mostafa Karimi[38] | 序列生成,给定折叠方式 | gcWGAN | - | 序列 | SCOPe v. 2.07 | - | 设计了一个从序列到折叠的预测器作为“oracle”,监督序列折叠成给定的折叠类型 | https://github.com/Shen-Lab/gcWGAN |
| ProteinMPNN[39] | 序列设计,结构约束 | 结构编码-序列解码的自回归模型 | 3D结构 | 序列 | CATH 4.2 | 单体、环状低聚物、蛋白质纳米颗粒 | 从结构中学习残基类型,将原子配对距离势融入到边的特征表示中,使序列恢复率直接提高约7.8% | https://github.com/dauparas/ProteinMPNN |
| ABACUS-R[40] | 序列设计,结构约束 | 结构编码-序列解码 | 3D结构 | 序列 | CATH 4.2 | PDB ID: 1r26, 1cy5 and 1ubq 3个骨架结构 | 从结构中学习残基类型,多任务学习 | https://github.com/liuyf020419/ABACUS-R |
| Transformer |
| David T. Jones[41] | 序列设计,结构约束 | 贪婪的半随机游走,逐步突变起始序列进行迭代的端到端设计 | 序列 | 序列 | - | Top7;Peak6; Foldit1; Ferredog-Diesel | 利用AlphaFold2预测生成序列的结构以及pLDDT打分,判断突变位点以及用距离图约束结构符合给定结构;对于最初始的序列,通过生成模型以及AlphaFold2结构约束产生初始序列 | |
| AlphaDesign[42] | 序列设计,结构约束 | 基于进化的遗传算法迭代生成序列 | 随机序列 | 序列 | - | 设计稳定的单体,二聚体直到六聚体 | 利用AlphaFold2预测的结构与要设计的骨架结构的差异来调整序列的优化 | - |
| trDesign[43] | 序列设计,结构约束 | trRosetta | 随机序列 | 序列 | - | - | 二维距离直方图的损失来更新梯度,更新被表示为PSSM的序列,可以理解为“折叠”的逆问题 | https://github.com/gjoni/trDesign |
| Hallucination[44] | 序列设计,结构约束,不固定骨架结构 | trRosetta | 随机序列 | 序列/结构 | PDB训练背景分布概率 | 设计2000条新的幻觉序列,聚类后129条表达后,62个蛋白可溶,高稳定 | 随机出发设计一条序列,通过最大化与随机背景序列的结构差异,约束该序列具有一个典型的2维结构特性 | https://github.com/gjoni/trDesign |
| Constrained hallucination2[45] | 序列设计,结构约束 | RoseTTAFold | 序列/结构 | 序列/结构 | RoseTTAFold训练集 | 免疫原;金属结合;新酶;特定结合的蛋白 | 设计具有给定motif的序列,通过神经网络不断迭代推理以及反向传播来设计序列 | https://github.com/RosettaCommons/RFDesign |
| RFjoint[45] | 序列设计,结构约束 | 训练RoseTTAFold | 序列/结构 | 序列/结构 | 微调,其中25%: PDB (2020-02-17);75%:AF2 预测结构 | 添加同时恢复序列和结构信息的损失,直接训练全新的模型 |
| PiFold[46] | 序列设计 | GNN | 3D结构 | 序列 | CATH | 序列恢复率:51.66%( CATH4.2),58.72%( TS50),60.42%( TS500) | 设计了新的残基特征器,PiGNN层学习多尺度(节点,边,全局)的残基相互作用信息 | https://github.com/A4Bio/PiFold |
| ProDESIGN-LE[47] | 序列设计 | Transformer+MLP | 3D结构 | 序列 | PDB40 | 设计CAT III酶新序列,3/5可表达且可溶;GFP | 通过Transformer学习当前残基在局部结构环境中的依赖性,使设计序列中的残基类型适配于当前的局部环境 | http://81.70.37.223/; https://github.com/bigict/ProDESIGN-LE |
1.2.1 主链结构设计主链结构设计,指的是在给定目标结构的拓扑下(例如:二级结构基本单元的组成以及顺序,相对位置等),设计出符合这种堆叠方式的蛋白质结构.这里介绍一个非常典型且由突破性的工作,SCUBA[32]. 该工作由中国科学技术大学刘海燕和陈泉团队提出,是一个具有高自主可设计性的主链设计算法,且并不依赖侧链类型.该算法在结构数据中基于核密度估计构造神经网络形式的能量函数来捕获高阶相关关系,可在不确定序列(即设计的能量函数不依赖于侧链,充分考虑柔性)的情况下,连续广泛搜索主链结构空间,突破之前方法仅限于已有模式的限制.再辅以该团队提出的给定主链设计序列的能量统计模型ABACUS[48],形成了一套全新的蛋白质自主设计新路线. ...
... 此外,Namrata Anand陆续提出基于生成对抗网络 (Generative Adversarial Network, GAN)[49] 实现蛋白质骨架设计的工作,从生成模型的角度考虑蛋白的骨架设计.第一个工作发表在2018年的NeurIPS,利用DCGAN (Deep Convolutional GANs)[50] 模型生成Cα原子之间的相对距离图(考虑到平移旋转不变性),将该配对距离约束引入到折叠成给定结构的可微问题中,并采用交替方向乘子法 (Alternating Direction Method of Multipliers, ADMM) 优化该凸规划问题[33].紧接着2019年发表的另一个工作也采用GAN实现给定距离约束下骨架设计,只是后面的精细化调整有所不同[34]. ...
Fully differentiable full-atom protein backbone generation
2
... Summary of protein design tools
Table 1| 方法名称/作者 | 类型 | 模型框架 | 输入 | 输出 | 训练集 | 应用 | 特点 | 网页/GitHub |
|---|
| SCUBA[32] | 骨架设计 | NC-NN | 二级结构motifs | 骨架 | PDB | 两层α/β蛋白;四螺旋束蛋白;EXTD | 突破之前方法仅限于已有模式的限制,基于核密度估计构造神经网络形式的能量函数 | https://doi.org/10.5281/zenodo.4533424 |
| Namrata Anand[33-34] | 骨架设计 | DCGAN | - | 距离图 | distance maps | 补齐完整的结构 | Cα原子之间的相对距离作为约束并优化 | - |
| Mire Zloh[35] | 序列生成 | LSTM | - | 序列 | CAMP+DBAASP+DRAMP+YADAMP | - | 设计对大肠杆菌具有潜在抗菌活性的短肽,并通过结构和表面性能与典型的AMP结构进行比较 | - |
| Gisbert Schneider[36] | 序列生成 | RNN | - | 序列 | ADAM/APD/DADP | 设计具有抗菌功能的肽 | 设计出的肽相比随机生成的肽具有抗菌活性的较高 | https://github.com/alexarnimueller/LSTM_peptides |
| ProteinGAN[37] | 序列生成 | GAN | - | 序列 | MDH 序列 | MDH酶 | 设计与苹果酸脱氢酶同样功能的酶,可同时出现100多个位点 | https://github.com/Biomatter-Designs/ProteinGAN |
| Mostafa Karimi[38] | 序列生成,给定折叠方式 | gcWGAN | - | 序列 | SCOPe v. 2.07 | - | 设计了一个从序列到折叠的预测器作为“oracle”,监督序列折叠成给定的折叠类型 | https://github.com/Shen-Lab/gcWGAN |
| ProteinMPNN[39] | 序列设计,结构约束 | 结构编码-序列解码的自回归模型 | 3D结构 | 序列 | CATH 4.2 | 单体、环状低聚物、蛋白质纳米颗粒 | 从结构中学习残基类型,将原子配对距离势融入到边的特征表示中,使序列恢复率直接提高约7.8% | https://github.com/dauparas/ProteinMPNN |
| ABACUS-R[40] | 序列设计,结构约束 | 结构编码-序列解码 | 3D结构 | 序列 | CATH 4.2 | PDB ID: 1r26, 1cy5 and 1ubq 3个骨架结构 | 从结构中学习残基类型,多任务学习 | https://github.com/liuyf020419/ABACUS-R |
| Transformer |
| David T. Jones[41] | 序列设计,结构约束 | 贪婪的半随机游走,逐步突变起始序列进行迭代的端到端设计 | 序列 | 序列 | - | Top7;Peak6; Foldit1; Ferredog-Diesel | 利用AlphaFold2预测生成序列的结构以及pLDDT打分,判断突变位点以及用距离图约束结构符合给定结构;对于最初始的序列,通过生成模型以及AlphaFold2结构约束产生初始序列 | |
| AlphaDesign[42] | 序列设计,结构约束 | 基于进化的遗传算法迭代生成序列 | 随机序列 | 序列 | - | 设计稳定的单体,二聚体直到六聚体 | 利用AlphaFold2预测的结构与要设计的骨架结构的差异来调整序列的优化 | - |
| trDesign[43] | 序列设计,结构约束 | trRosetta | 随机序列 | 序列 | - | - | 二维距离直方图的损失来更新梯度,更新被表示为PSSM的序列,可以理解为“折叠”的逆问题 | https://github.com/gjoni/trDesign |
| Hallucination[44] | 序列设计,结构约束,不固定骨架结构 | trRosetta | 随机序列 | 序列/结构 | PDB训练背景分布概率 | 设计2000条新的幻觉序列,聚类后129条表达后,62个蛋白可溶,高稳定 | 随机出发设计一条序列,通过最大化与随机背景序列的结构差异,约束该序列具有一个典型的2维结构特性 | https://github.com/gjoni/trDesign |
| Constrained hallucination2[45] | 序列设计,结构约束 | RoseTTAFold | 序列/结构 | 序列/结构 | RoseTTAFold训练集 | 免疫原;金属结合;新酶;特定结合的蛋白 | 设计具有给定motif的序列,通过神经网络不断迭代推理以及反向传播来设计序列 | https://github.com/RosettaCommons/RFDesign |
| RFjoint[45] | 序列设计,结构约束 | 训练RoseTTAFold | 序列/结构 | 序列/结构 | 微调,其中25%: PDB (2020-02-17);75%:AF2 预测结构 | 添加同时恢复序列和结构信息的损失,直接训练全新的模型 |
| PiFold[46] | 序列设计 | GNN | 3D结构 | 序列 | CATH | 序列恢复率:51.66%( CATH4.2),58.72%( TS50),60.42%( TS500) | 设计了新的残基特征器,PiGNN层学习多尺度(节点,边,全局)的残基相互作用信息 | https://github.com/A4Bio/PiFold |
| ProDESIGN-LE[47] | 序列设计 | Transformer+MLP | 3D结构 | 序列 | PDB40 | 设计CAT III酶新序列,3/5可表达且可溶;GFP | 通过Transformer学习当前残基在局部结构环境中的依赖性,使设计序列中的残基类型适配于当前的局部环境 | http://81.70.37.223/; https://github.com/bigict/ProDESIGN-LE |
1.2.1 主链结构设计主链结构设计,指的是在给定目标结构的拓扑下(例如:二级结构基本单元的组成以及顺序,相对位置等),设计出符合这种堆叠方式的蛋白质结构.这里介绍一个非常典型且由突破性的工作,SCUBA[32]. 该工作由中国科学技术大学刘海燕和陈泉团队提出,是一个具有高自主可设计性的主链设计算法,且并不依赖侧链类型.该算法在结构数据中基于核密度估计构造神经网络形式的能量函数来捕获高阶相关关系,可在不确定序列(即设计的能量函数不依赖于侧链,充分考虑柔性)的情况下,连续广泛搜索主链结构空间,突破之前方法仅限于已有模式的限制.再辅以该团队提出的给定主链设计序列的能量统计模型ABACUS[48],形成了一套全新的蛋白质自主设计新路线. ...
... 此外,Namrata Anand陆续提出基于生成对抗网络 (Generative Adversarial Network, GAN)[49] 实现蛋白质骨架设计的工作,从生成模型的角度考虑蛋白的骨架设计.第一个工作发表在2018年的NeurIPS,利用DCGAN (Deep Convolutional GANs)[50] 模型生成Cα原子之间的相对距离图(考虑到平移旋转不变性),将该配对距离约束引入到折叠成给定结构的可微问题中,并采用交替方向乘子法 (Alternating Direction Method of Multipliers, ADMM) 优化该凸规划问题[33].紧接着2019年发表的另一个工作也采用GAN实现给定距离约束下骨架设计,只是后面的精细化调整有所不同[34]. ...
Deep learning for novel antimicrobial peptide design
2
2021
... Summary of protein design tools
Table 1| 方法名称/作者 | 类型 | 模型框架 | 输入 | 输出 | 训练集 | 应用 | 特点 | 网页/GitHub |
|---|
| SCUBA[32] | 骨架设计 | NC-NN | 二级结构motifs | 骨架 | PDB | 两层α/β蛋白;四螺旋束蛋白;EXTD | 突破之前方法仅限于已有模式的限制,基于核密度估计构造神经网络形式的能量函数 | https://doi.org/10.5281/zenodo.4533424 |
| Namrata Anand[33-34] | 骨架设计 | DCGAN | - | 距离图 | distance maps | 补齐完整的结构 | Cα原子之间的相对距离作为约束并优化 | - |
| Mire Zloh[35] | 序列生成 | LSTM | - | 序列 | CAMP+DBAASP+DRAMP+YADAMP | - | 设计对大肠杆菌具有潜在抗菌活性的短肽,并通过结构和表面性能与典型的AMP结构进行比较 | - |
| Gisbert Schneider[36] | 序列生成 | RNN | - | 序列 | ADAM/APD/DADP | 设计具有抗菌功能的肽 | 设计出的肽相比随机生成的肽具有抗菌活性的较高 | https://github.com/alexarnimueller/LSTM_peptides |
| ProteinGAN[37] | 序列生成 | GAN | - | 序列 | MDH 序列 | MDH酶 | 设计与苹果酸脱氢酶同样功能的酶,可同时出现100多个位点 | https://github.com/Biomatter-Designs/ProteinGAN |
| Mostafa Karimi[38] | 序列生成,给定折叠方式 | gcWGAN | - | 序列 | SCOPe v. 2.07 | - | 设计了一个从序列到折叠的预测器作为“oracle”,监督序列折叠成给定的折叠类型 | https://github.com/Shen-Lab/gcWGAN |
| ProteinMPNN[39] | 序列设计,结构约束 | 结构编码-序列解码的自回归模型 | 3D结构 | 序列 | CATH 4.2 | 单体、环状低聚物、蛋白质纳米颗粒 | 从结构中学习残基类型,将原子配对距离势融入到边的特征表示中,使序列恢复率直接提高约7.8% | https://github.com/dauparas/ProteinMPNN |
| ABACUS-R[40] | 序列设计,结构约束 | 结构编码-序列解码 | 3D结构 | 序列 | CATH 4.2 | PDB ID: 1r26, 1cy5 and 1ubq 3个骨架结构 | 从结构中学习残基类型,多任务学习 | https://github.com/liuyf020419/ABACUS-R |
| Transformer |
| David T. Jones[41] | 序列设计,结构约束 | 贪婪的半随机游走,逐步突变起始序列进行迭代的端到端设计 | 序列 | 序列 | - | Top7;Peak6; Foldit1; Ferredog-Diesel | 利用AlphaFold2预测生成序列的结构以及pLDDT打分,判断突变位点以及用距离图约束结构符合给定结构;对于最初始的序列,通过生成模型以及AlphaFold2结构约束产生初始序列 | |
| AlphaDesign[42] | 序列设计,结构约束 | 基于进化的遗传算法迭代生成序列 | 随机序列 | 序列 | - | 设计稳定的单体,二聚体直到六聚体 | 利用AlphaFold2预测的结构与要设计的骨架结构的差异来调整序列的优化 | - |
| trDesign[43] | 序列设计,结构约束 | trRosetta | 随机序列 | 序列 | - | - | 二维距离直方图的损失来更新梯度,更新被表示为PSSM的序列,可以理解为“折叠”的逆问题 | https://github.com/gjoni/trDesign |
| Hallucination[44] | 序列设计,结构约束,不固定骨架结构 | trRosetta | 随机序列 | 序列/结构 | PDB训练背景分布概率 | 设计2000条新的幻觉序列,聚类后129条表达后,62个蛋白可溶,高稳定 | 随机出发设计一条序列,通过最大化与随机背景序列的结构差异,约束该序列具有一个典型的2维结构特性 | https://github.com/gjoni/trDesign |
| Constrained hallucination2[45] | 序列设计,结构约束 | RoseTTAFold | 序列/结构 | 序列/结构 | RoseTTAFold训练集 | 免疫原;金属结合;新酶;特定结合的蛋白 | 设计具有给定motif的序列,通过神经网络不断迭代推理以及反向传播来设计序列 | https://github.com/RosettaCommons/RFDesign |
| RFjoint[45] | 序列设计,结构约束 | 训练RoseTTAFold | 序列/结构 | 序列/结构 | 微调,其中25%: PDB (2020-02-17);75%:AF2 预测结构 | 添加同时恢复序列和结构信息的损失,直接训练全新的模型 |
| PiFold[46] | 序列设计 | GNN | 3D结构 | 序列 | CATH | 序列恢复率:51.66%( CATH4.2),58.72%( TS50),60.42%( TS500) | 设计了新的残基特征器,PiGNN层学习多尺度(节点,边,全局)的残基相互作用信息 | https://github.com/A4Bio/PiFold |
| ProDESIGN-LE[47] | 序列设计 | Transformer+MLP | 3D结构 | 序列 | PDB40 | 设计CAT III酶新序列,3/5可表达且可溶;GFP | 通过Transformer学习当前残基在局部结构环境中的依赖性,使设计序列中的残基类型适配于当前的局部环境 | http://81.70.37.223/; https://github.com/bigict/ProDESIGN-LE |
1.2.1 主链结构设计主链结构设计,指的是在给定目标结构的拓扑下(例如:二级结构基本单元的组成以及顺序,相对位置等),设计出符合这种堆叠方式的蛋白质结构.这里介绍一个非常典型且由突破性的工作,SCUBA[32]. 该工作由中国科学技术大学刘海燕和陈泉团队提出,是一个具有高自主可设计性的主链设计算法,且并不依赖侧链类型.该算法在结构数据中基于核密度估计构造神经网络形式的能量函数来捕获高阶相关关系,可在不确定序列(即设计的能量函数不依赖于侧链,充分考虑柔性)的情况下,连续广泛搜索主链结构空间,突破之前方法仅限于已有模式的限制.再辅以该团队提出的给定主链设计序列的能量统计模型ABACUS[48],形成了一套全新的蛋白质自主设计新路线. ...
... 当从功能上约束设计的序列时,可以采用序列生成方法,在具有给定功能的酶序列数据上挖掘残基间的模式直接生成新酶的序列.常用的生成模型有长短期记忆网络 (Long Short-Term Memory, LSTM) [51],GAN,变分自动编码器 (Variational Autoencoder, VAE)[52],Transformer[53]等.Mire Zloh课题组构建了基于LSTM的生成模型和双向LSTM分类模型,设计了对大肠杆菌具有潜在抗菌活性的新型的抗菌短肽序列,经过分类模型的预测发现设计出的肽序列被认为具有抗菌功能的概率在70.6-91.7%,且其三维构象表现出具有两亲性表面的α-螺旋结构[35].Gisbert Schneider课题组同样使用LSTM从螺旋抗菌肽序列上捕获数据的模式并将学习到的上下文信息运用于抗菌肽序列的生成[36].Aleksej Zelezniak课题组提出ProteinGAN,利用GAN学习到大量天然蛋白质序列的多样性并进而生成具有特定功能的酶序列[37].以苹果酸脱氢酶 (MDH) 为例,作者在该酶家族序列上进行训练并设计出具有相同功能酶的序列,其中有突变位点超过100个的设计序列,其活性与天然酶的活性相近. ...
Recurrent neural network model for constructive peptide design
2
2018
... Summary of protein design tools
Table 1| 方法名称/作者 | 类型 | 模型框架 | 输入 | 输出 | 训练集 | 应用 | 特点 | 网页/GitHub |
|---|
| SCUBA[32] | 骨架设计 | NC-NN | 二级结构motifs | 骨架 | PDB | 两层α/β蛋白;四螺旋束蛋白;EXTD | 突破之前方法仅限于已有模式的限制,基于核密度估计构造神经网络形式的能量函数 | https://doi.org/10.5281/zenodo.4533424 |
| Namrata Anand[33-34] | 骨架设计 | DCGAN | - | 距离图 | distance maps | 补齐完整的结构 | Cα原子之间的相对距离作为约束并优化 | - |
| Mire Zloh[35] | 序列生成 | LSTM | - | 序列 | CAMP+DBAASP+DRAMP+YADAMP | - | 设计对大肠杆菌具有潜在抗菌活性的短肽,并通过结构和表面性能与典型的AMP结构进行比较 | - |
| Gisbert Schneider[36] | 序列生成 | RNN | - | 序列 | ADAM/APD/DADP | 设计具有抗菌功能的肽 | 设计出的肽相比随机生成的肽具有抗菌活性的较高 | https://github.com/alexarnimueller/LSTM_peptides |
| ProteinGAN[37] | 序列生成 | GAN | - | 序列 | MDH 序列 | MDH酶 | 设计与苹果酸脱氢酶同样功能的酶,可同时出现100多个位点 | https://github.com/Biomatter-Designs/ProteinGAN |
| Mostafa Karimi[38] | 序列生成,给定折叠方式 | gcWGAN | - | 序列 | SCOPe v. 2.07 | - | 设计了一个从序列到折叠的预测器作为“oracle”,监督序列折叠成给定的折叠类型 | https://github.com/Shen-Lab/gcWGAN |
| ProteinMPNN[39] | 序列设计,结构约束 | 结构编码-序列解码的自回归模型 | 3D结构 | 序列 | CATH 4.2 | 单体、环状低聚物、蛋白质纳米颗粒 | 从结构中学习残基类型,将原子配对距离势融入到边的特征表示中,使序列恢复率直接提高约7.8% | https://github.com/dauparas/ProteinMPNN |
| ABACUS-R[40] | 序列设计,结构约束 | 结构编码-序列解码 | 3D结构 | 序列 | CATH 4.2 | PDB ID: 1r26, 1cy5 and 1ubq 3个骨架结构 | 从结构中学习残基类型,多任务学习 | https://github.com/liuyf020419/ABACUS-R |
| Transformer |
| David T. Jones[41] | 序列设计,结构约束 | 贪婪的半随机游走,逐步突变起始序列进行迭代的端到端设计 | 序列 | 序列 | - | Top7;Peak6; Foldit1; Ferredog-Diesel | 利用AlphaFold2预测生成序列的结构以及pLDDT打分,判断突变位点以及用距离图约束结构符合给定结构;对于最初始的序列,通过生成模型以及AlphaFold2结构约束产生初始序列 | |
| AlphaDesign[42] | 序列设计,结构约束 | 基于进化的遗传算法迭代生成序列 | 随机序列 | 序列 | - | 设计稳定的单体,二聚体直到六聚体 | 利用AlphaFold2预测的结构与要设计的骨架结构的差异来调整序列的优化 | - |
| trDesign[43] | 序列设计,结构约束 | trRosetta | 随机序列 | 序列 | - | - | 二维距离直方图的损失来更新梯度,更新被表示为PSSM的序列,可以理解为“折叠”的逆问题 | https://github.com/gjoni/trDesign |
| Hallucination[44] | 序列设计,结构约束,不固定骨架结构 | trRosetta | 随机序列 | 序列/结构 | PDB训练背景分布概率 | 设计2000条新的幻觉序列,聚类后129条表达后,62个蛋白可溶,高稳定 | 随机出发设计一条序列,通过最大化与随机背景序列的结构差异,约束该序列具有一个典型的2维结构特性 | https://github.com/gjoni/trDesign |
| Constrained hallucination2[45] | 序列设计,结构约束 | RoseTTAFold | 序列/结构 | 序列/结构 | RoseTTAFold训练集 | 免疫原;金属结合;新酶;特定结合的蛋白 | 设计具有给定motif的序列,通过神经网络不断迭代推理以及反向传播来设计序列 | https://github.com/RosettaCommons/RFDesign |
| RFjoint[45] | 序列设计,结构约束 | 训练RoseTTAFold | 序列/结构 | 序列/结构 | 微调,其中25%: PDB (2020-02-17);75%:AF2 预测结构 | 添加同时恢复序列和结构信息的损失,直接训练全新的模型 |
| PiFold[46] | 序列设计 | GNN | 3D结构 | 序列 | CATH | 序列恢复率:51.66%( CATH4.2),58.72%( TS50),60.42%( TS500) | 设计了新的残基特征器,PiGNN层学习多尺度(节点,边,全局)的残基相互作用信息 | https://github.com/A4Bio/PiFold |
| ProDESIGN-LE[47] | 序列设计 | Transformer+MLP | 3D结构 | 序列 | PDB40 | 设计CAT III酶新序列,3/5可表达且可溶;GFP | 通过Transformer学习当前残基在局部结构环境中的依赖性,使设计序列中的残基类型适配于当前的局部环境 | http://81.70.37.223/; https://github.com/bigict/ProDESIGN-LE |
1.2.1 主链结构设计主链结构设计,指的是在给定目标结构的拓扑下(例如:二级结构基本单元的组成以及顺序,相对位置等),设计出符合这种堆叠方式的蛋白质结构.这里介绍一个非常典型且由突破性的工作,SCUBA[32]. 该工作由中国科学技术大学刘海燕和陈泉团队提出,是一个具有高自主可设计性的主链设计算法,且并不依赖侧链类型.该算法在结构数据中基于核密度估计构造神经网络形式的能量函数来捕获高阶相关关系,可在不确定序列(即设计的能量函数不依赖于侧链,充分考虑柔性)的情况下,连续广泛搜索主链结构空间,突破之前方法仅限于已有模式的限制.再辅以该团队提出的给定主链设计序列的能量统计模型ABACUS[48],形成了一套全新的蛋白质自主设计新路线. ...
... 当从功能上约束设计的序列时,可以采用序列生成方法,在具有给定功能的酶序列数据上挖掘残基间的模式直接生成新酶的序列.常用的生成模型有长短期记忆网络 (Long Short-Term Memory, LSTM) [51],GAN,变分自动编码器 (Variational Autoencoder, VAE)[52],Transformer[53]等.Mire Zloh课题组构建了基于LSTM的生成模型和双向LSTM分类模型,设计了对大肠杆菌具有潜在抗菌活性的新型的抗菌短肽序列,经过分类模型的预测发现设计出的肽序列被认为具有抗菌功能的概率在70.6-91.7%,且其三维构象表现出具有两亲性表面的α-螺旋结构[35].Gisbert Schneider课题组同样使用LSTM从螺旋抗菌肽序列上捕获数据的模式并将学习到的上下文信息运用于抗菌肽序列的生成[36].Aleksej Zelezniak课题组提出ProteinGAN,利用GAN学习到大量天然蛋白质序列的多样性并进而生成具有特定功能的酶序列[37].以苹果酸脱氢酶 (MDH) 为例,作者在该酶家族序列上进行训练并设计出具有相同功能酶的序列,其中有突变位点超过100个的设计序列,其活性与天然酶的活性相近. ...
Expanding functional protein sequence spaces using generative adversarial networks
2
2021
... Summary of protein design tools
Table 1| 方法名称/作者 | 类型 | 模型框架 | 输入 | 输出 | 训练集 | 应用 | 特点 | 网页/GitHub |
|---|
| SCUBA[32] | 骨架设计 | NC-NN | 二级结构motifs | 骨架 | PDB | 两层α/β蛋白;四螺旋束蛋白;EXTD | 突破之前方法仅限于已有模式的限制,基于核密度估计构造神经网络形式的能量函数 | https://doi.org/10.5281/zenodo.4533424 |
| Namrata Anand[33-34] | 骨架设计 | DCGAN | - | 距离图 | distance maps | 补齐完整的结构 | Cα原子之间的相对距离作为约束并优化 | - |
| Mire Zloh[35] | 序列生成 | LSTM | - | 序列 | CAMP+DBAASP+DRAMP+YADAMP | - | 设计对大肠杆菌具有潜在抗菌活性的短肽,并通过结构和表面性能与典型的AMP结构进行比较 | - |
| Gisbert Schneider[36] | 序列生成 | RNN | - | 序列 | ADAM/APD/DADP | 设计具有抗菌功能的肽 | 设计出的肽相比随机生成的肽具有抗菌活性的较高 | https://github.com/alexarnimueller/LSTM_peptides |
| ProteinGAN[37] | 序列生成 | GAN | - | 序列 | MDH 序列 | MDH酶 | 设计与苹果酸脱氢酶同样功能的酶,可同时出现100多个位点 | https://github.com/Biomatter-Designs/ProteinGAN |
| Mostafa Karimi[38] | 序列生成,给定折叠方式 | gcWGAN | - | 序列 | SCOPe v. 2.07 | - | 设计了一个从序列到折叠的预测器作为“oracle”,监督序列折叠成给定的折叠类型 | https://github.com/Shen-Lab/gcWGAN |
| ProteinMPNN[39] | 序列设计,结构约束 | 结构编码-序列解码的自回归模型 | 3D结构 | 序列 | CATH 4.2 | 单体、环状低聚物、蛋白质纳米颗粒 | 从结构中学习残基类型,将原子配对距离势融入到边的特征表示中,使序列恢复率直接提高约7.8% | https://github.com/dauparas/ProteinMPNN |
| ABACUS-R[40] | 序列设计,结构约束 | 结构编码-序列解码 | 3D结构 | 序列 | CATH 4.2 | PDB ID: 1r26, 1cy5 and 1ubq 3个骨架结构 | 从结构中学习残基类型,多任务学习 | https://github.com/liuyf020419/ABACUS-R |
| Transformer |
| David T. Jones[41] | 序列设计,结构约束 | 贪婪的半随机游走,逐步突变起始序列进行迭代的端到端设计 | 序列 | 序列 | - | Top7;Peak6; Foldit1; Ferredog-Diesel | 利用AlphaFold2预测生成序列的结构以及pLDDT打分,判断突变位点以及用距离图约束结构符合给定结构;对于最初始的序列,通过生成模型以及AlphaFold2结构约束产生初始序列 | |
| AlphaDesign[42] | 序列设计,结构约束 | 基于进化的遗传算法迭代生成序列 | 随机序列 | 序列 | - | 设计稳定的单体,二聚体直到六聚体 | 利用AlphaFold2预测的结构与要设计的骨架结构的差异来调整序列的优化 | - |
| trDesign[43] | 序列设计,结构约束 | trRosetta | 随机序列 | 序列 | - | - | 二维距离直方图的损失来更新梯度,更新被表示为PSSM的序列,可以理解为“折叠”的逆问题 | https://github.com/gjoni/trDesign |
| Hallucination[44] | 序列设计,结构约束,不固定骨架结构 | trRosetta | 随机序列 | 序列/结构 | PDB训练背景分布概率 | 设计2000条新的幻觉序列,聚类后129条表达后,62个蛋白可溶,高稳定 | 随机出发设计一条序列,通过最大化与随机背景序列的结构差异,约束该序列具有一个典型的2维结构特性 | https://github.com/gjoni/trDesign |
| Constrained hallucination2[45] | 序列设计,结构约束 | RoseTTAFold | 序列/结构 | 序列/结构 | RoseTTAFold训练集 | 免疫原;金属结合;新酶;特定结合的蛋白 | 设计具有给定motif的序列,通过神经网络不断迭代推理以及反向传播来设计序列 | https://github.com/RosettaCommons/RFDesign |
| RFjoint[45] | 序列设计,结构约束 | 训练RoseTTAFold | 序列/结构 | 序列/结构 | 微调,其中25%: PDB (2020-02-17);75%:AF2 预测结构 | 添加同时恢复序列和结构信息的损失,直接训练全新的模型 |
| PiFold[46] | 序列设计 | GNN | 3D结构 | 序列 | CATH | 序列恢复率:51.66%( CATH4.2),58.72%( TS50),60.42%( TS500) | 设计了新的残基特征器,PiGNN层学习多尺度(节点,边,全局)的残基相互作用信息 | https://github.com/A4Bio/PiFold |
| ProDESIGN-LE[47] | 序列设计 | Transformer+MLP | 3D结构 | 序列 | PDB40 | 设计CAT III酶新序列,3/5可表达且可溶;GFP | 通过Transformer学习当前残基在局部结构环境中的依赖性,使设计序列中的残基类型适配于当前的局部环境 | http://81.70.37.223/; https://github.com/bigict/ProDESIGN-LE |
1.2.1 主链结构设计主链结构设计,指的是在给定目标结构的拓扑下(例如:二级结构基本单元的组成以及顺序,相对位置等),设计出符合这种堆叠方式的蛋白质结构.这里介绍一个非常典型且由突破性的工作,SCUBA[32]. 该工作由中国科学技术大学刘海燕和陈泉团队提出,是一个具有高自主可设计性的主链设计算法,且并不依赖侧链类型.该算法在结构数据中基于核密度估计构造神经网络形式的能量函数来捕获高阶相关关系,可在不确定序列(即设计的能量函数不依赖于侧链,充分考虑柔性)的情况下,连续广泛搜索主链结构空间,突破之前方法仅限于已有模式的限制.再辅以该团队提出的给定主链设计序列的能量统计模型ABACUS[48],形成了一套全新的蛋白质自主设计新路线. ...
... 当从功能上约束设计的序列时,可以采用序列生成方法,在具有给定功能的酶序列数据上挖掘残基间的模式直接生成新酶的序列.常用的生成模型有长短期记忆网络 (Long Short-Term Memory, LSTM) [51],GAN,变分自动编码器 (Variational Autoencoder, VAE)[52],Transformer[53]等.Mire Zloh课题组构建了基于LSTM的生成模型和双向LSTM分类模型,设计了对大肠杆菌具有潜在抗菌活性的新型的抗菌短肽序列,经过分类模型的预测发现设计出的肽序列被认为具有抗菌功能的概率在70.6-91.7%,且其三维构象表现出具有两亲性表面的α-螺旋结构[35].Gisbert Schneider课题组同样使用LSTM从螺旋抗菌肽序列上捕获数据的模式并将学习到的上下文信息运用于抗菌肽序列的生成[36].Aleksej Zelezniak课题组提出ProteinGAN,利用GAN学习到大量天然蛋白质序列的多样性并进而生成具有特定功能的酶序列[37].以苹果酸脱氢酶 (MDH) 为例,作者在该酶家族序列上进行训练并设计出具有相同功能酶的序列,其中有突变位点超过100个的设计序列,其活性与天然酶的活性相近. ...
De novo protein design for novel folds using guided conditional Wasserstein generative adversarial networks
2
2020
... Summary of protein design tools
Table 1| 方法名称/作者 | 类型 | 模型框架 | 输入 | 输出 | 训练集 | 应用 | 特点 | 网页/GitHub |
|---|
| SCUBA[32] | 骨架设计 | NC-NN | 二级结构motifs | 骨架 | PDB | 两层α/β蛋白;四螺旋束蛋白;EXTD | 突破之前方法仅限于已有模式的限制,基于核密度估计构造神经网络形式的能量函数 | https://doi.org/10.5281/zenodo.4533424 |
| Namrata Anand[33-34] | 骨架设计 | DCGAN | - | 距离图 | distance maps | 补齐完整的结构 | Cα原子之间的相对距离作为约束并优化 | - |
| Mire Zloh[35] | 序列生成 | LSTM | - | 序列 | CAMP+DBAASP+DRAMP+YADAMP | - | 设计对大肠杆菌具有潜在抗菌活性的短肽,并通过结构和表面性能与典型的AMP结构进行比较 | - |
| Gisbert Schneider[36] | 序列生成 | RNN | - | 序列 | ADAM/APD/DADP | 设计具有抗菌功能的肽 | 设计出的肽相比随机生成的肽具有抗菌活性的较高 | https://github.com/alexarnimueller/LSTM_peptides |
| ProteinGAN[37] | 序列生成 | GAN | - | 序列 | MDH 序列 | MDH酶 | 设计与苹果酸脱氢酶同样功能的酶,可同时出现100多个位点 | https://github.com/Biomatter-Designs/ProteinGAN |
| Mostafa Karimi[38] | 序列生成,给定折叠方式 | gcWGAN | - | 序列 | SCOPe v. 2.07 | - | 设计了一个从序列到折叠的预测器作为“oracle”,监督序列折叠成给定的折叠类型 | https://github.com/Shen-Lab/gcWGAN |
| ProteinMPNN[39] | 序列设计,结构约束 | 结构编码-序列解码的自回归模型 | 3D结构 | 序列 | CATH 4.2 | 单体、环状低聚物、蛋白质纳米颗粒 | 从结构中学习残基类型,将原子配对距离势融入到边的特征表示中,使序列恢复率直接提高约7.8% | https://github.com/dauparas/ProteinMPNN |
| ABACUS-R[40] | 序列设计,结构约束 | 结构编码-序列解码 | 3D结构 | 序列 | CATH 4.2 | PDB ID: 1r26, 1cy5 and 1ubq 3个骨架结构 | 从结构中学习残基类型,多任务学习 | https://github.com/liuyf020419/ABACUS-R |
| Transformer |
| David T. Jones[41] | 序列设计,结构约束 | 贪婪的半随机游走,逐步突变起始序列进行迭代的端到端设计 | 序列 | 序列 | - | Top7;Peak6; Foldit1; Ferredog-Diesel | 利用AlphaFold2预测生成序列的结构以及pLDDT打分,判断突变位点以及用距离图约束结构符合给定结构;对于最初始的序列,通过生成模型以及AlphaFold2结构约束产生初始序列 | |
| AlphaDesign[42] | 序列设计,结构约束 | 基于进化的遗传算法迭代生成序列 | 随机序列 | 序列 | - | 设计稳定的单体,二聚体直到六聚体 | 利用AlphaFold2预测的结构与要设计的骨架结构的差异来调整序列的优化 | - |
| trDesign[43] | 序列设计,结构约束 | trRosetta | 随机序列 | 序列 | - | - | 二维距离直方图的损失来更新梯度,更新被表示为PSSM的序列,可以理解为“折叠”的逆问题 | https://github.com/gjoni/trDesign |
| Hallucination[44] | 序列设计,结构约束,不固定骨架结构 | trRosetta | 随机序列 | 序列/结构 | PDB训练背景分布概率 | 设计2000条新的幻觉序列,聚类后129条表达后,62个蛋白可溶,高稳定 | 随机出发设计一条序列,通过最大化与随机背景序列的结构差异,约束该序列具有一个典型的2维结构特性 | https://github.com/gjoni/trDesign |
| Constrained hallucination2[45] | 序列设计,结构约束 | RoseTTAFold | 序列/结构 | 序列/结构 | RoseTTAFold训练集 | 免疫原;金属结合;新酶;特定结合的蛋白 | 设计具有给定motif的序列,通过神经网络不断迭代推理以及反向传播来设计序列 | https://github.com/RosettaCommons/RFDesign |
| RFjoint[45] | 序列设计,结构约束 | 训练RoseTTAFold | 序列/结构 | 序列/结构 | 微调,其中25%: PDB (2020-02-17);75%:AF2 预测结构 | 添加同时恢复序列和结构信息的损失,直接训练全新的模型 |
| PiFold[46] | 序列设计 | GNN | 3D结构 | 序列 | CATH | 序列恢复率:51.66%( CATH4.2),58.72%( TS50),60.42%( TS500) | 设计了新的残基特征器,PiGNN层学习多尺度(节点,边,全局)的残基相互作用信息 | https://github.com/A4Bio/PiFold |
| ProDESIGN-LE[47] | 序列设计 | Transformer+MLP | 3D结构 | 序列 | PDB40 | 设计CAT III酶新序列,3/5可表达且可溶;GFP | 通过Transformer学习当前残基在局部结构环境中的依赖性,使设计序列中的残基类型适配于当前的局部环境 | http://81.70.37.223/; https://github.com/bigict/ProDESIGN-LE |
1.2.1 主链结构设计主链结构设计,指的是在给定目标结构的拓扑下(例如:二级结构基本单元的组成以及顺序,相对位置等),设计出符合这种堆叠方式的蛋白质结构.这里介绍一个非常典型且由突破性的工作,SCUBA[32]. 该工作由中国科学技术大学刘海燕和陈泉团队提出,是一个具有高自主可设计性的主链设计算法,且并不依赖侧链类型.该算法在结构数据中基于核密度估计构造神经网络形式的能量函数来捕获高阶相关关系,可在不确定序列(即设计的能量函数不依赖于侧链,充分考虑柔性)的情况下,连续广泛搜索主链结构空间,突破之前方法仅限于已有模式的限制.再辅以该团队提出的给定主链设计序列的能量统计模型ABACUS[48],形成了一套全新的蛋白质自主设计新路线. ...
... 同样,可以采用结构约束来指导进而设计氨基酸序列.这种情况下,设计的氨基酸序列能否折叠成目标的蛋白质结构是至关重要的指标.最近被称为新一代Rosetta蛋白设计内核的Rosetta MPNN “Mover”,突破了传统的Rosetta设计范式“Inside-out”模式.该方法ProteinMPNN由David Baker组提出,基于structured-transformer[54],采用了结构编码-序列解码的自回归模型框架,将原子配对距离势融入到边的特征表示中,使序列恢复率提高约7.8%[39].ProteinMPNN对根据幻想的主链进行蛋白设计(500条),其中96条蛋白质序列在大肠杆菌体系中可以被大量的可溶表达,且成功结晶一个与设计结构高度一致的设计蛋白.同时,ProteinMPNN对单体,同源二聚体,异二聚体结构进行设计,其序列恢复率均在50%以上,其中核心区域的恢复率高达90%-95%.中国科学技术大学刘海燕和陈泉团队提出的ABACUS-R完全基于深度学习算法实现给定骨架设计氨基酸序列,不再依赖于传统能量项构建,并且序列恢复率高于ABACUS计算的,在测试集上基本可以达到50%[40].其主要思路是在给定骨架的情况下,通过编码-解码 (encoder-decoder) 框架学习在给定残基的结构特征以及周边结构环境的特性预测该残基的序列类型(侧链).值得一提的是,ABACUS-R采用多任务学习,不仅仅学习该残基的类型,还同时预测其二级结构,溶剂可达面积,B-factor以及一些结构构象扭转角任务.这些辅助任务的设计不仅提高了模型设计序列的能力,还隐式的在序列设计中加入了实时的结构约束.实验验证设计了3个天然骨架的蛋白序列设计并做了相应的实验验证.最后通过ABACUS-R设计出了可以成功表达且折叠成相应的三维结构的蛋白质序列,充分证明蛋白质设计可以绕过侧链模型的显示建模的可行性.卜东波课题组提出ProDESIGN-LE也是基于Transformer框架,通过计算序列类型是否符合给定的局部结构环境来设计蛋白序列[47].在实验中为CAT III酶设计的5条序列中,有3条可以成功表达且可溶.许锦波课题组提出的一种基于骨架设计蛋白序列的方法,基于生成SE (3) 等变模型,显著改进了现有的自回归方法[55].Mostafa Karimi组[38]提出gcWGAN探索生成给定折叠条件下的序列,使序列折叠成给定的方式.要是构造了一个基于DeepSF[56]的快速从序列预测折叠模式的模型并实时反馈监督序列是否可以正确折叠,这个模型被称为“Oracle”.Po-Ssu Huang组的Namrata Anand[57]直接从蛋白质骨架结构信息中学习侧链氨基酸类型,从而学习到一个基于自回归的自动的神经网络能量来指导后续的序列设计.在实际的TIM-barrel设计中,设计出的序列中有两个成功结晶且与设计的骨架高度一致. ...
Robust deep learning–based protein sequence design using ProteinMPNN
2
2022
... Summary of protein design tools
Table 1| 方法名称/作者 | 类型 | 模型框架 | 输入 | 输出 | 训练集 | 应用 | 特点 | 网页/GitHub |
|---|
| SCUBA[32] | 骨架设计 | NC-NN | 二级结构motifs | 骨架 | PDB | 两层α/β蛋白;四螺旋束蛋白;EXTD | 突破之前方法仅限于已有模式的限制,基于核密度估计构造神经网络形式的能量函数 | https://doi.org/10.5281/zenodo.4533424 |
| Namrata Anand[33-34] | 骨架设计 | DCGAN | - | 距离图 | distance maps | 补齐完整的结构 | Cα原子之间的相对距离作为约束并优化 | - |
| Mire Zloh[35] | 序列生成 | LSTM | - | 序列 | CAMP+DBAASP+DRAMP+YADAMP | - | 设计对大肠杆菌具有潜在抗菌活性的短肽,并通过结构和表面性能与典型的AMP结构进行比较 | - |
| Gisbert Schneider[36] | 序列生成 | RNN | - | 序列 | ADAM/APD/DADP | 设计具有抗菌功能的肽 | 设计出的肽相比随机生成的肽具有抗菌活性的较高 | https://github.com/alexarnimueller/LSTM_peptides |
| ProteinGAN[37] | 序列生成 | GAN | - | 序列 | MDH 序列 | MDH酶 | 设计与苹果酸脱氢酶同样功能的酶,可同时出现100多个位点 | https://github.com/Biomatter-Designs/ProteinGAN |
| Mostafa Karimi[38] | 序列生成,给定折叠方式 | gcWGAN | - | 序列 | SCOPe v. 2.07 | - | 设计了一个从序列到折叠的预测器作为“oracle”,监督序列折叠成给定的折叠类型 | https://github.com/Shen-Lab/gcWGAN |
| ProteinMPNN[39] | 序列设计,结构约束 | 结构编码-序列解码的自回归模型 | 3D结构 | 序列 | CATH 4.2 | 单体、环状低聚物、蛋白质纳米颗粒 | 从结构中学习残基类型,将原子配对距离势融入到边的特征表示中,使序列恢复率直接提高约7.8% | https://github.com/dauparas/ProteinMPNN |
| ABACUS-R[40] | 序列设计,结构约束 | 结构编码-序列解码 | 3D结构 | 序列 | CATH 4.2 | PDB ID: 1r26, 1cy5 and 1ubq 3个骨架结构 | 从结构中学习残基类型,多任务学习 | https://github.com/liuyf020419/ABACUS-R |
| Transformer |
| David T. Jones[41] | 序列设计,结构约束 | 贪婪的半随机游走,逐步突变起始序列进行迭代的端到端设计 | 序列 | 序列 | - | Top7;Peak6; Foldit1; Ferredog-Diesel | 利用AlphaFold2预测生成序列的结构以及pLDDT打分,判断突变位点以及用距离图约束结构符合给定结构;对于最初始的序列,通过生成模型以及AlphaFold2结构约束产生初始序列 | |
| AlphaDesign[42] | 序列设计,结构约束 | 基于进化的遗传算法迭代生成序列 | 随机序列 | 序列 | - | 设计稳定的单体,二聚体直到六聚体 | 利用AlphaFold2预测的结构与要设计的骨架结构的差异来调整序列的优化 | - |
| trDesign[43] | 序列设计,结构约束 | trRosetta | 随机序列 | 序列 | - | - | 二维距离直方图的损失来更新梯度,更新被表示为PSSM的序列,可以理解为“折叠”的逆问题 | https://github.com/gjoni/trDesign |
| Hallucination[44] | 序列设计,结构约束,不固定骨架结构 | trRosetta | 随机序列 | 序列/结构 | PDB训练背景分布概率 | 设计2000条新的幻觉序列,聚类后129条表达后,62个蛋白可溶,高稳定 | 随机出发设计一条序列,通过最大化与随机背景序列的结构差异,约束该序列具有一个典型的2维结构特性 | https://github.com/gjoni/trDesign |
| Constrained hallucination2[45] | 序列设计,结构约束 | RoseTTAFold | 序列/结构 | 序列/结构 | RoseTTAFold训练集 | 免疫原;金属结合;新酶;特定结合的蛋白 | 设计具有给定motif的序列,通过神经网络不断迭代推理以及反向传播来设计序列 | https://github.com/RosettaCommons/RFDesign |
| RFjoint[45] | 序列设计,结构约束 | 训练RoseTTAFold | 序列/结构 | 序列/结构 | 微调,其中25%: PDB (2020-02-17);75%:AF2 预测结构 | 添加同时恢复序列和结构信息的损失,直接训练全新的模型 |
| PiFold[46] | 序列设计 | GNN | 3D结构 | 序列 | CATH | 序列恢复率:51.66%( CATH4.2),58.72%( TS50),60.42%( TS500) | 设计了新的残基特征器,PiGNN层学习多尺度(节点,边,全局)的残基相互作用信息 | https://github.com/A4Bio/PiFold |
| ProDESIGN-LE[47] | 序列设计 | Transformer+MLP | 3D结构 | 序列 | PDB40 | 设计CAT III酶新序列,3/5可表达且可溶;GFP | 通过Transformer学习当前残基在局部结构环境中的依赖性,使设计序列中的残基类型适配于当前的局部环境 | http://81.70.37.223/; https://github.com/bigict/ProDESIGN-LE |
1.2.1 主链结构设计主链结构设计,指的是在给定目标结构的拓扑下(例如:二级结构基本单元的组成以及顺序,相对位置等),设计出符合这种堆叠方式的蛋白质结构.这里介绍一个非常典型且由突破性的工作,SCUBA[32]. 该工作由中国科学技术大学刘海燕和陈泉团队提出,是一个具有高自主可设计性的主链设计算法,且并不依赖侧链类型.该算法在结构数据中基于核密度估计构造神经网络形式的能量函数来捕获高阶相关关系,可在不确定序列(即设计的能量函数不依赖于侧链,充分考虑柔性)的情况下,连续广泛搜索主链结构空间,突破之前方法仅限于已有模式的限制.再辅以该团队提出的给定主链设计序列的能量统计模型ABACUS[48],形成了一套全新的蛋白质自主设计新路线. ...
... 同样,可以采用结构约束来指导进而设计氨基酸序列.这种情况下,设计的氨基酸序列能否折叠成目标的蛋白质结构是至关重要的指标.最近被称为新一代Rosetta蛋白设计内核的Rosetta MPNN “Mover”,突破了传统的Rosetta设计范式“Inside-out”模式.该方法ProteinMPNN由David Baker组提出,基于structured-transformer[54],采用了结构编码-序列解码的自回归模型框架,将原子配对距离势融入到边的特征表示中,使序列恢复率提高约7.8%[39].ProteinMPNN对根据幻想的主链进行蛋白设计(500条),其中96条蛋白质序列在大肠杆菌体系中可以被大量的可溶表达,且成功结晶一个与设计结构高度一致的设计蛋白.同时,ProteinMPNN对单体,同源二聚体,异二聚体结构进行设计,其序列恢复率均在50%以上,其中核心区域的恢复率高达90%-95%.中国科学技术大学刘海燕和陈泉团队提出的ABACUS-R完全基于深度学习算法实现给定骨架设计氨基酸序列,不再依赖于传统能量项构建,并且序列恢复率高于ABACUS计算的,在测试集上基本可以达到50%[40].其主要思路是在给定骨架的情况下,通过编码-解码 (encoder-decoder) 框架学习在给定残基的结构特征以及周边结构环境的特性预测该残基的序列类型(侧链).值得一提的是,ABACUS-R采用多任务学习,不仅仅学习该残基的类型,还同时预测其二级结构,溶剂可达面积,B-factor以及一些结构构象扭转角任务.这些辅助任务的设计不仅提高了模型设计序列的能力,还隐式的在序列设计中加入了实时的结构约束.实验验证设计了3个天然骨架的蛋白序列设计并做了相应的实验验证.最后通过ABACUS-R设计出了可以成功表达且折叠成相应的三维结构的蛋白质序列,充分证明蛋白质设计可以绕过侧链模型的显示建模的可行性.卜东波课题组提出ProDESIGN-LE也是基于Transformer框架,通过计算序列类型是否符合给定的局部结构环境来设计蛋白序列[47].在实验中为CAT III酶设计的5条序列中,有3条可以成功表达且可溶.许锦波课题组提出的一种基于骨架设计蛋白序列的方法,基于生成SE (3) 等变模型,显著改进了现有的自回归方法[55].Mostafa Karimi组[38]提出gcWGAN探索生成给定折叠条件下的序列,使序列折叠成给定的方式.要是构造了一个基于DeepSF[56]的快速从序列预测折叠模式的模型并实时反馈监督序列是否可以正确折叠,这个模型被称为“Oracle”.Po-Ssu Huang组的Namrata Anand[57]直接从蛋白质骨架结构信息中学习侧链氨基酸类型,从而学习到一个基于自回归的自动的神经网络能量来指导后续的序列设计.在实际的TIM-barrel设计中,设计出的序列中有两个成功结晶且与设计的骨架高度一致. ...
Rotamer-free protein sequence design based on deep learning and self-consistency
2
2022
... Summary of protein design tools
Table 1| 方法名称/作者 | 类型 | 模型框架 | 输入 | 输出 | 训练集 | 应用 | 特点 | 网页/GitHub |
|---|
| SCUBA[32] | 骨架设计 | NC-NN | 二级结构motifs | 骨架 | PDB | 两层α/β蛋白;四螺旋束蛋白;EXTD | 突破之前方法仅限于已有模式的限制,基于核密度估计构造神经网络形式的能量函数 | https://doi.org/10.5281/zenodo.4533424 |
| Namrata Anand[33-34] | 骨架设计 | DCGAN | - | 距离图 | distance maps | 补齐完整的结构 | Cα原子之间的相对距离作为约束并优化 | - |
| Mire Zloh[35] | 序列生成 | LSTM | - | 序列 | CAMP+DBAASP+DRAMP+YADAMP | - | 设计对大肠杆菌具有潜在抗菌活性的短肽,并通过结构和表面性能与典型的AMP结构进行比较 | - |
| Gisbert Schneider[36] | 序列生成 | RNN | - | 序列 | ADAM/APD/DADP | 设计具有抗菌功能的肽 | 设计出的肽相比随机生成的肽具有抗菌活性的较高 | https://github.com/alexarnimueller/LSTM_peptides |
| ProteinGAN[37] | 序列生成 | GAN | - | 序列 | MDH 序列 | MDH酶 | 设计与苹果酸脱氢酶同样功能的酶,可同时出现100多个位点 | https://github.com/Biomatter-Designs/ProteinGAN |
| Mostafa Karimi[38] | 序列生成,给定折叠方式 | gcWGAN | - | 序列 | SCOPe v. 2.07 | - | 设计了一个从序列到折叠的预测器作为“oracle”,监督序列折叠成给定的折叠类型 | https://github.com/Shen-Lab/gcWGAN |
| ProteinMPNN[39] | 序列设计,结构约束 | 结构编码-序列解码的自回归模型 | 3D结构 | 序列 | CATH 4.2 | 单体、环状低聚物、蛋白质纳米颗粒 | 从结构中学习残基类型,将原子配对距离势融入到边的特征表示中,使序列恢复率直接提高约7.8% | https://github.com/dauparas/ProteinMPNN |
| ABACUS-R[40] | 序列设计,结构约束 | 结构编码-序列解码 | 3D结构 | 序列 | CATH 4.2 | PDB ID: 1r26, 1cy5 and 1ubq 3个骨架结构 | 从结构中学习残基类型,多任务学习 | https://github.com/liuyf020419/ABACUS-R |
| Transformer |
| David T. Jones[41] | 序列设计,结构约束 | 贪婪的半随机游走,逐步突变起始序列进行迭代的端到端设计 | 序列 | 序列 | - | Top7;Peak6; Foldit1; Ferredog-Diesel | 利用AlphaFold2预测生成序列的结构以及pLDDT打分,判断突变位点以及用距离图约束结构符合给定结构;对于最初始的序列,通过生成模型以及AlphaFold2结构约束产生初始序列 | |
| AlphaDesign[42] | 序列设计,结构约束 | 基于进化的遗传算法迭代生成序列 | 随机序列 | 序列 | - | 设计稳定的单体,二聚体直到六聚体 | 利用AlphaFold2预测的结构与要设计的骨架结构的差异来调整序列的优化 | - |
| trDesign[43] | 序列设计,结构约束 | trRosetta | 随机序列 | 序列 | - | - | 二维距离直方图的损失来更新梯度,更新被表示为PSSM的序列,可以理解为“折叠”的逆问题 | https://github.com/gjoni/trDesign |
| Hallucination[44] | 序列设计,结构约束,不固定骨架结构 | trRosetta | 随机序列 | 序列/结构 | PDB训练背景分布概率 | 设计2000条新的幻觉序列,聚类后129条表达后,62个蛋白可溶,高稳定 | 随机出发设计一条序列,通过最大化与随机背景序列的结构差异,约束该序列具有一个典型的2维结构特性 | https://github.com/gjoni/trDesign |
| Constrained hallucination2[45] | 序列设计,结构约束 | RoseTTAFold | 序列/结构 | 序列/结构 | RoseTTAFold训练集 | 免疫原;金属结合;新酶;特定结合的蛋白 | 设计具有给定motif的序列,通过神经网络不断迭代推理以及反向传播来设计序列 | https://github.com/RosettaCommons/RFDesign |
| RFjoint[45] | 序列设计,结构约束 | 训练RoseTTAFold | 序列/结构 | 序列/结构 | 微调,其中25%: PDB (2020-02-17);75%:AF2 预测结构 | 添加同时恢复序列和结构信息的损失,直接训练全新的模型 |
| PiFold[46] | 序列设计 | GNN | 3D结构 | 序列 | CATH | 序列恢复率:51.66%( CATH4.2),58.72%( TS50),60.42%( TS500) | 设计了新的残基特征器,PiGNN层学习多尺度(节点,边,全局)的残基相互作用信息 | https://github.com/A4Bio/PiFold |
| ProDESIGN-LE[47] | 序列设计 | Transformer+MLP | 3D结构 | 序列 | PDB40 | 设计CAT III酶新序列,3/5可表达且可溶;GFP | 通过Transformer学习当前残基在局部结构环境中的依赖性,使设计序列中的残基类型适配于当前的局部环境 | http://81.70.37.223/; https://github.com/bigict/ProDESIGN-LE |
1.2.1 主链结构设计主链结构设计,指的是在给定目标结构的拓扑下(例如:二级结构基本单元的组成以及顺序,相对位置等),设计出符合这种堆叠方式的蛋白质结构.这里介绍一个非常典型且由突破性的工作,SCUBA[32]. 该工作由中国科学技术大学刘海燕和陈泉团队提出,是一个具有高自主可设计性的主链设计算法,且并不依赖侧链类型.该算法在结构数据中基于核密度估计构造神经网络形式的能量函数来捕获高阶相关关系,可在不确定序列(即设计的能量函数不依赖于侧链,充分考虑柔性)的情况下,连续广泛搜索主链结构空间,突破之前方法仅限于已有模式的限制.再辅以该团队提出的给定主链设计序列的能量统计模型ABACUS[48],形成了一套全新的蛋白质自主设计新路线. ...
... 同样,可以采用结构约束来指导进而设计氨基酸序列.这种情况下,设计的氨基酸序列能否折叠成目标的蛋白质结构是至关重要的指标.最近被称为新一代Rosetta蛋白设计内核的Rosetta MPNN “Mover”,突破了传统的Rosetta设计范式“Inside-out”模式.该方法ProteinMPNN由David Baker组提出,基于structured-transformer[54],采用了结构编码-序列解码的自回归模型框架,将原子配对距离势融入到边的特征表示中,使序列恢复率提高约7.8%[39].ProteinMPNN对根据幻想的主链进行蛋白设计(500条),其中96条蛋白质序列在大肠杆菌体系中可以被大量的可溶表达,且成功结晶一个与设计结构高度一致的设计蛋白.同时,ProteinMPNN对单体,同源二聚体,异二聚体结构进行设计,其序列恢复率均在50%以上,其中核心区域的恢复率高达90%-95%.中国科学技术大学刘海燕和陈泉团队提出的ABACUS-R完全基于深度学习算法实现给定骨架设计氨基酸序列,不再依赖于传统能量项构建,并且序列恢复率高于ABACUS计算的,在测试集上基本可以达到50%[40].其主要思路是在给定骨架的情况下,通过编码-解码 (encoder-decoder) 框架学习在给定残基的结构特征以及周边结构环境的特性预测该残基的序列类型(侧链).值得一提的是,ABACUS-R采用多任务学习,不仅仅学习该残基的类型,还同时预测其二级结构,溶剂可达面积,B-factor以及一些结构构象扭转角任务.这些辅助任务的设计不仅提高了模型设计序列的能力,还隐式的在序列设计中加入了实时的结构约束.实验验证设计了3个天然骨架的蛋白序列设计并做了相应的实验验证.最后通过ABACUS-R设计出了可以成功表达且折叠成相应的三维结构的蛋白质序列,充分证明蛋白质设计可以绕过侧链模型的显示建模的可行性.卜东波课题组提出ProDESIGN-LE也是基于Transformer框架,通过计算序列类型是否符合给定的局部结构环境来设计蛋白序列[47].在实验中为CAT III酶设计的5条序列中,有3条可以成功表达且可溶.许锦波课题组提出的一种基于骨架设计蛋白序列的方法,基于生成SE (3) 等变模型,显著改进了现有的自回归方法[55].Mostafa Karimi组[38]提出gcWGAN探索生成给定折叠条件下的序列,使序列折叠成给定的方式.要是构造了一个基于DeepSF[56]的快速从序列预测折叠模式的模型并实时反馈监督序列是否可以正确折叠,这个模型被称为“Oracle”.Po-Ssu Huang组的Namrata Anand[57]直接从蛋白质骨架结构信息中学习侧链氨基酸类型,从而学习到一个基于自回归的自动的神经网络能量来指导后续的序列设计.在实际的TIM-barrel设计中,设计出的序列中有两个成功结晶且与设计的骨架高度一致. ...
Using AlphaFold for rapid and accurate fixed backbone protein design
3
2021
... Summary of protein design tools
Table 1| 方法名称/作者 | 类型 | 模型框架 | 输入 | 输出 | 训练集 | 应用 | 特点 | 网页/GitHub |
|---|
| SCUBA[32] | 骨架设计 | NC-NN | 二级结构motifs | 骨架 | PDB | 两层α/β蛋白;四螺旋束蛋白;EXTD | 突破之前方法仅限于已有模式的限制,基于核密度估计构造神经网络形式的能量函数 | https://doi.org/10.5281/zenodo.4533424 |
| Namrata Anand[33-34] | 骨架设计 | DCGAN | - | 距离图 | distance maps | 补齐完整的结构 | Cα原子之间的相对距离作为约束并优化 | - |
| Mire Zloh[35] | 序列生成 | LSTM | - | 序列 | CAMP+DBAASP+DRAMP+YADAMP | - | 设计对大肠杆菌具有潜在抗菌活性的短肽,并通过结构和表面性能与典型的AMP结构进行比较 | - |
| Gisbert Schneider[36] | 序列生成 | RNN | - | 序列 | ADAM/APD/DADP | 设计具有抗菌功能的肽 | 设计出的肽相比随机生成的肽具有抗菌活性的较高 | https://github.com/alexarnimueller/LSTM_peptides |
| ProteinGAN[37] | 序列生成 | GAN | - | 序列 | MDH 序列 | MDH酶 | 设计与苹果酸脱氢酶同样功能的酶,可同时出现100多个位点 | https://github.com/Biomatter-Designs/ProteinGAN |
| Mostafa Karimi[38] | 序列生成,给定折叠方式 | gcWGAN | - | 序列 | SCOPe v. 2.07 | - | 设计了一个从序列到折叠的预测器作为“oracle”,监督序列折叠成给定的折叠类型 | https://github.com/Shen-Lab/gcWGAN |
| ProteinMPNN[39] | 序列设计,结构约束 | 结构编码-序列解码的自回归模型 | 3D结构 | 序列 | CATH 4.2 | 单体、环状低聚物、蛋白质纳米颗粒 | 从结构中学习残基类型,将原子配对距离势融入到边的特征表示中,使序列恢复率直接提高约7.8% | https://github.com/dauparas/ProteinMPNN |
| ABACUS-R[40] | 序列设计,结构约束 | 结构编码-序列解码 | 3D结构 | 序列 | CATH 4.2 | PDB ID: 1r26, 1cy5 and 1ubq 3个骨架结构 | 从结构中学习残基类型,多任务学习 | https://github.com/liuyf020419/ABACUS-R |
| Transformer |
| David T. Jones[41] | 序列设计,结构约束 | 贪婪的半随机游走,逐步突变起始序列进行迭代的端到端设计 | 序列 | 序列 | - | Top7;Peak6; Foldit1; Ferredog-Diesel | 利用AlphaFold2预测生成序列的结构以及pLDDT打分,判断突变位点以及用距离图约束结构符合给定结构;对于最初始的序列,通过生成模型以及AlphaFold2结构约束产生初始序列 | |
| AlphaDesign[42] | 序列设计,结构约束 | 基于进化的遗传算法迭代生成序列 | 随机序列 | 序列 | - | 设计稳定的单体,二聚体直到六聚体 | 利用AlphaFold2预测的结构与要设计的骨架结构的差异来调整序列的优化 | - |
| trDesign[43] | 序列设计,结构约束 | trRosetta | 随机序列 | 序列 | - | - | 二维距离直方图的损失来更新梯度,更新被表示为PSSM的序列,可以理解为“折叠”的逆问题 | https://github.com/gjoni/trDesign |
| Hallucination[44] | 序列设计,结构约束,不固定骨架结构 | trRosetta | 随机序列 | 序列/结构 | PDB训练背景分布概率 | 设计2000条新的幻觉序列,聚类后129条表达后,62个蛋白可溶,高稳定 | 随机出发设计一条序列,通过最大化与随机背景序列的结构差异,约束该序列具有一个典型的2维结构特性 | https://github.com/gjoni/trDesign |
| Constrained hallucination2[45] | 序列设计,结构约束 | RoseTTAFold | 序列/结构 | 序列/结构 | RoseTTAFold训练集 | 免疫原;金属结合;新酶;特定结合的蛋白 | 设计具有给定motif的序列,通过神经网络不断迭代推理以及反向传播来设计序列 | https://github.com/RosettaCommons/RFDesign |
| RFjoint[45] | 序列设计,结构约束 | 训练RoseTTAFold | 序列/结构 | 序列/结构 | 微调,其中25%: PDB (2020-02-17);75%:AF2 预测结构 | 添加同时恢复序列和结构信息的损失,直接训练全新的模型 |
| PiFold[46] | 序列设计 | GNN | 3D结构 | 序列 | CATH | 序列恢复率:51.66%( CATH4.2),58.72%( TS50),60.42%( TS500) | 设计了新的残基特征器,PiGNN层学习多尺度(节点,边,全局)的残基相互作用信息 | https://github.com/A4Bio/PiFold |
| ProDESIGN-LE[47] | 序列设计 | Transformer+MLP | 3D结构 | 序列 | PDB40 | 设计CAT III酶新序列,3/5可表达且可溶;GFP | 通过Transformer学习当前残基在局部结构环境中的依赖性,使设计序列中的残基类型适配于当前的局部环境 | http://81.70.37.223/; https://github.com/bigict/ProDESIGN-LE |
1.2.1 主链结构设计主链结构设计,指的是在给定目标结构的拓扑下(例如:二级结构基本单元的组成以及顺序,相对位置等),设计出符合这种堆叠方式的蛋白质结构.这里介绍一个非常典型且由突破性的工作,SCUBA[32]. 该工作由中国科学技术大学刘海燕和陈泉团队提出,是一个具有高自主可设计性的主链设计算法,且并不依赖侧链类型.该算法在结构数据中基于核密度估计构造神经网络形式的能量函数来捕获高阶相关关系,可在不确定序列(即设计的能量函数不依赖于侧链,充分考虑柔性)的情况下,连续广泛搜索主链结构空间,突破之前方法仅限于已有模式的限制.再辅以该团队提出的给定主链设计序列的能量统计模型ABACUS[48],形成了一套全新的蛋白质自主设计新路线. ...
... 考虑酶的“可折叠性”,最直观的解决办法是快速预测设计的新酶的结构,检验其是否具有给定结构.因此,第一种预测是将蛋白结构预测工具作为一个监督者,约束生成的序列具有折叠成给定结构的能力(如图1左上角)[41-45,85].这个思路实施起来的最大难点是从序列预测结构的精度限制.但是现在得益于结构预测的突破性进展,使的这种设计新酶成为可能.其基本思路是在设计序列的时候,加入一个辅助的“监督者”对于生成的序列是否可以折叠且具有给定的构象进行评分.根据得分对蛋白质序列通过基于梯度的,梯度自由的,或者神经网络构造的优化方法来更新序列.通过不断重复迭代这一过程,最终得到构象约束下的收敛序列.设计序列的时候一般遵从最小能量的原则.但是,我们不清楚给定的构象就一定是设计的这条序列折叠后的最低能量构象.因此结构预测做为“监督器”实际上去计算了在给定结构情况下蛋白质序列的最大联合概率. ...
... David T. Jones尝试将AlphaFold2引入固定骨架设计序列的过程中,以约束生成的序列能够折叠成给定的骨架,并且正交实验中也验证了分子动力学方法模拟的结构对AlphaFold2监督后的实验结构高度支持[41].其具体流程是:(1)生成初始蛋白序列.基于研究者之前提出的基于自回归的Transformer蛋白质序列生成模型[88]生成1000条初始序列.同时对于得到的序列用AlphaFold2预测其结构,并与要设计的骨架结构用TM-align[89]做结构比对.最后选择结构比对得分最高的那部分结构的序列为初始序列,不具有高结构置信度的序列则用丙氨酸填充.这样做的好处是保证初始的序列是可收敛的,否则可能序列太随机导致最后没办法折叠.(2)在序列空间中执行贪婪的半随机游走,逐步突变起始序列进行迭代的端到端的设计.这里面AlphaFold2的作用有两个,一个是预测序列结构,比较与要设计结构的距离直方图损失.根据损失是否减小来判断突变序列是否合理.另一个是确定该序列中哪一部分残基位点要被突变,修改.举例来说,从起始序列出发并通过AlphaFold2预测其结构以及每一个残基的pLDDT打分(衡量每个残基的局部结构合理性).这里,计算预测结构中的距离直方图并与要设计的骨架结构的直方图计算损失.同时,利用每个残基的pLDDT打分设置为序列位点是否要被采样的概率.得分较高代表此处残基是稳定的,反之则是下一次迭代序列设计采样的点.在下次迭代采样中,对于选定的采样位点进行饱和突变,直到距离直方图损失减小,才接受序列的突变采样.这样设置的好处是对于与要设计结构的高度匹配的序列不再改变,大量减少采样时间尽快收敛以及可能引起的负协同效应.作者在人工设计的Top7上进行测试,得到的序列结构不论是通过AlphaFold2,trRosetta还是基于片段从头折叠的方法,均被证实与要设计的骨架可能是同一种折叠.该工作应用AlphaFold2在初始序列设计上保证了与目标结构的局部高结构匹配度,同时在序列设计过程中利用AlphaFold2预测的结构与目标结构的距离直方图损失约束其设计序列保持全局结构相似性以及利用残基位点可信度增强局部残基结构稳定性.同年,S. Kashif Sadiq也在bioRxiv上提交AlphaDesign工作,基本思路也是利用AlphaFold2预测的结构与要设计的骨架结构的差异来限制调整序列的优化,采用的优化函数是基于进化的遗传算法来迭代生成序列[42].主要差别在于该方法利用预测结构的三维坐标信息差异构建目标函数优化而不仅仅是二维的配对距离直方图约束,可能在结构约束上更加有效.而且该方法扩展了可能的设计任务的范围,设计了一些长度在32到256的结构稳定的从头设计的具有不同折叠的单体蛋白,同源二聚体,异源二聚体,同源低聚物(三聚体到六聚体).Baker组提出的trDesign是第一个提出将结构预测工具trRosetta应用到蛋白质序列设计中的工作,考虑的也是二维距离直方图的损失来更新梯度,更新被表示为PSSM的序列[43].但是受限于trRosetta利用的是二维的结构约束,在正交验证中发现基于这种反向传播的方式设计序列不能很好的对三维结构进行编码.且上述三个工作均是基于给定骨架设计序列,限制了实际设计酶的应用需求.后来Baker组提出的“幻想” (hallucination) 的方法,不从给定骨架结构出发设计序列,而是考虑在这种目标结构缺失的条件下,是否能随机产生结构和序列.其实现是通过最大化设计序列的结构与随机背景序列的差异约束,从而约束该序列折叠后的结构具有一个典型的2维结构特性[44].实验中幻想了2000条序列,聚类后发现均可以在已有的PDB结构库中寻找到相似的折叠.实验验证的时候有62条是可溶表达的(实验验证了129条),且CD的圆二色谱和目标结构的二级结构分布吻合.相比传统设计验证的方法,仅仅129条实验验证且48%的成功率,极大的减少了人工验证的成本和时间.但是由于trRosetta精度有限以及二维结构约束的不足,在接下来的工作中将RoseTTAFold嵌入到具有给定motif的序列设计中[45].RoseTTAFold显示利用SE-3 Transformer预测三维结构坐标以及二维距离分布,大大提高了序列设计的准确性.在免疫相关蛋白中,成功设计出携带中和性抗体表位的蛋白以及与新冠病毒S突刺蛋白受体结合的ACE2类似物蛋白.后续提出的RFjoint,不再通过神经网络不断迭代推理以及反向传播来设计序列,而是将结构预测和序列设计两大任务结合起来,直接训练全新的模型[45].这样的好处是减少了反向推理时间大大减少了设计的时间成本. ...
AlphaDesign: a de novo protein design framework based on AlphaFold
3
2021
... Summary of protein design tools
Table 1| 方法名称/作者 | 类型 | 模型框架 | 输入 | 输出 | 训练集 | 应用 | 特点 | 网页/GitHub |
|---|
| SCUBA[32] | 骨架设计 | NC-NN | 二级结构motifs | 骨架 | PDB | 两层α/β蛋白;四螺旋束蛋白;EXTD | 突破之前方法仅限于已有模式的限制,基于核密度估计构造神经网络形式的能量函数 | https://doi.org/10.5281/zenodo.4533424 |
| Namrata Anand[33-34] | 骨架设计 | DCGAN | - | 距离图 | distance maps | 补齐完整的结构 | Cα原子之间的相对距离作为约束并优化 | - |
| Mire Zloh[35] | 序列生成 | LSTM | - | 序列 | CAMP+DBAASP+DRAMP+YADAMP | - | 设计对大肠杆菌具有潜在抗菌活性的短肽,并通过结构和表面性能与典型的AMP结构进行比较 | - |
| Gisbert Schneider[36] | 序列生成 | RNN | - | 序列 | ADAM/APD/DADP | 设计具有抗菌功能的肽 | 设计出的肽相比随机生成的肽具有抗菌活性的较高 | https://github.com/alexarnimueller/LSTM_peptides |
| ProteinGAN[37] | 序列生成 | GAN | - | 序列 | MDH 序列 | MDH酶 | 设计与苹果酸脱氢酶同样功能的酶,可同时出现100多个位点 | https://github.com/Biomatter-Designs/ProteinGAN |
| Mostafa Karimi[38] | 序列生成,给定折叠方式 | gcWGAN | - | 序列 | SCOPe v. 2.07 | - | 设计了一个从序列到折叠的预测器作为“oracle”,监督序列折叠成给定的折叠类型 | https://github.com/Shen-Lab/gcWGAN |
| ProteinMPNN[39] | 序列设计,结构约束 | 结构编码-序列解码的自回归模型 | 3D结构 | 序列 | CATH 4.2 | 单体、环状低聚物、蛋白质纳米颗粒 | 从结构中学习残基类型,将原子配对距离势融入到边的特征表示中,使序列恢复率直接提高约7.8% | https://github.com/dauparas/ProteinMPNN |
| ABACUS-R[40] | 序列设计,结构约束 | 结构编码-序列解码 | 3D结构 | 序列 | CATH 4.2 | PDB ID: 1r26, 1cy5 and 1ubq 3个骨架结构 | 从结构中学习残基类型,多任务学习 | https://github.com/liuyf020419/ABACUS-R |
| Transformer |
| David T. Jones[41] | 序列设计,结构约束 | 贪婪的半随机游走,逐步突变起始序列进行迭代的端到端设计 | 序列 | 序列 | - | Top7;Peak6; Foldit1; Ferredog-Diesel | 利用AlphaFold2预测生成序列的结构以及pLDDT打分,判断突变位点以及用距离图约束结构符合给定结构;对于最初始的序列,通过生成模型以及AlphaFold2结构约束产生初始序列 | |
| AlphaDesign[42] | 序列设计,结构约束 | 基于进化的遗传算法迭代生成序列 | 随机序列 | 序列 | - | 设计稳定的单体,二聚体直到六聚体 | 利用AlphaFold2预测的结构与要设计的骨架结构的差异来调整序列的优化 | - |
| trDesign[43] | 序列设计,结构约束 | trRosetta | 随机序列 | 序列 | - | - | 二维距离直方图的损失来更新梯度,更新被表示为PSSM的序列,可以理解为“折叠”的逆问题 | https://github.com/gjoni/trDesign |
| Hallucination[44] | 序列设计,结构约束,不固定骨架结构 | trRosetta | 随机序列 | 序列/结构 | PDB训练背景分布概率 | 设计2000条新的幻觉序列,聚类后129条表达后,62个蛋白可溶,高稳定 | 随机出发设计一条序列,通过最大化与随机背景序列的结构差异,约束该序列具有一个典型的2维结构特性 | https://github.com/gjoni/trDesign |
| Constrained hallucination2[45] | 序列设计,结构约束 | RoseTTAFold | 序列/结构 | 序列/结构 | RoseTTAFold训练集 | 免疫原;金属结合;新酶;特定结合的蛋白 | 设计具有给定motif的序列,通过神经网络不断迭代推理以及反向传播来设计序列 | https://github.com/RosettaCommons/RFDesign |
| RFjoint[45] | 序列设计,结构约束 | 训练RoseTTAFold | 序列/结构 | 序列/结构 | 微调,其中25%: PDB (2020-02-17);75%:AF2 预测结构 | 添加同时恢复序列和结构信息的损失,直接训练全新的模型 |
| PiFold[46] | 序列设计 | GNN | 3D结构 | 序列 | CATH | 序列恢复率:51.66%( CATH4.2),58.72%( TS50),60.42%( TS500) | 设计了新的残基特征器,PiGNN层学习多尺度(节点,边,全局)的残基相互作用信息 | https://github.com/A4Bio/PiFold |
| ProDESIGN-LE[47] | 序列设计 | Transformer+MLP | 3D结构 | 序列 | PDB40 | 设计CAT III酶新序列,3/5可表达且可溶;GFP | 通过Transformer学习当前残基在局部结构环境中的依赖性,使设计序列中的残基类型适配于当前的局部环境 | http://81.70.37.223/; https://github.com/bigict/ProDESIGN-LE |
1.2.1 主链结构设计主链结构设计,指的是在给定目标结构的拓扑下(例如:二级结构基本单元的组成以及顺序,相对位置等),设计出符合这种堆叠方式的蛋白质结构.这里介绍一个非常典型且由突破性的工作,SCUBA[32]. 该工作由中国科学技术大学刘海燕和陈泉团队提出,是一个具有高自主可设计性的主链设计算法,且并不依赖侧链类型.该算法在结构数据中基于核密度估计构造神经网络形式的能量函数来捕获高阶相关关系,可在不确定序列(即设计的能量函数不依赖于侧链,充分考虑柔性)的情况下,连续广泛搜索主链结构空间,突破之前方法仅限于已有模式的限制.再辅以该团队提出的给定主链设计序列的能量统计模型ABACUS[48],形成了一套全新的蛋白质自主设计新路线. ...
... (1PLDDT (predicted local distance difference test): 描述每个残基预测的局部置信度,局部结构指标,0-100,越大越好2PAE (predicted aligned error): 估计氨基酸在每一个位置上的误差,成对计算指标3Disfavoured: 反应预测序列类型于当前结构微环境的“适配性”4TMscore (template modeling score): 衡量蛋白质结构间的相似度,全局结构指标,0-1,越大越好5RMSD (root-mean-square deviation): 表示两个蛋白结构对齐后的结构差异,一般认为小于2Å预测准确.越小越好(图中展示的代表性方法分别按照1,2,3三部分从上至下摘自文献[43,45],[42-43],[86-87])) ...
... David T. Jones尝试将AlphaFold2引入固定骨架设计序列的过程中,以约束生成的序列能够折叠成给定的骨架,并且正交实验中也验证了分子动力学方法模拟的结构对AlphaFold2监督后的实验结构高度支持[41].其具体流程是:(1)生成初始蛋白序列.基于研究者之前提出的基于自回归的Transformer蛋白质序列生成模型[88]生成1000条初始序列.同时对于得到的序列用AlphaFold2预测其结构,并与要设计的骨架结构用TM-align[89]做结构比对.最后选择结构比对得分最高的那部分结构的序列为初始序列,不具有高结构置信度的序列则用丙氨酸填充.这样做的好处是保证初始的序列是可收敛的,否则可能序列太随机导致最后没办法折叠.(2)在序列空间中执行贪婪的半随机游走,逐步突变起始序列进行迭代的端到端的设计.这里面AlphaFold2的作用有两个,一个是预测序列结构,比较与要设计结构的距离直方图损失.根据损失是否减小来判断突变序列是否合理.另一个是确定该序列中哪一部分残基位点要被突变,修改.举例来说,从起始序列出发并通过AlphaFold2预测其结构以及每一个残基的pLDDT打分(衡量每个残基的局部结构合理性).这里,计算预测结构中的距离直方图并与要设计的骨架结构的直方图计算损失.同时,利用每个残基的pLDDT打分设置为序列位点是否要被采样的概率.得分较高代表此处残基是稳定的,反之则是下一次迭代序列设计采样的点.在下次迭代采样中,对于选定的采样位点进行饱和突变,直到距离直方图损失减小,才接受序列的突变采样.这样设置的好处是对于与要设计结构的高度匹配的序列不再改变,大量减少采样时间尽快收敛以及可能引起的负协同效应.作者在人工设计的Top7上进行测试,得到的序列结构不论是通过AlphaFold2,trRosetta还是基于片段从头折叠的方法,均被证实与要设计的骨架可能是同一种折叠.该工作应用AlphaFold2在初始序列设计上保证了与目标结构的局部高结构匹配度,同时在序列设计过程中利用AlphaFold2预测的结构与目标结构的距离直方图损失约束其设计序列保持全局结构相似性以及利用残基位点可信度增强局部残基结构稳定性.同年,S. Kashif Sadiq也在bioRxiv上提交AlphaDesign工作,基本思路也是利用AlphaFold2预测的结构与要设计的骨架结构的差异来限制调整序列的优化,采用的优化函数是基于进化的遗传算法来迭代生成序列[42].主要差别在于该方法利用预测结构的三维坐标信息差异构建目标函数优化而不仅仅是二维的配对距离直方图约束,可能在结构约束上更加有效.而且该方法扩展了可能的设计任务的范围,设计了一些长度在32到256的结构稳定的从头设计的具有不同折叠的单体蛋白,同源二聚体,异源二聚体,同源低聚物(三聚体到六聚体).Baker组提出的trDesign是第一个提出将结构预测工具trRosetta应用到蛋白质序列设计中的工作,考虑的也是二维距离直方图的损失来更新梯度,更新被表示为PSSM的序列[43].但是受限于trRosetta利用的是二维的结构约束,在正交验证中发现基于这种反向传播的方式设计序列不能很好的对三维结构进行编码.且上述三个工作均是基于给定骨架设计序列,限制了实际设计酶的应用需求.后来Baker组提出的“幻想” (hallucination) 的方法,不从给定骨架结构出发设计序列,而是考虑在这种目标结构缺失的条件下,是否能随机产生结构和序列.其实现是通过最大化设计序列的结构与随机背景序列的差异约束,从而约束该序列折叠后的结构具有一个典型的2维结构特性[44].实验中幻想了2000条序列,聚类后发现均可以在已有的PDB结构库中寻找到相似的折叠.实验验证的时候有62条是可溶表达的(实验验证了129条),且CD的圆二色谱和目标结构的二级结构分布吻合.相比传统设计验证的方法,仅仅129条实验验证且48%的成功率,极大的减少了人工验证的成本和时间.但是由于trRosetta精度有限以及二维结构约束的不足,在接下来的工作中将RoseTTAFold嵌入到具有给定motif的序列设计中[45].RoseTTAFold显示利用SE-3 Transformer预测三维结构坐标以及二维距离分布,大大提高了序列设计的准确性.在免疫相关蛋白中,成功设计出携带中和性抗体表位的蛋白以及与新冠病毒S突刺蛋白受体结合的ACE2类似物蛋白.后续提出的RFjoint,不再通过神经网络不断迭代推理以及反向传播来设计序列,而是将结构预测和序列设计两大任务结合起来,直接训练全新的模型[45].这样的好处是减少了反向推理时间大大减少了设计的时间成本. ...
Protein sequence design by explicit energy landscape optimization
4
2020
... Summary of protein design tools
Table 1| 方法名称/作者 | 类型 | 模型框架 | 输入 | 输出 | 训练集 | 应用 | 特点 | 网页/GitHub |
|---|
| SCUBA[32] | 骨架设计 | NC-NN | 二级结构motifs | 骨架 | PDB | 两层α/β蛋白;四螺旋束蛋白;EXTD | 突破之前方法仅限于已有模式的限制,基于核密度估计构造神经网络形式的能量函数 | https://doi.org/10.5281/zenodo.4533424 |
| Namrata Anand[33-34] | 骨架设计 | DCGAN | - | 距离图 | distance maps | 补齐完整的结构 | Cα原子之间的相对距离作为约束并优化 | - |
| Mire Zloh[35] | 序列生成 | LSTM | - | 序列 | CAMP+DBAASP+DRAMP+YADAMP | - | 设计对大肠杆菌具有潜在抗菌活性的短肽,并通过结构和表面性能与典型的AMP结构进行比较 | - |
| Gisbert Schneider[36] | 序列生成 | RNN | - | 序列 | ADAM/APD/DADP | 设计具有抗菌功能的肽 | 设计出的肽相比随机生成的肽具有抗菌活性的较高 | https://github.com/alexarnimueller/LSTM_peptides |
| ProteinGAN[37] | 序列生成 | GAN | - | 序列 | MDH 序列 | MDH酶 | 设计与苹果酸脱氢酶同样功能的酶,可同时出现100多个位点 | https://github.com/Biomatter-Designs/ProteinGAN |
| Mostafa Karimi[38] | 序列生成,给定折叠方式 | gcWGAN | - | 序列 | SCOPe v. 2.07 | - | 设计了一个从序列到折叠的预测器作为“oracle”,监督序列折叠成给定的折叠类型 | https://github.com/Shen-Lab/gcWGAN |
| ProteinMPNN[39] | 序列设计,结构约束 | 结构编码-序列解码的自回归模型 | 3D结构 | 序列 | CATH 4.2 | 单体、环状低聚物、蛋白质纳米颗粒 | 从结构中学习残基类型,将原子配对距离势融入到边的特征表示中,使序列恢复率直接提高约7.8% | https://github.com/dauparas/ProteinMPNN |
| ABACUS-R[40] | 序列设计,结构约束 | 结构编码-序列解码 | 3D结构 | 序列 | CATH 4.2 | PDB ID: 1r26, 1cy5 and 1ubq 3个骨架结构 | 从结构中学习残基类型,多任务学习 | https://github.com/liuyf020419/ABACUS-R |
| Transformer |
| David T. Jones[41] | 序列设计,结构约束 | 贪婪的半随机游走,逐步突变起始序列进行迭代的端到端设计 | 序列 | 序列 | - | Top7;Peak6; Foldit1; Ferredog-Diesel | 利用AlphaFold2预测生成序列的结构以及pLDDT打分,判断突变位点以及用距离图约束结构符合给定结构;对于最初始的序列,通过生成模型以及AlphaFold2结构约束产生初始序列 | |
| AlphaDesign[42] | 序列设计,结构约束 | 基于进化的遗传算法迭代生成序列 | 随机序列 | 序列 | - | 设计稳定的单体,二聚体直到六聚体 | 利用AlphaFold2预测的结构与要设计的骨架结构的差异来调整序列的优化 | - |
| trDesign[43] | 序列设计,结构约束 | trRosetta | 随机序列 | 序列 | - | - | 二维距离直方图的损失来更新梯度,更新被表示为PSSM的序列,可以理解为“折叠”的逆问题 | https://github.com/gjoni/trDesign |
| Hallucination[44] | 序列设计,结构约束,不固定骨架结构 | trRosetta | 随机序列 | 序列/结构 | PDB训练背景分布概率 | 设计2000条新的幻觉序列,聚类后129条表达后,62个蛋白可溶,高稳定 | 随机出发设计一条序列,通过最大化与随机背景序列的结构差异,约束该序列具有一个典型的2维结构特性 | https://github.com/gjoni/trDesign |
| Constrained hallucination2[45] | 序列设计,结构约束 | RoseTTAFold | 序列/结构 | 序列/结构 | RoseTTAFold训练集 | 免疫原;金属结合;新酶;特定结合的蛋白 | 设计具有给定motif的序列,通过神经网络不断迭代推理以及反向传播来设计序列 | https://github.com/RosettaCommons/RFDesign |
| RFjoint[45] | 序列设计,结构约束 | 训练RoseTTAFold | 序列/结构 | 序列/结构 | 微调,其中25%: PDB (2020-02-17);75%:AF2 预测结构 | 添加同时恢复序列和结构信息的损失,直接训练全新的模型 |
| PiFold[46] | 序列设计 | GNN | 3D结构 | 序列 | CATH | 序列恢复率:51.66%( CATH4.2),58.72%( TS50),60.42%( TS500) | 设计了新的残基特征器,PiGNN层学习多尺度(节点,边,全局)的残基相互作用信息 | https://github.com/A4Bio/PiFold |
| ProDESIGN-LE[47] | 序列设计 | Transformer+MLP | 3D结构 | 序列 | PDB40 | 设计CAT III酶新序列,3/5可表达且可溶;GFP | 通过Transformer学习当前残基在局部结构环境中的依赖性,使设计序列中的残基类型适配于当前的局部环境 | http://81.70.37.223/; https://github.com/bigict/ProDESIGN-LE |
1.2.1 主链结构设计主链结构设计,指的是在给定目标结构的拓扑下(例如:二级结构基本单元的组成以及顺序,相对位置等),设计出符合这种堆叠方式的蛋白质结构.这里介绍一个非常典型且由突破性的工作,SCUBA[32]. 该工作由中国科学技术大学刘海燕和陈泉团队提出,是一个具有高自主可设计性的主链设计算法,且并不依赖侧链类型.该算法在结构数据中基于核密度估计构造神经网络形式的能量函数来捕获高阶相关关系,可在不确定序列(即设计的能量函数不依赖于侧链,充分考虑柔性)的情况下,连续广泛搜索主链结构空间,突破之前方法仅限于已有模式的限制.再辅以该团队提出的给定主链设计序列的能量统计模型ABACUS[48],形成了一套全新的蛋白质自主设计新路线. ...
... (1PLDDT (predicted local distance difference test): 描述每个残基预测的局部置信度,局部结构指标,0-100,越大越好2PAE (predicted aligned error): 估计氨基酸在每一个位置上的误差,成对计算指标3Disfavoured: 反应预测序列类型于当前结构微环境的“适配性”4TMscore (template modeling score): 衡量蛋白质结构间的相似度,全局结构指标,0-1,越大越好5RMSD (root-mean-square deviation): 表示两个蛋白结构对齐后的结构差异,一般认为小于2Å预测准确.越小越好(图中展示的代表性方法分别按照1,2,3三部分从上至下摘自文献[43,45],[42-43],[86-87])) ...
... -43],[86-87])) ...
... David T. Jones尝试将AlphaFold2引入固定骨架设计序列的过程中,以约束生成的序列能够折叠成给定的骨架,并且正交实验中也验证了分子动力学方法模拟的结构对AlphaFold2监督后的实验结构高度支持[41].其具体流程是:(1)生成初始蛋白序列.基于研究者之前提出的基于自回归的Transformer蛋白质序列生成模型[88]生成1000条初始序列.同时对于得到的序列用AlphaFold2预测其结构,并与要设计的骨架结构用TM-align[89]做结构比对.最后选择结构比对得分最高的那部分结构的序列为初始序列,不具有高结构置信度的序列则用丙氨酸填充.这样做的好处是保证初始的序列是可收敛的,否则可能序列太随机导致最后没办法折叠.(2)在序列空间中执行贪婪的半随机游走,逐步突变起始序列进行迭代的端到端的设计.这里面AlphaFold2的作用有两个,一个是预测序列结构,比较与要设计结构的距离直方图损失.根据损失是否减小来判断突变序列是否合理.另一个是确定该序列中哪一部分残基位点要被突变,修改.举例来说,从起始序列出发并通过AlphaFold2预测其结构以及每一个残基的pLDDT打分(衡量每个残基的局部结构合理性).这里,计算预测结构中的距离直方图并与要设计的骨架结构的直方图计算损失.同时,利用每个残基的pLDDT打分设置为序列位点是否要被采样的概率.得分较高代表此处残基是稳定的,反之则是下一次迭代序列设计采样的点.在下次迭代采样中,对于选定的采样位点进行饱和突变,直到距离直方图损失减小,才接受序列的突变采样.这样设置的好处是对于与要设计结构的高度匹配的序列不再改变,大量减少采样时间尽快收敛以及可能引起的负协同效应.作者在人工设计的Top7上进行测试,得到的序列结构不论是通过AlphaFold2,trRosetta还是基于片段从头折叠的方法,均被证实与要设计的骨架可能是同一种折叠.该工作应用AlphaFold2在初始序列设计上保证了与目标结构的局部高结构匹配度,同时在序列设计过程中利用AlphaFold2预测的结构与目标结构的距离直方图损失约束其设计序列保持全局结构相似性以及利用残基位点可信度增强局部残基结构稳定性.同年,S. Kashif Sadiq也在bioRxiv上提交AlphaDesign工作,基本思路也是利用AlphaFold2预测的结构与要设计的骨架结构的差异来限制调整序列的优化,采用的优化函数是基于进化的遗传算法来迭代生成序列[42].主要差别在于该方法利用预测结构的三维坐标信息差异构建目标函数优化而不仅仅是二维的配对距离直方图约束,可能在结构约束上更加有效.而且该方法扩展了可能的设计任务的范围,设计了一些长度在32到256的结构稳定的从头设计的具有不同折叠的单体蛋白,同源二聚体,异源二聚体,同源低聚物(三聚体到六聚体).Baker组提出的trDesign是第一个提出将结构预测工具trRosetta应用到蛋白质序列设计中的工作,考虑的也是二维距离直方图的损失来更新梯度,更新被表示为PSSM的序列[43].但是受限于trRosetta利用的是二维的结构约束,在正交验证中发现基于这种反向传播的方式设计序列不能很好的对三维结构进行编码.且上述三个工作均是基于给定骨架设计序列,限制了实际设计酶的应用需求.后来Baker组提出的“幻想” (hallucination) 的方法,不从给定骨架结构出发设计序列,而是考虑在这种目标结构缺失的条件下,是否能随机产生结构和序列.其实现是通过最大化设计序列的结构与随机背景序列的差异约束,从而约束该序列折叠后的结构具有一个典型的2维结构特性[44].实验中幻想了2000条序列,聚类后发现均可以在已有的PDB结构库中寻找到相似的折叠.实验验证的时候有62条是可溶表达的(实验验证了129条),且CD的圆二色谱和目标结构的二级结构分布吻合.相比传统设计验证的方法,仅仅129条实验验证且48%的成功率,极大的减少了人工验证的成本和时间.但是由于trRosetta精度有限以及二维结构约束的不足,在接下来的工作中将RoseTTAFold嵌入到具有给定motif的序列设计中[45].RoseTTAFold显示利用SE-3 Transformer预测三维结构坐标以及二维距离分布,大大提高了序列设计的准确性.在免疫相关蛋白中,成功设计出携带中和性抗体表位的蛋白以及与新冠病毒S突刺蛋白受体结合的ACE2类似物蛋白.后续提出的RFjoint,不再通过神经网络不断迭代推理以及反向传播来设计序列,而是将结构预测和序列设计两大任务结合起来,直接训练全新的模型[45].这样的好处是减少了反向推理时间大大减少了设计的时间成本. ...
De novo protein design by deep network hallucination
2
2021
... Summary of protein design tools
Table 1| 方法名称/作者 | 类型 | 模型框架 | 输入 | 输出 | 训练集 | 应用 | 特点 | 网页/GitHub |
|---|
| SCUBA[32] | 骨架设计 | NC-NN | 二级结构motifs | 骨架 | PDB | 两层α/β蛋白;四螺旋束蛋白;EXTD | 突破之前方法仅限于已有模式的限制,基于核密度估计构造神经网络形式的能量函数 | https://doi.org/10.5281/zenodo.4533424 |
| Namrata Anand[33-34] | 骨架设计 | DCGAN | - | 距离图 | distance maps | 补齐完整的结构 | Cα原子之间的相对距离作为约束并优化 | - |
| Mire Zloh[35] | 序列生成 | LSTM | - | 序列 | CAMP+DBAASP+DRAMP+YADAMP | - | 设计对大肠杆菌具有潜在抗菌活性的短肽,并通过结构和表面性能与典型的AMP结构进行比较 | - |
| Gisbert Schneider[36] | 序列生成 | RNN | - | 序列 | ADAM/APD/DADP | 设计具有抗菌功能的肽 | 设计出的肽相比随机生成的肽具有抗菌活性的较高 | https://github.com/alexarnimueller/LSTM_peptides |
| ProteinGAN[37] | 序列生成 | GAN | - | 序列 | MDH 序列 | MDH酶 | 设计与苹果酸脱氢酶同样功能的酶,可同时出现100多个位点 | https://github.com/Biomatter-Designs/ProteinGAN |
| Mostafa Karimi[38] | 序列生成,给定折叠方式 | gcWGAN | - | 序列 | SCOPe v. 2.07 | - | 设计了一个从序列到折叠的预测器作为“oracle”,监督序列折叠成给定的折叠类型 | https://github.com/Shen-Lab/gcWGAN |
| ProteinMPNN[39] | 序列设计,结构约束 | 结构编码-序列解码的自回归模型 | 3D结构 | 序列 | CATH 4.2 | 单体、环状低聚物、蛋白质纳米颗粒 | 从结构中学习残基类型,将原子配对距离势融入到边的特征表示中,使序列恢复率直接提高约7.8% | https://github.com/dauparas/ProteinMPNN |
| ABACUS-R[40] | 序列设计,结构约束 | 结构编码-序列解码 | 3D结构 | 序列 | CATH 4.2 | PDB ID: 1r26, 1cy5 and 1ubq 3个骨架结构 | 从结构中学习残基类型,多任务学习 | https://github.com/liuyf020419/ABACUS-R |
| Transformer |
| David T. Jones[41] | 序列设计,结构约束 | 贪婪的半随机游走,逐步突变起始序列进行迭代的端到端设计 | 序列 | 序列 | - | Top7;Peak6; Foldit1; Ferredog-Diesel | 利用AlphaFold2预测生成序列的结构以及pLDDT打分,判断突变位点以及用距离图约束结构符合给定结构;对于最初始的序列,通过生成模型以及AlphaFold2结构约束产生初始序列 | |
| AlphaDesign[42] | 序列设计,结构约束 | 基于进化的遗传算法迭代生成序列 | 随机序列 | 序列 | - | 设计稳定的单体,二聚体直到六聚体 | 利用AlphaFold2预测的结构与要设计的骨架结构的差异来调整序列的优化 | - |
| trDesign[43] | 序列设计,结构约束 | trRosetta | 随机序列 | 序列 | - | - | 二维距离直方图的损失来更新梯度,更新被表示为PSSM的序列,可以理解为“折叠”的逆问题 | https://github.com/gjoni/trDesign |
| Hallucination[44] | 序列设计,结构约束,不固定骨架结构 | trRosetta | 随机序列 | 序列/结构 | PDB训练背景分布概率 | 设计2000条新的幻觉序列,聚类后129条表达后,62个蛋白可溶,高稳定 | 随机出发设计一条序列,通过最大化与随机背景序列的结构差异,约束该序列具有一个典型的2维结构特性 | https://github.com/gjoni/trDesign |
| Constrained hallucination2[45] | 序列设计,结构约束 | RoseTTAFold | 序列/结构 | 序列/结构 | RoseTTAFold训练集 | 免疫原;金属结合;新酶;特定结合的蛋白 | 设计具有给定motif的序列,通过神经网络不断迭代推理以及反向传播来设计序列 | https://github.com/RosettaCommons/RFDesign |
| RFjoint[45] | 序列设计,结构约束 | 训练RoseTTAFold | 序列/结构 | 序列/结构 | 微调,其中25%: PDB (2020-02-17);75%:AF2 预测结构 | 添加同时恢复序列和结构信息的损失,直接训练全新的模型 |
| PiFold[46] | 序列设计 | GNN | 3D结构 | 序列 | CATH | 序列恢复率:51.66%( CATH4.2),58.72%( TS50),60.42%( TS500) | 设计了新的残基特征器,PiGNN层学习多尺度(节点,边,全局)的残基相互作用信息 | https://github.com/A4Bio/PiFold |
| ProDESIGN-LE[47] | 序列设计 | Transformer+MLP | 3D结构 | 序列 | PDB40 | 设计CAT III酶新序列,3/5可表达且可溶;GFP | 通过Transformer学习当前残基在局部结构环境中的依赖性,使设计序列中的残基类型适配于当前的局部环境 | http://81.70.37.223/; https://github.com/bigict/ProDESIGN-LE |
1.2.1 主链结构设计主链结构设计,指的是在给定目标结构的拓扑下(例如:二级结构基本单元的组成以及顺序,相对位置等),设计出符合这种堆叠方式的蛋白质结构.这里介绍一个非常典型且由突破性的工作,SCUBA[32]. 该工作由中国科学技术大学刘海燕和陈泉团队提出,是一个具有高自主可设计性的主链设计算法,且并不依赖侧链类型.该算法在结构数据中基于核密度估计构造神经网络形式的能量函数来捕获高阶相关关系,可在不确定序列(即设计的能量函数不依赖于侧链,充分考虑柔性)的情况下,连续广泛搜索主链结构空间,突破之前方法仅限于已有模式的限制.再辅以该团队提出的给定主链设计序列的能量统计模型ABACUS[48],形成了一套全新的蛋白质自主设计新路线. ...
... David T. Jones尝试将AlphaFold2引入固定骨架设计序列的过程中,以约束生成的序列能够折叠成给定的骨架,并且正交实验中也验证了分子动力学方法模拟的结构对AlphaFold2监督后的实验结构高度支持[41].其具体流程是:(1)生成初始蛋白序列.基于研究者之前提出的基于自回归的Transformer蛋白质序列生成模型[88]生成1000条初始序列.同时对于得到的序列用AlphaFold2预测其结构,并与要设计的骨架结构用TM-align[89]做结构比对.最后选择结构比对得分最高的那部分结构的序列为初始序列,不具有高结构置信度的序列则用丙氨酸填充.这样做的好处是保证初始的序列是可收敛的,否则可能序列太随机导致最后没办法折叠.(2)在序列空间中执行贪婪的半随机游走,逐步突变起始序列进行迭代的端到端的设计.这里面AlphaFold2的作用有两个,一个是预测序列结构,比较与要设计结构的距离直方图损失.根据损失是否减小来判断突变序列是否合理.另一个是确定该序列中哪一部分残基位点要被突变,修改.举例来说,从起始序列出发并通过AlphaFold2预测其结构以及每一个残基的pLDDT打分(衡量每个残基的局部结构合理性).这里,计算预测结构中的距离直方图并与要设计的骨架结构的直方图计算损失.同时,利用每个残基的pLDDT打分设置为序列位点是否要被采样的概率.得分较高代表此处残基是稳定的,反之则是下一次迭代序列设计采样的点.在下次迭代采样中,对于选定的采样位点进行饱和突变,直到距离直方图损失减小,才接受序列的突变采样.这样设置的好处是对于与要设计结构的高度匹配的序列不再改变,大量减少采样时间尽快收敛以及可能引起的负协同效应.作者在人工设计的Top7上进行测试,得到的序列结构不论是通过AlphaFold2,trRosetta还是基于片段从头折叠的方法,均被证实与要设计的骨架可能是同一种折叠.该工作应用AlphaFold2在初始序列设计上保证了与目标结构的局部高结构匹配度,同时在序列设计过程中利用AlphaFold2预测的结构与目标结构的距离直方图损失约束其设计序列保持全局结构相似性以及利用残基位点可信度增强局部残基结构稳定性.同年,S. Kashif Sadiq也在bioRxiv上提交AlphaDesign工作,基本思路也是利用AlphaFold2预测的结构与要设计的骨架结构的差异来限制调整序列的优化,采用的优化函数是基于进化的遗传算法来迭代生成序列[42].主要差别在于该方法利用预测结构的三维坐标信息差异构建目标函数优化而不仅仅是二维的配对距离直方图约束,可能在结构约束上更加有效.而且该方法扩展了可能的设计任务的范围,设计了一些长度在32到256的结构稳定的从头设计的具有不同折叠的单体蛋白,同源二聚体,异源二聚体,同源低聚物(三聚体到六聚体).Baker组提出的trDesign是第一个提出将结构预测工具trRosetta应用到蛋白质序列设计中的工作,考虑的也是二维距离直方图的损失来更新梯度,更新被表示为PSSM的序列[43].但是受限于trRosetta利用的是二维的结构约束,在正交验证中发现基于这种反向传播的方式设计序列不能很好的对三维结构进行编码.且上述三个工作均是基于给定骨架设计序列,限制了实际设计酶的应用需求.后来Baker组提出的“幻想” (hallucination) 的方法,不从给定骨架结构出发设计序列,而是考虑在这种目标结构缺失的条件下,是否能随机产生结构和序列.其实现是通过最大化设计序列的结构与随机背景序列的差异约束,从而约束该序列折叠后的结构具有一个典型的2维结构特性[44].实验中幻想了2000条序列,聚类后发现均可以在已有的PDB结构库中寻找到相似的折叠.实验验证的时候有62条是可溶表达的(实验验证了129条),且CD的圆二色谱和目标结构的二级结构分布吻合.相比传统设计验证的方法,仅仅129条实验验证且48%的成功率,极大的减少了人工验证的成本和时间.但是由于trRosetta精度有限以及二维结构约束的不足,在接下来的工作中将RoseTTAFold嵌入到具有给定motif的序列设计中[45].RoseTTAFold显示利用SE-3 Transformer预测三维结构坐标以及二维距离分布,大大提高了序列设计的准确性.在免疫相关蛋白中,成功设计出携带中和性抗体表位的蛋白以及与新冠病毒S突刺蛋白受体结合的ACE2类似物蛋白.后续提出的RFjoint,不再通过神经网络不断迭代推理以及反向传播来设计序列,而是将结构预测和序列设计两大任务结合起来,直接训练全新的模型[45].这样的好处是减少了反向推理时间大大减少了设计的时间成本. ...
Scaffolding protein functional sites using deep learning
7
2022
... Summary of protein design tools
Table 1| 方法名称/作者 | 类型 | 模型框架 | 输入 | 输出 | 训练集 | 应用 | 特点 | 网页/GitHub |
|---|
| SCUBA[32] | 骨架设计 | NC-NN | 二级结构motifs | 骨架 | PDB | 两层α/β蛋白;四螺旋束蛋白;EXTD | 突破之前方法仅限于已有模式的限制,基于核密度估计构造神经网络形式的能量函数 | https://doi.org/10.5281/zenodo.4533424 |
| Namrata Anand[33-34] | 骨架设计 | DCGAN | - | 距离图 | distance maps | 补齐完整的结构 | Cα原子之间的相对距离作为约束并优化 | - |
| Mire Zloh[35] | 序列生成 | LSTM | - | 序列 | CAMP+DBAASP+DRAMP+YADAMP | - | 设计对大肠杆菌具有潜在抗菌活性的短肽,并通过结构和表面性能与典型的AMP结构进行比较 | - |
| Gisbert Schneider[36] | 序列生成 | RNN | - | 序列 | ADAM/APD/DADP | 设计具有抗菌功能的肽 | 设计出的肽相比随机生成的肽具有抗菌活性的较高 | https://github.com/alexarnimueller/LSTM_peptides |
| ProteinGAN[37] | 序列生成 | GAN | - | 序列 | MDH 序列 | MDH酶 | 设计与苹果酸脱氢酶同样功能的酶,可同时出现100多个位点 | https://github.com/Biomatter-Designs/ProteinGAN |
| Mostafa Karimi[38] | 序列生成,给定折叠方式 | gcWGAN | - | 序列 | SCOPe v. 2.07 | - | 设计了一个从序列到折叠的预测器作为“oracle”,监督序列折叠成给定的折叠类型 | https://github.com/Shen-Lab/gcWGAN |
| ProteinMPNN[39] | 序列设计,结构约束 | 结构编码-序列解码的自回归模型 | 3D结构 | 序列 | CATH 4.2 | 单体、环状低聚物、蛋白质纳米颗粒 | 从结构中学习残基类型,将原子配对距离势融入到边的特征表示中,使序列恢复率直接提高约7.8% | https://github.com/dauparas/ProteinMPNN |
| ABACUS-R[40] | 序列设计,结构约束 | 结构编码-序列解码 | 3D结构 | 序列 | CATH 4.2 | PDB ID: 1r26, 1cy5 and 1ubq 3个骨架结构 | 从结构中学习残基类型,多任务学习 | https://github.com/liuyf020419/ABACUS-R |
| Transformer |
| David T. Jones[41] | 序列设计,结构约束 | 贪婪的半随机游走,逐步突变起始序列进行迭代的端到端设计 | 序列 | 序列 | - | Top7;Peak6; Foldit1; Ferredog-Diesel | 利用AlphaFold2预测生成序列的结构以及pLDDT打分,判断突变位点以及用距离图约束结构符合给定结构;对于最初始的序列,通过生成模型以及AlphaFold2结构约束产生初始序列 | |
| AlphaDesign[42] | 序列设计,结构约束 | 基于进化的遗传算法迭代生成序列 | 随机序列 | 序列 | - | 设计稳定的单体,二聚体直到六聚体 | 利用AlphaFold2预测的结构与要设计的骨架结构的差异来调整序列的优化 | - |
| trDesign[43] | 序列设计,结构约束 | trRosetta | 随机序列 | 序列 | - | - | 二维距离直方图的损失来更新梯度,更新被表示为PSSM的序列,可以理解为“折叠”的逆问题 | https://github.com/gjoni/trDesign |
| Hallucination[44] | 序列设计,结构约束,不固定骨架结构 | trRosetta | 随机序列 | 序列/结构 | PDB训练背景分布概率 | 设计2000条新的幻觉序列,聚类后129条表达后,62个蛋白可溶,高稳定 | 随机出发设计一条序列,通过最大化与随机背景序列的结构差异,约束该序列具有一个典型的2维结构特性 | https://github.com/gjoni/trDesign |
| Constrained hallucination2[45] | 序列设计,结构约束 | RoseTTAFold | 序列/结构 | 序列/结构 | RoseTTAFold训练集 | 免疫原;金属结合;新酶;特定结合的蛋白 | 设计具有给定motif的序列,通过神经网络不断迭代推理以及反向传播来设计序列 | https://github.com/RosettaCommons/RFDesign |
| RFjoint[45] | 序列设计,结构约束 | 训练RoseTTAFold | 序列/结构 | 序列/结构 | 微调,其中25%: PDB (2020-02-17);75%:AF2 预测结构 | 添加同时恢复序列和结构信息的损失,直接训练全新的模型 |
| PiFold[46] | 序列设计 | GNN | 3D结构 | 序列 | CATH | 序列恢复率:51.66%( CATH4.2),58.72%( TS50),60.42%( TS500) | 设计了新的残基特征器,PiGNN层学习多尺度(节点,边,全局)的残基相互作用信息 | https://github.com/A4Bio/PiFold |
| ProDESIGN-LE[47] | 序列设计 | Transformer+MLP | 3D结构 | 序列 | PDB40 | 设计CAT III酶新序列,3/5可表达且可溶;GFP | 通过Transformer学习当前残基在局部结构环境中的依赖性,使设计序列中的残基类型适配于当前的局部环境 | http://81.70.37.223/; https://github.com/bigict/ProDESIGN-LE |
1.2.1 主链结构设计主链结构设计,指的是在给定目标结构的拓扑下(例如:二级结构基本单元的组成以及顺序,相对位置等),设计出符合这种堆叠方式的蛋白质结构.这里介绍一个非常典型且由突破性的工作,SCUBA[32]. 该工作由中国科学技术大学刘海燕和陈泉团队提出,是一个具有高自主可设计性的主链设计算法,且并不依赖侧链类型.该算法在结构数据中基于核密度估计构造神经网络形式的能量函数来捕获高阶相关关系,可在不确定序列(即设计的能量函数不依赖于侧链,充分考虑柔性)的情况下,连续广泛搜索主链结构空间,突破之前方法仅限于已有模式的限制.再辅以该团队提出的给定主链设计序列的能量统计模型ABACUS[48],形成了一套全新的蛋白质自主设计新路线. ...
... [
45]
序列设计,结构约束 | 训练RoseTTAFold | 序列/结构 | 序列/结构 | 微调,其中25%: PDB (2020-02-17);75%:AF2 预测结构 | 添加同时恢复序列和结构信息的损失,直接训练全新的模型 | | PiFold[46] | 序列设计 | GNN | 3D结构 | 序列 | CATH | 序列恢复率:51.66%( CATH4.2),58.72%( TS50),60.42%( TS500) | 设计了新的残基特征器,PiGNN层学习多尺度(节点,边,全局)的残基相互作用信息 | https://github.com/A4Bio/PiFold |
| ProDESIGN-LE[47] | 序列设计 | Transformer+MLP | 3D结构 | 序列 | PDB40 | 设计CAT III酶新序列,3/5可表达且可溶;GFP | 通过Transformer学习当前残基在局部结构环境中的依赖性,使设计序列中的残基类型适配于当前的局部环境 | http://81.70.37.223/; https://github.com/bigict/ProDESIGN-LE |
1.2.1 主链结构设计主链结构设计,指的是在给定目标结构的拓扑下(例如:二级结构基本单元的组成以及顺序,相对位置等),设计出符合这种堆叠方式的蛋白质结构.这里介绍一个非常典型且由突破性的工作,SCUBA[32]. 该工作由中国科学技术大学刘海燕和陈泉团队提出,是一个具有高自主可设计性的主链设计算法,且并不依赖侧链类型.该算法在结构数据中基于核密度估计构造神经网络形式的能量函数来捕获高阶相关关系,可在不确定序列(即设计的能量函数不依赖于侧链,充分考虑柔性)的情况下,连续广泛搜索主链结构空间,突破之前方法仅限于已有模式的限制.再辅以该团队提出的给定主链设计序列的能量统计模型ABACUS[48],形成了一套全新的蛋白质自主设计新路线. ...
... 考虑酶的“可折叠性”,最直观的解决办法是快速预测设计的新酶的结构,检验其是否具有给定结构.因此,第一种预测是将蛋白结构预测工具作为一个监督者,约束生成的序列具有折叠成给定结构的能力(如图1左上角)[41-45,85].这个思路实施起来的最大难点是从序列预测结构的精度限制.但是现在得益于结构预测的突破性进展,使的这种设计新酶成为可能.其基本思路是在设计序列的时候,加入一个辅助的“监督者”对于生成的序列是否可以折叠且具有给定的构象进行评分.根据得分对蛋白质序列通过基于梯度的,梯度自由的,或者神经网络构造的优化方法来更新序列.通过不断重复迭代这一过程,最终得到构象约束下的收敛序列.设计序列的时候一般遵从最小能量的原则.但是,我们不清楚给定的构象就一定是设计的这条序列折叠后的最低能量构象.因此结构预测做为“监督器”实际上去计算了在给定结构情况下蛋白质序列的最大联合概率. ...
... (1PLDDT (predicted local distance difference test): 描述每个残基预测的局部置信度,局部结构指标,0-100,越大越好2PAE (predicted aligned error): 估计氨基酸在每一个位置上的误差,成对计算指标3Disfavoured: 反应预测序列类型于当前结构微环境的“适配性”4TMscore (template modeling score): 衡量蛋白质结构间的相似度,全局结构指标,0-1,越大越好5RMSD (root-mean-square deviation): 表示两个蛋白结构对齐后的结构差异,一般认为小于2Å预测准确.越小越好(图中展示的代表性方法分别按照1,2,3三部分从上至下摘自文献[43,45],[42-43],[86-87])) ...
... David T. Jones尝试将AlphaFold2引入固定骨架设计序列的过程中,以约束生成的序列能够折叠成给定的骨架,并且正交实验中也验证了分子动力学方法模拟的结构对AlphaFold2监督后的实验结构高度支持[41].其具体流程是:(1)生成初始蛋白序列.基于研究者之前提出的基于自回归的Transformer蛋白质序列生成模型[88]生成1000条初始序列.同时对于得到的序列用AlphaFold2预测其结构,并与要设计的骨架结构用TM-align[89]做结构比对.最后选择结构比对得分最高的那部分结构的序列为初始序列,不具有高结构置信度的序列则用丙氨酸填充.这样做的好处是保证初始的序列是可收敛的,否则可能序列太随机导致最后没办法折叠.(2)在序列空间中执行贪婪的半随机游走,逐步突变起始序列进行迭代的端到端的设计.这里面AlphaFold2的作用有两个,一个是预测序列结构,比较与要设计结构的距离直方图损失.根据损失是否减小来判断突变序列是否合理.另一个是确定该序列中哪一部分残基位点要被突变,修改.举例来说,从起始序列出发并通过AlphaFold2预测其结构以及每一个残基的pLDDT打分(衡量每个残基的局部结构合理性).这里,计算预测结构中的距离直方图并与要设计的骨架结构的直方图计算损失.同时,利用每个残基的pLDDT打分设置为序列位点是否要被采样的概率.得分较高代表此处残基是稳定的,反之则是下一次迭代序列设计采样的点.在下次迭代采样中,对于选定的采样位点进行饱和突变,直到距离直方图损失减小,才接受序列的突变采样.这样设置的好处是对于与要设计结构的高度匹配的序列不再改变,大量减少采样时间尽快收敛以及可能引起的负协同效应.作者在人工设计的Top7上进行测试,得到的序列结构不论是通过AlphaFold2,trRosetta还是基于片段从头折叠的方法,均被证实与要设计的骨架可能是同一种折叠.该工作应用AlphaFold2在初始序列设计上保证了与目标结构的局部高结构匹配度,同时在序列设计过程中利用AlphaFold2预测的结构与目标结构的距离直方图损失约束其设计序列保持全局结构相似性以及利用残基位点可信度增强局部残基结构稳定性.同年,S. Kashif Sadiq也在bioRxiv上提交AlphaDesign工作,基本思路也是利用AlphaFold2预测的结构与要设计的骨架结构的差异来限制调整序列的优化,采用的优化函数是基于进化的遗传算法来迭代生成序列[42].主要差别在于该方法利用预测结构的三维坐标信息差异构建目标函数优化而不仅仅是二维的配对距离直方图约束,可能在结构约束上更加有效.而且该方法扩展了可能的设计任务的范围,设计了一些长度在32到256的结构稳定的从头设计的具有不同折叠的单体蛋白,同源二聚体,异源二聚体,同源低聚物(三聚体到六聚体).Baker组提出的trDesign是第一个提出将结构预测工具trRosetta应用到蛋白质序列设计中的工作,考虑的也是二维距离直方图的损失来更新梯度,更新被表示为PSSM的序列[43].但是受限于trRosetta利用的是二维的结构约束,在正交验证中发现基于这种反向传播的方式设计序列不能很好的对三维结构进行编码.且上述三个工作均是基于给定骨架设计序列,限制了实际设计酶的应用需求.后来Baker组提出的“幻想” (hallucination) 的方法,不从给定骨架结构出发设计序列,而是考虑在这种目标结构缺失的条件下,是否能随机产生结构和序列.其实现是通过最大化设计序列的结构与随机背景序列的差异约束,从而约束该序列折叠后的结构具有一个典型的2维结构特性[44].实验中幻想了2000条序列,聚类后发现均可以在已有的PDB结构库中寻找到相似的折叠.实验验证的时候有62条是可溶表达的(实验验证了129条),且CD的圆二色谱和目标结构的二级结构分布吻合.相比传统设计验证的方法,仅仅129条实验验证且48%的成功率,极大的减少了人工验证的成本和时间.但是由于trRosetta精度有限以及二维结构约束的不足,在接下来的工作中将RoseTTAFold嵌入到具有给定motif的序列设计中[45].RoseTTAFold显示利用SE-3 Transformer预测三维结构坐标以及二维距离分布,大大提高了序列设计的准确性.在免疫相关蛋白中,成功设计出携带中和性抗体表位的蛋白以及与新冠病毒S突刺蛋白受体结合的ACE2类似物蛋白.后续提出的RFjoint,不再通过神经网络不断迭代推理以及反向传播来设计序列,而是将结构预测和序列设计两大任务结合起来,直接训练全新的模型[45].这样的好处是减少了反向推理时间大大减少了设计的时间成本. ...
... [45].这样的好处是减少了反向推理时间大大减少了设计的时间成本. ...
... 在介绍应用的时候,我们提出三种应用方式.这三种应用的前提均是认为AlphaFold2这类蛋白质结构预测工具学习到了蛋白质序列到结构的复杂关系,对蛋白质结构的全局以及局部结构预测的准确度是可信的.随着越来越多结构预测工具的开发,根据不同任务(无同源序列),数据(α螺旋结构比例较高)等,可以将AlphaFold2替换成其他的结构预测工具.例如上面提到的David Baker组提出的RFjoint[45]采用的就是他们组的另一个非常优秀的工具RoseTTAFold[84]. ...
PiFold: Toward effective and efficient protein inverse folding
1
2022
... Summary of protein design tools
Table 1| 方法名称/作者 | 类型 | 模型框架 | 输入 | 输出 | 训练集 | 应用 | 特点 | 网页/GitHub |
|---|
| SCUBA[32] | 骨架设计 | NC-NN | 二级结构motifs | 骨架 | PDB | 两层α/β蛋白;四螺旋束蛋白;EXTD | 突破之前方法仅限于已有模式的限制,基于核密度估计构造神经网络形式的能量函数 | https://doi.org/10.5281/zenodo.4533424 |
| Namrata Anand[33-34] | 骨架设计 | DCGAN | - | 距离图 | distance maps | 补齐完整的结构 | Cα原子之间的相对距离作为约束并优化 | - |
| Mire Zloh[35] | 序列生成 | LSTM | - | 序列 | CAMP+DBAASP+DRAMP+YADAMP | - | 设计对大肠杆菌具有潜在抗菌活性的短肽,并通过结构和表面性能与典型的AMP结构进行比较 | - |
| Gisbert Schneider[36] | 序列生成 | RNN | - | 序列 | ADAM/APD/DADP | 设计具有抗菌功能的肽 | 设计出的肽相比随机生成的肽具有抗菌活性的较高 | https://github.com/alexarnimueller/LSTM_peptides |
| ProteinGAN[37] | 序列生成 | GAN | - | 序列 | MDH 序列 | MDH酶 | 设计与苹果酸脱氢酶同样功能的酶,可同时出现100多个位点 | https://github.com/Biomatter-Designs/ProteinGAN |
| Mostafa Karimi[38] | 序列生成,给定折叠方式 | gcWGAN | - | 序列 | SCOPe v. 2.07 | - | 设计了一个从序列到折叠的预测器作为“oracle”,监督序列折叠成给定的折叠类型 | https://github.com/Shen-Lab/gcWGAN |
| ProteinMPNN[39] | 序列设计,结构约束 | 结构编码-序列解码的自回归模型 | 3D结构 | 序列 | CATH 4.2 | 单体、环状低聚物、蛋白质纳米颗粒 | 从结构中学习残基类型,将原子配对距离势融入到边的特征表示中,使序列恢复率直接提高约7.8% | https://github.com/dauparas/ProteinMPNN |
| ABACUS-R[40] | 序列设计,结构约束 | 结构编码-序列解码 | 3D结构 | 序列 | CATH 4.2 | PDB ID: 1r26, 1cy5 and 1ubq 3个骨架结构 | 从结构中学习残基类型,多任务学习 | https://github.com/liuyf020419/ABACUS-R |
| Transformer |
| David T. Jones[41] | 序列设计,结构约束 | 贪婪的半随机游走,逐步突变起始序列进行迭代的端到端设计 | 序列 | 序列 | - | Top7;Peak6; Foldit1; Ferredog-Diesel | 利用AlphaFold2预测生成序列的结构以及pLDDT打分,判断突变位点以及用距离图约束结构符合给定结构;对于最初始的序列,通过生成模型以及AlphaFold2结构约束产生初始序列 | |
| AlphaDesign[42] | 序列设计,结构约束 | 基于进化的遗传算法迭代生成序列 | 随机序列 | 序列 | - | 设计稳定的单体,二聚体直到六聚体 | 利用AlphaFold2预测的结构与要设计的骨架结构的差异来调整序列的优化 | - |
| trDesign[43] | 序列设计,结构约束 | trRosetta | 随机序列 | 序列 | - | - | 二维距离直方图的损失来更新梯度,更新被表示为PSSM的序列,可以理解为“折叠”的逆问题 | https://github.com/gjoni/trDesign |
| Hallucination[44] | 序列设计,结构约束,不固定骨架结构 | trRosetta | 随机序列 | 序列/结构 | PDB训练背景分布概率 | 设计2000条新的幻觉序列,聚类后129条表达后,62个蛋白可溶,高稳定 | 随机出发设计一条序列,通过最大化与随机背景序列的结构差异,约束该序列具有一个典型的2维结构特性 | https://github.com/gjoni/trDesign |
| Constrained hallucination2[45] | 序列设计,结构约束 | RoseTTAFold | 序列/结构 | 序列/结构 | RoseTTAFold训练集 | 免疫原;金属结合;新酶;特定结合的蛋白 | 设计具有给定motif的序列,通过神经网络不断迭代推理以及反向传播来设计序列 | https://github.com/RosettaCommons/RFDesign |
| RFjoint[45] | 序列设计,结构约束 | 训练RoseTTAFold | 序列/结构 | 序列/结构 | 微调,其中25%: PDB (2020-02-17);75%:AF2 预测结构 | 添加同时恢复序列和结构信息的损失,直接训练全新的模型 |
| PiFold[46] | 序列设计 | GNN | 3D结构 | 序列 | CATH | 序列恢复率:51.66%( CATH4.2),58.72%( TS50),60.42%( TS500) | 设计了新的残基特征器,PiGNN层学习多尺度(节点,边,全局)的残基相互作用信息 | https://github.com/A4Bio/PiFold |
| ProDESIGN-LE[47] | 序列设计 | Transformer+MLP | 3D结构 | 序列 | PDB40 | 设计CAT III酶新序列,3/5可表达且可溶;GFP | 通过Transformer学习当前残基在局部结构环境中的依赖性,使设计序列中的残基类型适配于当前的局部环境 | http://81.70.37.223/; https://github.com/bigict/ProDESIGN-LE |
1.2.1 主链结构设计主链结构设计,指的是在给定目标结构的拓扑下(例如:二级结构基本单元的组成以及顺序,相对位置等),设计出符合这种堆叠方式的蛋白质结构.这里介绍一个非常典型且由突破性的工作,SCUBA[32]. 该工作由中国科学技术大学刘海燕和陈泉团队提出,是一个具有高自主可设计性的主链设计算法,且并不依赖侧链类型.该算法在结构数据中基于核密度估计构造神经网络形式的能量函数来捕获高阶相关关系,可在不确定序列(即设计的能量函数不依赖于侧链,充分考虑柔性)的情况下,连续广泛搜索主链结构空间,突破之前方法仅限于已有模式的限制.再辅以该团队提出的给定主链设计序列的能量统计模型ABACUS[48],形成了一套全新的蛋白质自主设计新路线. ...
Accurate and efficient protein sequence design through learning concise local environment of residues
3
2023
... Summary of protein design tools
Table 1| 方法名称/作者 | 类型 | 模型框架 | 输入 | 输出 | 训练集 | 应用 | 特点 | 网页/GitHub |
|---|
| SCUBA[32] | 骨架设计 | NC-NN | 二级结构motifs | 骨架 | PDB | 两层α/β蛋白;四螺旋束蛋白;EXTD | 突破之前方法仅限于已有模式的限制,基于核密度估计构造神经网络形式的能量函数 | https://doi.org/10.5281/zenodo.4533424 |
| Namrata Anand[33-34] | 骨架设计 | DCGAN | - | 距离图 | distance maps | 补齐完整的结构 | Cα原子之间的相对距离作为约束并优化 | - |
| Mire Zloh[35] | 序列生成 | LSTM | - | 序列 | CAMP+DBAASP+DRAMP+YADAMP | - | 设计对大肠杆菌具有潜在抗菌活性的短肽,并通过结构和表面性能与典型的AMP结构进行比较 | - |
| Gisbert Schneider[36] | 序列生成 | RNN | - | 序列 | ADAM/APD/DADP | 设计具有抗菌功能的肽 | 设计出的肽相比随机生成的肽具有抗菌活性的较高 | https://github.com/alexarnimueller/LSTM_peptides |
| ProteinGAN[37] | 序列生成 | GAN | - | 序列 | MDH 序列 | MDH酶 | 设计与苹果酸脱氢酶同样功能的酶,可同时出现100多个位点 | https://github.com/Biomatter-Designs/ProteinGAN |
| Mostafa Karimi[38] | 序列生成,给定折叠方式 | gcWGAN | - | 序列 | SCOPe v. 2.07 | - | 设计了一个从序列到折叠的预测器作为“oracle”,监督序列折叠成给定的折叠类型 | https://github.com/Shen-Lab/gcWGAN |
| ProteinMPNN[39] | 序列设计,结构约束 | 结构编码-序列解码的自回归模型 | 3D结构 | 序列 | CATH 4.2 | 单体、环状低聚物、蛋白质纳米颗粒 | 从结构中学习残基类型,将原子配对距离势融入到边的特征表示中,使序列恢复率直接提高约7.8% | https://github.com/dauparas/ProteinMPNN |
| ABACUS-R[40] | 序列设计,结构约束 | 结构编码-序列解码 | 3D结构 | 序列 | CATH 4.2 | PDB ID: 1r26, 1cy5 and 1ubq 3个骨架结构 | 从结构中学习残基类型,多任务学习 | https://github.com/liuyf020419/ABACUS-R |
| Transformer |
| David T. Jones[41] | 序列设计,结构约束 | 贪婪的半随机游走,逐步突变起始序列进行迭代的端到端设计 | 序列 | 序列 | - | Top7;Peak6; Foldit1; Ferredog-Diesel | 利用AlphaFold2预测生成序列的结构以及pLDDT打分,判断突变位点以及用距离图约束结构符合给定结构;对于最初始的序列,通过生成模型以及AlphaFold2结构约束产生初始序列 | |
| AlphaDesign[42] | 序列设计,结构约束 | 基于进化的遗传算法迭代生成序列 | 随机序列 | 序列 | - | 设计稳定的单体,二聚体直到六聚体 | 利用AlphaFold2预测的结构与要设计的骨架结构的差异来调整序列的优化 | - |
| trDesign[43] | 序列设计,结构约束 | trRosetta | 随机序列 | 序列 | - | - | 二维距离直方图的损失来更新梯度,更新被表示为PSSM的序列,可以理解为“折叠”的逆问题 | https://github.com/gjoni/trDesign |
| Hallucination[44] | 序列设计,结构约束,不固定骨架结构 | trRosetta | 随机序列 | 序列/结构 | PDB训练背景分布概率 | 设计2000条新的幻觉序列,聚类后129条表达后,62个蛋白可溶,高稳定 | 随机出发设计一条序列,通过最大化与随机背景序列的结构差异,约束该序列具有一个典型的2维结构特性 | https://github.com/gjoni/trDesign |
| Constrained hallucination2[45] | 序列设计,结构约束 | RoseTTAFold | 序列/结构 | 序列/结构 | RoseTTAFold训练集 | 免疫原;金属结合;新酶;特定结合的蛋白 | 设计具有给定motif的序列,通过神经网络不断迭代推理以及反向传播来设计序列 | https://github.com/RosettaCommons/RFDesign |
| RFjoint[45] | 序列设计,结构约束 | 训练RoseTTAFold | 序列/结构 | 序列/结构 | 微调,其中25%: PDB (2020-02-17);75%:AF2 预测结构 | 添加同时恢复序列和结构信息的损失,直接训练全新的模型 |
| PiFold[46] | 序列设计 | GNN | 3D结构 | 序列 | CATH | 序列恢复率:51.66%( CATH4.2),58.72%( TS50),60.42%( TS500) | 设计了新的残基特征器,PiGNN层学习多尺度(节点,边,全局)的残基相互作用信息 | https://github.com/A4Bio/PiFold |
| ProDESIGN-LE[47] | 序列设计 | Transformer+MLP | 3D结构 | 序列 | PDB40 | 设计CAT III酶新序列,3/5可表达且可溶;GFP | 通过Transformer学习当前残基在局部结构环境中的依赖性,使设计序列中的残基类型适配于当前的局部环境 | http://81.70.37.223/; https://github.com/bigict/ProDESIGN-LE |
1.2.1 主链结构设计主链结构设计,指的是在给定目标结构的拓扑下(例如:二级结构基本单元的组成以及顺序,相对位置等),设计出符合这种堆叠方式的蛋白质结构.这里介绍一个非常典型且由突破性的工作,SCUBA[32]. 该工作由中国科学技术大学刘海燕和陈泉团队提出,是一个具有高自主可设计性的主链设计算法,且并不依赖侧链类型.该算法在结构数据中基于核密度估计构造神经网络形式的能量函数来捕获高阶相关关系,可在不确定序列(即设计的能量函数不依赖于侧链,充分考虑柔性)的情况下,连续广泛搜索主链结构空间,突破之前方法仅限于已有模式的限制.再辅以该团队提出的给定主链设计序列的能量统计模型ABACUS[48],形成了一套全新的蛋白质自主设计新路线. ...
... 同样,可以采用结构约束来指导进而设计氨基酸序列.这种情况下,设计的氨基酸序列能否折叠成目标的蛋白质结构是至关重要的指标.最近被称为新一代Rosetta蛋白设计内核的Rosetta MPNN “Mover”,突破了传统的Rosetta设计范式“Inside-out”模式.该方法ProteinMPNN由David Baker组提出,基于structured-transformer[54],采用了结构编码-序列解码的自回归模型框架,将原子配对距离势融入到边的特征表示中,使序列恢复率提高约7.8%[39].ProteinMPNN对根据幻想的主链进行蛋白设计(500条),其中96条蛋白质序列在大肠杆菌体系中可以被大量的可溶表达,且成功结晶一个与设计结构高度一致的设计蛋白.同时,ProteinMPNN对单体,同源二聚体,异二聚体结构进行设计,其序列恢复率均在50%以上,其中核心区域的恢复率高达90%-95%.中国科学技术大学刘海燕和陈泉团队提出的ABACUS-R完全基于深度学习算法实现给定骨架设计氨基酸序列,不再依赖于传统能量项构建,并且序列恢复率高于ABACUS计算的,在测试集上基本可以达到50%[40].其主要思路是在给定骨架的情况下,通过编码-解码 (encoder-decoder) 框架学习在给定残基的结构特征以及周边结构环境的特性预测该残基的序列类型(侧链).值得一提的是,ABACUS-R采用多任务学习,不仅仅学习该残基的类型,还同时预测其二级结构,溶剂可达面积,B-factor以及一些结构构象扭转角任务.这些辅助任务的设计不仅提高了模型设计序列的能力,还隐式的在序列设计中加入了实时的结构约束.实验验证设计了3个天然骨架的蛋白序列设计并做了相应的实验验证.最后通过ABACUS-R设计出了可以成功表达且折叠成相应的三维结构的蛋白质序列,充分证明蛋白质设计可以绕过侧链模型的显示建模的可行性.卜东波课题组提出ProDESIGN-LE也是基于Transformer框架,通过计算序列类型是否符合给定的局部结构环境来设计蛋白序列[47].在实验中为CAT III酶设计的5条序列中,有3条可以成功表达且可溶.许锦波课题组提出的一种基于骨架设计蛋白序列的方法,基于生成SE (3) 等变模型,显著改进了现有的自回归方法[55].Mostafa Karimi组[38]提出gcWGAN探索生成给定折叠条件下的序列,使序列折叠成给定的方式.要是构造了一个基于DeepSF[56]的快速从序列预测折叠模式的模型并实时反馈监督序列是否可以正确折叠,这个模型被称为“Oracle”.Po-Ssu Huang组的Namrata Anand[57]直接从蛋白质骨架结构信息中学习侧链氨基酸类型,从而学习到一个基于自回归的自动的神经网络能量来指导后续的序列设计.在实际的TIM-barrel设计中,设计出的序列中有两个成功结晶且与设计的骨架高度一致. ...
... 总的来说,对于酶的智能设计,人工智能方法的设计相比传统基于力场的模式带来更高的成功率且更加快速(ProDesign[47]仅需30秒即可设计一条少于100长的蛋白序列).根据不同任务需求,可以实现酶的全新骨架设计和酶序列的从头设计.同时将二者结合起来可以形成一套按需从头设计酶的流程.酶设计中直接从给定结构建模设计序列的方法(类似于MPNN),本质上是为了寻求一条序列使结构能量最低.但是给定一条序列,其所能折叠成的状态有很多.目标结构不一定是设计的序列所能折叠成的最低的能量结构.因此现今从头酶设计中最关键的是后续对新酶的折叠能力评估.设计的新酶序列在后续的实验中评估能否折叠或者折叠成给定的目标构象,这是在实际应用中最关注的问题.因此,在设计酶的过程中,利用“可折叠性”作为指标过滤设计序列,有助于设计更高质量的酶,减少了实验室的后续验证酶序列从而降低成本. ...
Protein design with a comprehensive statistical energy function and boosted by experimental selection for foldability
1
2014
... 主链结构设计,指的是在给定目标结构的拓扑下(例如:二级结构基本单元的组成以及顺序,相对位置等),设计出符合这种堆叠方式的蛋白质结构.这里介绍一个非常典型且由突破性的工作,SCUBA[32]. 该工作由中国科学技术大学刘海燕和陈泉团队提出,是一个具有高自主可设计性的主链设计算法,且并不依赖侧链类型.该算法在结构数据中基于核密度估计构造神经网络形式的能量函数来捕获高阶相关关系,可在不确定序列(即设计的能量函数不依赖于侧链,充分考虑柔性)的情况下,连续广泛搜索主链结构空间,突破之前方法仅限于已有模式的限制.再辅以该团队提出的给定主链设计序列的能量统计模型ABACUS[48],形成了一套全新的蛋白质自主设计新路线. ...
Generative adversarial networks
1
2020
... 此外,Namrata Anand陆续提出基于生成对抗网络 (Generative Adversarial Network, GAN)[49] 实现蛋白质骨架设计的工作,从生成模型的角度考虑蛋白的骨架设计.第一个工作发表在2018年的NeurIPS,利用DCGAN (Deep Convolutional GANs)[50] 模型生成Cα原子之间的相对距离图(考虑到平移旋转不变性),将该配对距离约束引入到折叠成给定结构的可微问题中,并采用交替方向乘子法 (Alternating Direction Method of Multipliers, ADMM) 优化该凸规划问题[33].紧接着2019年发表的另一个工作也采用GAN实现给定距离约束下骨架设计,只是后面的精细化调整有所不同[34]. ...
Unsupervised representation learning with deep convolutional generative adversarial networks
1
2015
... 此外,Namrata Anand陆续提出基于生成对抗网络 (Generative Adversarial Network, GAN)[49] 实现蛋白质骨架设计的工作,从生成模型的角度考虑蛋白的骨架设计.第一个工作发表在2018年的NeurIPS,利用DCGAN (Deep Convolutional GANs)[50] 模型生成Cα原子之间的相对距离图(考虑到平移旋转不变性),将该配对距离约束引入到折叠成给定结构的可微问题中,并采用交替方向乘子法 (Alternating Direction Method of Multipliers, ADMM) 优化该凸规划问题[33].紧接着2019年发表的另一个工作也采用GAN实现给定距离约束下骨架设计,只是后面的精细化调整有所不同[34]. ...
Long short-term memory
1
1997
... 当从功能上约束设计的序列时,可以采用序列生成方法,在具有给定功能的酶序列数据上挖掘残基间的模式直接生成新酶的序列.常用的生成模型有长短期记忆网络 (Long Short-Term Memory, LSTM) [51],GAN,变分自动编码器 (Variational Autoencoder, VAE)[52],Transformer[53]等.Mire Zloh课题组构建了基于LSTM的生成模型和双向LSTM分类模型,设计了对大肠杆菌具有潜在抗菌活性的新型的抗菌短肽序列,经过分类模型的预测发现设计出的肽序列被认为具有抗菌功能的概率在70.6-91.7%,且其三维构象表现出具有两亲性表面的α-螺旋结构[35].Gisbert Schneider课题组同样使用LSTM从螺旋抗菌肽序列上捕获数据的模式并将学习到的上下文信息运用于抗菌肽序列的生成[36].Aleksej Zelezniak课题组提出ProteinGAN,利用GAN学习到大量天然蛋白质序列的多样性并进而生成具有特定功能的酶序列[37].以苹果酸脱氢酶 (MDH) 为例,作者在该酶家族序列上进行训练并设计出具有相同功能酶的序列,其中有突变位点超过100个的设计序列,其活性与天然酶的活性相近. ...
Auto-encoding variational bayes
1
2013
... 当从功能上约束设计的序列时,可以采用序列生成方法,在具有给定功能的酶序列数据上挖掘残基间的模式直接生成新酶的序列.常用的生成模型有长短期记忆网络 (Long Short-Term Memory, LSTM) [51],GAN,变分自动编码器 (Variational Autoencoder, VAE)[52],Transformer[53]等.Mire Zloh课题组构建了基于LSTM的生成模型和双向LSTM分类模型,设计了对大肠杆菌具有潜在抗菌活性的新型的抗菌短肽序列,经过分类模型的预测发现设计出的肽序列被认为具有抗菌功能的概率在70.6-91.7%,且其三维构象表现出具有两亲性表面的α-螺旋结构[35].Gisbert Schneider课题组同样使用LSTM从螺旋抗菌肽序列上捕获数据的模式并将学习到的上下文信息运用于抗菌肽序列的生成[36].Aleksej Zelezniak课题组提出ProteinGAN,利用GAN学习到大量天然蛋白质序列的多样性并进而生成具有特定功能的酶序列[37].以苹果酸脱氢酶 (MDH) 为例,作者在该酶家族序列上进行训练并设计出具有相同功能酶的序列,其中有突变位点超过100个的设计序列,其活性与天然酶的活性相近. ...
Attention is all You need
1
2017
... 当从功能上约束设计的序列时,可以采用序列生成方法,在具有给定功能的酶序列数据上挖掘残基间的模式直接生成新酶的序列.常用的生成模型有长短期记忆网络 (Long Short-Term Memory, LSTM) [51],GAN,变分自动编码器 (Variational Autoencoder, VAE)[52],Transformer[53]等.Mire Zloh课题组构建了基于LSTM的生成模型和双向LSTM分类模型,设计了对大肠杆菌具有潜在抗菌活性的新型的抗菌短肽序列,经过分类模型的预测发现设计出的肽序列被认为具有抗菌功能的概率在70.6-91.7%,且其三维构象表现出具有两亲性表面的α-螺旋结构[35].Gisbert Schneider课题组同样使用LSTM从螺旋抗菌肽序列上捕获数据的模式并将学习到的上下文信息运用于抗菌肽序列的生成[36].Aleksej Zelezniak课题组提出ProteinGAN,利用GAN学习到大量天然蛋白质序列的多样性并进而生成具有特定功能的酶序列[37].以苹果酸脱氢酶 (MDH) 为例,作者在该酶家族序列上进行训练并设计出具有相同功能酶的序列,其中有突变位点超过100个的设计序列,其活性与天然酶的活性相近. ...
Generative models for graph-based protein design
1
2019
... 同样,可以采用结构约束来指导进而设计氨基酸序列.这种情况下,设计的氨基酸序列能否折叠成目标的蛋白质结构是至关重要的指标.最近被称为新一代Rosetta蛋白设计内核的Rosetta MPNN “Mover”,突破了传统的Rosetta设计范式“Inside-out”模式.该方法ProteinMPNN由David Baker组提出,基于structured-transformer[54],采用了结构编码-序列解码的自回归模型框架,将原子配对距离势融入到边的特征表示中,使序列恢复率提高约7.8%[39].ProteinMPNN对根据幻想的主链进行蛋白设计(500条),其中96条蛋白质序列在大肠杆菌体系中可以被大量的可溶表达,且成功结晶一个与设计结构高度一致的设计蛋白.同时,ProteinMPNN对单体,同源二聚体,异二聚体结构进行设计,其序列恢复率均在50%以上,其中核心区域的恢复率高达90%-95%.中国科学技术大学刘海燕和陈泉团队提出的ABACUS-R完全基于深度学习算法实现给定骨架设计氨基酸序列,不再依赖于传统能量项构建,并且序列恢复率高于ABACUS计算的,在测试集上基本可以达到50%[40].其主要思路是在给定骨架的情况下,通过编码-解码 (encoder-decoder) 框架学习在给定残基的结构特征以及周边结构环境的特性预测该残基的序列类型(侧链).值得一提的是,ABACUS-R采用多任务学习,不仅仅学习该残基的类型,还同时预测其二级结构,溶剂可达面积,B-factor以及一些结构构象扭转角任务.这些辅助任务的设计不仅提高了模型设计序列的能力,还隐式的在序列设计中加入了实时的结构约束.实验验证设计了3个天然骨架的蛋白序列设计并做了相应的实验验证.最后通过ABACUS-R设计出了可以成功表达且折叠成相应的三维结构的蛋白质序列,充分证明蛋白质设计可以绕过侧链模型的显示建模的可行性.卜东波课题组提出ProDESIGN-LE也是基于Transformer框架,通过计算序列类型是否符合给定的局部结构环境来设计蛋白序列[47].在实验中为CAT III酶设计的5条序列中,有3条可以成功表达且可溶.许锦波课题组提出的一种基于骨架设计蛋白序列的方法,基于生成SE (3) 等变模型,显著改进了现有的自回归方法[55].Mostafa Karimi组[38]提出gcWGAN探索生成给定折叠条件下的序列,使序列折叠成给定的方式.要是构造了一个基于DeepSF[56]的快速从序列预测折叠模式的模型并实时反馈监督序列是否可以正确折叠,这个模型被称为“Oracle”.Po-Ssu Huang组的Namrata Anand[57]直接从蛋白质骨架结构信息中学习侧链氨基酸类型,从而学习到一个基于自回归的自动的神经网络能量来指导后续的序列设计.在实际的TIM-barrel设计中,设计出的序列中有两个成功结晶且与设计的骨架高度一致. ...
A deep SE(3)-equivariant model for learning inverse protein folding
1
2022
... 同样,可以采用结构约束来指导进而设计氨基酸序列.这种情况下,设计的氨基酸序列能否折叠成目标的蛋白质结构是至关重要的指标.最近被称为新一代Rosetta蛋白设计内核的Rosetta MPNN “Mover”,突破了传统的Rosetta设计范式“Inside-out”模式.该方法ProteinMPNN由David Baker组提出,基于structured-transformer[54],采用了结构编码-序列解码的自回归模型框架,将原子配对距离势融入到边的特征表示中,使序列恢复率提高约7.8%[39].ProteinMPNN对根据幻想的主链进行蛋白设计(500条),其中96条蛋白质序列在大肠杆菌体系中可以被大量的可溶表达,且成功结晶一个与设计结构高度一致的设计蛋白.同时,ProteinMPNN对单体,同源二聚体,异二聚体结构进行设计,其序列恢复率均在50%以上,其中核心区域的恢复率高达90%-95%.中国科学技术大学刘海燕和陈泉团队提出的ABACUS-R完全基于深度学习算法实现给定骨架设计氨基酸序列,不再依赖于传统能量项构建,并且序列恢复率高于ABACUS计算的,在测试集上基本可以达到50%[40].其主要思路是在给定骨架的情况下,通过编码-解码 (encoder-decoder) 框架学习在给定残基的结构特征以及周边结构环境的特性预测该残基的序列类型(侧链).值得一提的是,ABACUS-R采用多任务学习,不仅仅学习该残基的类型,还同时预测其二级结构,溶剂可达面积,B-factor以及一些结构构象扭转角任务.这些辅助任务的设计不仅提高了模型设计序列的能力,还隐式的在序列设计中加入了实时的结构约束.实验验证设计了3个天然骨架的蛋白序列设计并做了相应的实验验证.最后通过ABACUS-R设计出了可以成功表达且折叠成相应的三维结构的蛋白质序列,充分证明蛋白质设计可以绕过侧链模型的显示建模的可行性.卜东波课题组提出ProDESIGN-LE也是基于Transformer框架,通过计算序列类型是否符合给定的局部结构环境来设计蛋白序列[47].在实验中为CAT III酶设计的5条序列中,有3条可以成功表达且可溶.许锦波课题组提出的一种基于骨架设计蛋白序列的方法,基于生成SE (3) 等变模型,显著改进了现有的自回归方法[55].Mostafa Karimi组[38]提出gcWGAN探索生成给定折叠条件下的序列,使序列折叠成给定的方式.要是构造了一个基于DeepSF[56]的快速从序列预测折叠模式的模型并实时反馈监督序列是否可以正确折叠,这个模型被称为“Oracle”.Po-Ssu Huang组的Namrata Anand[57]直接从蛋白质骨架结构信息中学习侧链氨基酸类型,从而学习到一个基于自回归的自动的神经网络能量来指导后续的序列设计.在实际的TIM-barrel设计中,设计出的序列中有两个成功结晶且与设计的骨架高度一致. ...
DeepSF: deep convolutional neural network for mapping protein sequences to folds
1
2018
... 同样,可以采用结构约束来指导进而设计氨基酸序列.这种情况下,设计的氨基酸序列能否折叠成目标的蛋白质结构是至关重要的指标.最近被称为新一代Rosetta蛋白设计内核的Rosetta MPNN “Mover”,突破了传统的Rosetta设计范式“Inside-out”模式.该方法ProteinMPNN由David Baker组提出,基于structured-transformer[54],采用了结构编码-序列解码的自回归模型框架,将原子配对距离势融入到边的特征表示中,使序列恢复率提高约7.8%[39].ProteinMPNN对根据幻想的主链进行蛋白设计(500条),其中96条蛋白质序列在大肠杆菌体系中可以被大量的可溶表达,且成功结晶一个与设计结构高度一致的设计蛋白.同时,ProteinMPNN对单体,同源二聚体,异二聚体结构进行设计,其序列恢复率均在50%以上,其中核心区域的恢复率高达90%-95%.中国科学技术大学刘海燕和陈泉团队提出的ABACUS-R完全基于深度学习算法实现给定骨架设计氨基酸序列,不再依赖于传统能量项构建,并且序列恢复率高于ABACUS计算的,在测试集上基本可以达到50%[40].其主要思路是在给定骨架的情况下,通过编码-解码 (encoder-decoder) 框架学习在给定残基的结构特征以及周边结构环境的特性预测该残基的序列类型(侧链).值得一提的是,ABACUS-R采用多任务学习,不仅仅学习该残基的类型,还同时预测其二级结构,溶剂可达面积,B-factor以及一些结构构象扭转角任务.这些辅助任务的设计不仅提高了模型设计序列的能力,还隐式的在序列设计中加入了实时的结构约束.实验验证设计了3个天然骨架的蛋白序列设计并做了相应的实验验证.最后通过ABACUS-R设计出了可以成功表达且折叠成相应的三维结构的蛋白质序列,充分证明蛋白质设计可以绕过侧链模型的显示建模的可行性.卜东波课题组提出ProDESIGN-LE也是基于Transformer框架,通过计算序列类型是否符合给定的局部结构环境来设计蛋白序列[47].在实验中为CAT III酶设计的5条序列中,有3条可以成功表达且可溶.许锦波课题组提出的一种基于骨架设计蛋白序列的方法,基于生成SE (3) 等变模型,显著改进了现有的自回归方法[55].Mostafa Karimi组[38]提出gcWGAN探索生成给定折叠条件下的序列,使序列折叠成给定的方式.要是构造了一个基于DeepSF[56]的快速从序列预测折叠模式的模型并实时反馈监督序列是否可以正确折叠,这个模型被称为“Oracle”.Po-Ssu Huang组的Namrata Anand[57]直接从蛋白质骨架结构信息中学习侧链氨基酸类型,从而学习到一个基于自回归的自动的神经网络能量来指导后续的序列设计.在实际的TIM-barrel设计中,设计出的序列中有两个成功结晶且与设计的骨架高度一致. ...
Protein sequence design with a learned potential
1
2022
... 同样,可以采用结构约束来指导进而设计氨基酸序列.这种情况下,设计的氨基酸序列能否折叠成目标的蛋白质结构是至关重要的指标.最近被称为新一代Rosetta蛋白设计内核的Rosetta MPNN “Mover”,突破了传统的Rosetta设计范式“Inside-out”模式.该方法ProteinMPNN由David Baker组提出,基于structured-transformer[54],采用了结构编码-序列解码的自回归模型框架,将原子配对距离势融入到边的特征表示中,使序列恢复率提高约7.8%[39].ProteinMPNN对根据幻想的主链进行蛋白设计(500条),其中96条蛋白质序列在大肠杆菌体系中可以被大量的可溶表达,且成功结晶一个与设计结构高度一致的设计蛋白.同时,ProteinMPNN对单体,同源二聚体,异二聚体结构进行设计,其序列恢复率均在50%以上,其中核心区域的恢复率高达90%-95%.中国科学技术大学刘海燕和陈泉团队提出的ABACUS-R完全基于深度学习算法实现给定骨架设计氨基酸序列,不再依赖于传统能量项构建,并且序列恢复率高于ABACUS计算的,在测试集上基本可以达到50%[40].其主要思路是在给定骨架的情况下,通过编码-解码 (encoder-decoder) 框架学习在给定残基的结构特征以及周边结构环境的特性预测该残基的序列类型(侧链).值得一提的是,ABACUS-R采用多任务学习,不仅仅学习该残基的类型,还同时预测其二级结构,溶剂可达面积,B-factor以及一些结构构象扭转角任务.这些辅助任务的设计不仅提高了模型设计序列的能力,还隐式的在序列设计中加入了实时的结构约束.实验验证设计了3个天然骨架的蛋白序列设计并做了相应的实验验证.最后通过ABACUS-R设计出了可以成功表达且折叠成相应的三维结构的蛋白质序列,充分证明蛋白质设计可以绕过侧链模型的显示建模的可行性.卜东波课题组提出ProDESIGN-LE也是基于Transformer框架,通过计算序列类型是否符合给定的局部结构环境来设计蛋白序列[47].在实验中为CAT III酶设计的5条序列中,有3条可以成功表达且可溶.许锦波课题组提出的一种基于骨架设计蛋白序列的方法,基于生成SE (3) 等变模型,显著改进了现有的自回归方法[55].Mostafa Karimi组[38]提出gcWGAN探索生成给定折叠条件下的序列,使序列折叠成给定的方式.要是构造了一个基于DeepSF[56]的快速从序列预测折叠模式的模型并实时反馈监督序列是否可以正确折叠,这个模型被称为“Oracle”.Po-Ssu Huang组的Namrata Anand[57]直接从蛋白质骨架结构信息中学习侧链氨基酸类型,从而学习到一个基于自回归的自动的神经网络能量来指导后续的序列设计.在实际的TIM-barrel设计中,设计出的序列中有两个成功结晶且与设计的骨架高度一致. ...
Recent applications of deep learning methods on evolution- and contact-based protein structure prediction
1
2021
... 基于物理的能量项,通过描述原子在折叠过程中,原子内部之间相互作用以及蛋白质分子与溶剂分子之间的相互作用,来模拟构象的最终能量.一般包括成键作用和非成键作用[58].后者主要包括氢键,范德华力,静电力等,前者则包含一些二面角,键角,键长等势能[59-61].但是在实际过程中,由于我们对蛋白质折叠机制尚未完全理解,例如哪些相互作用力对折叠是重要的,不同相互作用力的叠加是否是有益的,这就导致在设计能量函数的时候并不一定合适. ...
CHARMM: a program for macromolecular energy, minimization, and dynamics calculations
1
1983
... 基于物理的能量项,通过描述原子在折叠过程中,原子内部之间相互作用以及蛋白质分子与溶剂分子之间的相互作用,来模拟构象的最终能量.一般包括成键作用和非成键作用[58].后者主要包括氢键,范德华力,静电力等,前者则包含一些二面角,键角,键长等势能[59-61].但是在实际过程中,由于我们对蛋白质折叠机制尚未完全理解,例如哪些相互作用力对折叠是重要的,不同相互作用力的叠加是否是有益的,这就导致在设计能量函数的时候并不一定合适. ...
ASTRO-FOLD: a combinatorial and global optimization framework for Ab initio prediction of three-dimensional structures of proteins from the amino acid sequence
0
2003
ASTRO-FOLD 2.0: An enhanced framework for protein structure prediction
1
2012
... 基于物理的能量项,通过描述原子在折叠过程中,原子内部之间相互作用以及蛋白质分子与溶剂分子之间的相互作用,来模拟构象的最终能量.一般包括成键作用和非成键作用[58].后者主要包括氢键,范德华力,静电力等,前者则包含一些二面角,键角,键长等势能[59-61].但是在实际过程中,由于我们对蛋白质折叠机制尚未完全理解,例如哪些相互作用力对折叠是重要的,不同相互作用力的叠加是否是有益的,这就导致在设计能量函数的时候并不一定合适. ...
Protein data bank (PDB): the single global macromolecular structure archive
1
2017
... 基于知识统计的方法,一般要求有一个大型结构数据集(类似于PDB[62]),从中统计不同原子对之间的相对位置,进而构造一个打分矩阵,得到原子对之间的打分函数.例如,在打分矩阵中,发现某种氨基酸在其相邻的3.6Å范围内经常有一种氨基酸出现,且对方的相邻打分矩阵中也显示经常与之相邻,则能量值打分一定是较低的.从中,其实可以看出该方法要求预测的这个蛋白质结构在已有的蛋白质库中存在相似的蛋白质结构区域,即局部的某些构象出现的次数一定是不低的.否则这个能量项即使很高,也是有一定“偏见”的.美国密西根大学张阳实验室开发的从头预测蛋白质结构预测工具QUARK是典型的基于统计能量项的工作[63].QUARK分别从原子层面,残基层面,拓扑层面统计了11种基于知识的能量项,利用副本交换的蒙特卡洛搜索算法实现仅从序列出发预测蛋白质结构的工作.另一个同样由张阳实验室开发的I-TASSER,采用基于统计的能量项迭代的基于线程结构模板装配方法在近几年的Community-Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction (CASP7-CASP15) 大赛上均位列服务器组第一名[64-65].I-TASSER采用的是基于统计的势能,包含三种类型:1)通用的统计势能:特定方向(平行,反平行,垂直方向)的接触特征,手性局部结构的短程Cα原子的距离关系,相隔5个残基的局部结构特征规律等.2)氢键网络.3)基于线程模板的约束,包含Cα原子之间的距离约束以及侧链质心原子的接触距离约束.而与I-TASSER并驾齐驱的由美国华盛顿大学的David Baker组开发的Rosetta方法,则同时采用了基于物理能量项和基于统计的能量项,运用蒙特卡洛算法在构象空间中基于Metropolis准则随机搜索最低能量构象[66]. ...
Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field
1
2012
... 基于知识统计的方法,一般要求有一个大型结构数据集(类似于PDB[62]),从中统计不同原子对之间的相对位置,进而构造一个打分矩阵,得到原子对之间的打分函数.例如,在打分矩阵中,发现某种氨基酸在其相邻的3.6Å范围内经常有一种氨基酸出现,且对方的相邻打分矩阵中也显示经常与之相邻,则能量值打分一定是较低的.从中,其实可以看出该方法要求预测的这个蛋白质结构在已有的蛋白质库中存在相似的蛋白质结构区域,即局部的某些构象出现的次数一定是不低的.否则这个能量项即使很高,也是有一定“偏见”的.美国密西根大学张阳实验室开发的从头预测蛋白质结构预测工具QUARK是典型的基于统计能量项的工作[63].QUARK分别从原子层面,残基层面,拓扑层面统计了11种基于知识的能量项,利用副本交换的蒙特卡洛搜索算法实现仅从序列出发预测蛋白质结构的工作.另一个同样由张阳实验室开发的I-TASSER,采用基于统计的能量项迭代的基于线程结构模板装配方法在近几年的Community-Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction (CASP7-CASP15) 大赛上均位列服务器组第一名[64-65].I-TASSER采用的是基于统计的势能,包含三种类型:1)通用的统计势能:特定方向(平行,反平行,垂直方向)的接触特征,手性局部结构的短程Cα原子的距离关系,相隔5个残基的局部结构特征规律等.2)氢键网络.3)基于线程模板的约束,包含Cα原子之间的距离约束以及侧链质心原子的接触距离约束.而与I-TASSER并驾齐驱的由美国华盛顿大学的David Baker组开发的Rosetta方法,则同时采用了基于物理能量项和基于统计的能量项,运用蒙特卡洛算法在构象空间中基于Metropolis准则随机搜索最低能量构象[66]. ...
I-TASSER server: new development for protein structure and function predictions
1
2015
... 基于知识统计的方法,一般要求有一个大型结构数据集(类似于PDB[62]),从中统计不同原子对之间的相对位置,进而构造一个打分矩阵,得到原子对之间的打分函数.例如,在打分矩阵中,发现某种氨基酸在其相邻的3.6Å范围内经常有一种氨基酸出现,且对方的相邻打分矩阵中也显示经常与之相邻,则能量值打分一定是较低的.从中,其实可以看出该方法要求预测的这个蛋白质结构在已有的蛋白质库中存在相似的蛋白质结构区域,即局部的某些构象出现的次数一定是不低的.否则这个能量项即使很高,也是有一定“偏见”的.美国密西根大学张阳实验室开发的从头预测蛋白质结构预测工具QUARK是典型的基于统计能量项的工作[63].QUARK分别从原子层面,残基层面,拓扑层面统计了11种基于知识的能量项,利用副本交换的蒙特卡洛搜索算法实现仅从序列出发预测蛋白质结构的工作.另一个同样由张阳实验室开发的I-TASSER,采用基于统计的能量项迭代的基于线程结构模板装配方法在近几年的Community-Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction (CASP7-CASP15) 大赛上均位列服务器组第一名[64-65].I-TASSER采用的是基于统计的势能,包含三种类型:1)通用的统计势能:特定方向(平行,反平行,垂直方向)的接触特征,手性局部结构的短程Cα原子的距离关系,相隔5个残基的局部结构特征规律等.2)氢键网络.3)基于线程模板的约束,包含Cα原子之间的距离约束以及侧链质心原子的接触距离约束.而与I-TASSER并驾齐驱的由美国华盛顿大学的David Baker组开发的Rosetta方法,则同时采用了基于物理能量项和基于统计的能量项,运用蒙特卡洛算法在构象空间中基于Metropolis准则随机搜索最低能量构象[66]. ...
The I-TASSER Suite: protein structure and function prediction
1
2015
... 基于知识统计的方法,一般要求有一个大型结构数据集(类似于PDB[62]),从中统计不同原子对之间的相对位置,进而构造一个打分矩阵,得到原子对之间的打分函数.例如,在打分矩阵中,发现某种氨基酸在其相邻的3.6Å范围内经常有一种氨基酸出现,且对方的相邻打分矩阵中也显示经常与之相邻,则能量值打分一定是较低的.从中,其实可以看出该方法要求预测的这个蛋白质结构在已有的蛋白质库中存在相似的蛋白质结构区域,即局部的某些构象出现的次数一定是不低的.否则这个能量项即使很高,也是有一定“偏见”的.美国密西根大学张阳实验室开发的从头预测蛋白质结构预测工具QUARK是典型的基于统计能量项的工作[63].QUARK分别从原子层面,残基层面,拓扑层面统计了11种基于知识的能量项,利用副本交换的蒙特卡洛搜索算法实现仅从序列出发预测蛋白质结构的工作.另一个同样由张阳实验室开发的I-TASSER,采用基于统计的能量项迭代的基于线程结构模板装配方法在近几年的Community-Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction (CASP7-CASP15) 大赛上均位列服务器组第一名[64-65].I-TASSER采用的是基于统计的势能,包含三种类型:1)通用的统计势能:特定方向(平行,反平行,垂直方向)的接触特征,手性局部结构的短程Cα原子的距离关系,相隔5个残基的局部结构特征规律等.2)氢键网络.3)基于线程模板的约束,包含Cα原子之间的距离约束以及侧链质心原子的接触距离约束.而与I-TASSER并驾齐驱的由美国华盛顿大学的David Baker组开发的Rosetta方法,则同时采用了基于物理能量项和基于统计的能量项,运用蒙特卡洛算法在构象空间中基于Metropolis准则随机搜索最低能量构象[66]. ...
ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules
1
2011
... 基于知识统计的方法,一般要求有一个大型结构数据集(类似于PDB[62]),从中统计不同原子对之间的相对位置,进而构造一个打分矩阵,得到原子对之间的打分函数.例如,在打分矩阵中,发现某种氨基酸在其相邻的3.6Å范围内经常有一种氨基酸出现,且对方的相邻打分矩阵中也显示经常与之相邻,则能量值打分一定是较低的.从中,其实可以看出该方法要求预测的这个蛋白质结构在已有的蛋白质库中存在相似的蛋白质结构区域,即局部的某些构象出现的次数一定是不低的.否则这个能量项即使很高,也是有一定“偏见”的.美国密西根大学张阳实验室开发的从头预测蛋白质结构预测工具QUARK是典型的基于统计能量项的工作[63].QUARK分别从原子层面,残基层面,拓扑层面统计了11种基于知识的能量项,利用副本交换的蒙特卡洛搜索算法实现仅从序列出发预测蛋白质结构的工作.另一个同样由张阳实验室开发的I-TASSER,采用基于统计的能量项迭代的基于线程结构模板装配方法在近几年的Community-Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction (CASP7-CASP15) 大赛上均位列服务器组第一名[64-65].I-TASSER采用的是基于统计的势能,包含三种类型:1)通用的统计势能:特定方向(平行,反平行,垂直方向)的接触特征,手性局部结构的短程Cα原子的距离关系,相隔5个残基的局部结构特征规律等.2)氢键网络.3)基于线程模板的约束,包含Cα原子之间的距离约束以及侧链质心原子的接触距离约束.而与I-TASSER并驾齐驱的由美国华盛顿大学的David Baker组开发的Rosetta方法,则同时采用了基于物理能量项和基于统计的能量项,运用蒙特卡洛算法在构象空间中基于Metropolis准则随机搜索最低能量构象[66]. ...
PSICOV: precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments
1
2012
... 早期的蛋白质折叠将蛋白质三维结构中的物理接触(contact)作为约束.通过分析蛋白质序列残基的共进化信息,将序列中残基的共变关系映射到蛋白质三维空间结构中的物理接触中.共进化指的是在蛋白质家族的进化演变中,由于环境以及自身进化的需要,某些残基发生突变后,为了维持某些主要的功能或者结构不变,其他残基随之发生共同变化 (co-evolution) 的现象.从蛋白质家族的多序列比对 (Multiple Sequence Alignment, MSA) 中统计不同位置上不同残基对共同出现的频率大小进而估计它们之间的相互作用,根据相互作用大小判断在空间结构上是否接触还是排斥.主要的估计方法有:稀疏逆协方差方差估计[67],互信息最大化[68],直接耦合分析 (Direct Coupling Analysis, DCA) [69].这样基于概率统计模型得到残基相互作用对估计量的方法,显而易见依赖于MSA的丰富程度并且难以达到满意的精度(主要是噪声以及信息的不足).但是由于结合了全局信息,相比“孤立”预测残基对的方法,还是有了很大的突破[70-72].随着人工智能技术的发展,解决手段就变得而更为丰富多样起来,预测精度也有了突破性的进展.2016年许锦波课题组提出的“RaptorX-Contact”方法首次将深度神经网络应用在蛋白质结构领域[73],在CASP12比赛中一举夺冠,证明了深度学习算法在该领域的可行性.该方法将残基对之间的相互作用关系看作图像问题,提取一维的序列保守性特征,结构特征以及二维的共进化特征,然后采用2D的深度残差网络 (ResNet) 块对特征进行提取融合捕获上下文来预测残基对是否接触,协助蛋白质的从头折叠.该方法使用的ResNet网络相比前面提到的早期研究方法,学习到了更高阶 (high-order) 的残基对关系而且训练数据从单一到大量蛋白质家族上挖掘,因而精度有了明显的提升[74]. ...
Inferring interaction partners from protein sequences
1
2016
... 早期的蛋白质折叠将蛋白质三维结构中的物理接触(contact)作为约束.通过分析蛋白质序列残基的共进化信息,将序列中残基的共变关系映射到蛋白质三维空间结构中的物理接触中.共进化指的是在蛋白质家族的进化演变中,由于环境以及自身进化的需要,某些残基发生突变后,为了维持某些主要的功能或者结构不变,其他残基随之发生共同变化 (co-evolution) 的现象.从蛋白质家族的多序列比对 (Multiple Sequence Alignment, MSA) 中统计不同位置上不同残基对共同出现的频率大小进而估计它们之间的相互作用,根据相互作用大小判断在空间结构上是否接触还是排斥.主要的估计方法有:稀疏逆协方差方差估计[67],互信息最大化[68],直接耦合分析 (Direct Coupling Analysis, DCA) [69].这样基于概率统计模型得到残基相互作用对估计量的方法,显而易见依赖于MSA的丰富程度并且难以达到满意的精度(主要是噪声以及信息的不足).但是由于结合了全局信息,相比“孤立”预测残基对的方法,还是有了很大的突破[70-72].随着人工智能技术的发展,解决手段就变得而更为丰富多样起来,预测精度也有了突破性的进展.2016年许锦波课题组提出的“RaptorX-Contact”方法首次将深度神经网络应用在蛋白质结构领域[73],在CASP12比赛中一举夺冠,证明了深度学习算法在该领域的可行性.该方法将残基对之间的相互作用关系看作图像问题,提取一维的序列保守性特征,结构特征以及二维的共进化特征,然后采用2D的深度残差网络 (ResNet) 块对特征进行提取融合捕获上下文来预测残基对是否接触,协助蛋白质的从头折叠.该方法使用的ResNet网络相比前面提到的早期研究方法,学习到了更高阶 (high-order) 的残基对关系而且训练数据从单一到大量蛋白质家族上挖掘,因而精度有了明显的提升[74]. ...
Direct-coupling analysis of residue coevolution captures native contacts across many protein families
1
2011
... 早期的蛋白质折叠将蛋白质三维结构中的物理接触(contact)作为约束.通过分析蛋白质序列残基的共进化信息,将序列中残基的共变关系映射到蛋白质三维空间结构中的物理接触中.共进化指的是在蛋白质家族的进化演变中,由于环境以及自身进化的需要,某些残基发生突变后,为了维持某些主要的功能或者结构不变,其他残基随之发生共同变化 (co-evolution) 的现象.从蛋白质家族的多序列比对 (Multiple Sequence Alignment, MSA) 中统计不同位置上不同残基对共同出现的频率大小进而估计它们之间的相互作用,根据相互作用大小判断在空间结构上是否接触还是排斥.主要的估计方法有:稀疏逆协方差方差估计[67],互信息最大化[68],直接耦合分析 (Direct Coupling Analysis, DCA) [69].这样基于概率统计模型得到残基相互作用对估计量的方法,显而易见依赖于MSA的丰富程度并且难以达到满意的精度(主要是噪声以及信息的不足).但是由于结合了全局信息,相比“孤立”预测残基对的方法,还是有了很大的突破[70-72].随着人工智能技术的发展,解决手段就变得而更为丰富多样起来,预测精度也有了突破性的进展.2016年许锦波课题组提出的“RaptorX-Contact”方法首次将深度神经网络应用在蛋白质结构领域[73],在CASP12比赛中一举夺冠,证明了深度学习算法在该领域的可行性.该方法将残基对之间的相互作用关系看作图像问题,提取一维的序列保守性特征,结构特征以及二维的共进化特征,然后采用2D的深度残差网络 (ResNet) 块对特征进行提取融合捕获上下文来预测残基对是否接触,协助蛋白质的从头折叠.该方法使用的ResNet网络相比前面提到的早期研究方法,学习到了更高阶 (high-order) 的残基对关系而且训练数据从单一到大量蛋白质家族上挖掘,因而精度有了明显的提升[74]. ...
CCMpred—fast and precise prediction of protein residue-residue contacts from correlated mutations
1
2014
... 早期的蛋白质折叠将蛋白质三维结构中的物理接触(contact)作为约束.通过分析蛋白质序列残基的共进化信息,将序列中残基的共变关系映射到蛋白质三维空间结构中的物理接触中.共进化指的是在蛋白质家族的进化演变中,由于环境以及自身进化的需要,某些残基发生突变后,为了维持某些主要的功能或者结构不变,其他残基随之发生共同变化 (co-evolution) 的现象.从蛋白质家族的多序列比对 (Multiple Sequence Alignment, MSA) 中统计不同位置上不同残基对共同出现的频率大小进而估计它们之间的相互作用,根据相互作用大小判断在空间结构上是否接触还是排斥.主要的估计方法有:稀疏逆协方差方差估计[67],互信息最大化[68],直接耦合分析 (Direct Coupling Analysis, DCA) [69].这样基于概率统计模型得到残基相互作用对估计量的方法,显而易见依赖于MSA的丰富程度并且难以达到满意的精度(主要是噪声以及信息的不足).但是由于结合了全局信息,相比“孤立”预测残基对的方法,还是有了很大的突破[70-72].随着人工智能技术的发展,解决手段就变得而更为丰富多样起来,预测精度也有了突破性的进展.2016年许锦波课题组提出的“RaptorX-Contact”方法首次将深度神经网络应用在蛋白质结构领域[73],在CASP12比赛中一举夺冠,证明了深度学习算法在该领域的可行性.该方法将残基对之间的相互作用关系看作图像问题,提取一维的序列保守性特征,结构特征以及二维的共进化特征,然后采用2D的深度残差网络 (ResNet) 块对特征进行提取融合捕获上下文来预测残基对是否接触,协助蛋白质的从头折叠.该方法使用的ResNet网络相比前面提到的早期研究方法,学习到了更高阶 (high-order) 的残基对关系而且训练数据从单一到大量蛋白质家族上挖掘,因而精度有了明显的提升[74]. ...
Identification of direct residue contacts in protein-protein interaction by message passing
0
2009
Assessing the utility of coevolution-based residue-residue contact predictions in a sequence- and structure-rich era
1
2013
... 早期的蛋白质折叠将蛋白质三维结构中的物理接触(contact)作为约束.通过分析蛋白质序列残基的共进化信息,将序列中残基的共变关系映射到蛋白质三维空间结构中的物理接触中.共进化指的是在蛋白质家族的进化演变中,由于环境以及自身进化的需要,某些残基发生突变后,为了维持某些主要的功能或者结构不变,其他残基随之发生共同变化 (co-evolution) 的现象.从蛋白质家族的多序列比对 (Multiple Sequence Alignment, MSA) 中统计不同位置上不同残基对共同出现的频率大小进而估计它们之间的相互作用,根据相互作用大小判断在空间结构上是否接触还是排斥.主要的估计方法有:稀疏逆协方差方差估计[67],互信息最大化[68],直接耦合分析 (Direct Coupling Analysis, DCA) [69].这样基于概率统计模型得到残基相互作用对估计量的方法,显而易见依赖于MSA的丰富程度并且难以达到满意的精度(主要是噪声以及信息的不足).但是由于结合了全局信息,相比“孤立”预测残基对的方法,还是有了很大的突破[70-72].随着人工智能技术的发展,解决手段就变得而更为丰富多样起来,预测精度也有了突破性的进展.2016年许锦波课题组提出的“RaptorX-Contact”方法首次将深度神经网络应用在蛋白质结构领域[73],在CASP12比赛中一举夺冠,证明了深度学习算法在该领域的可行性.该方法将残基对之间的相互作用关系看作图像问题,提取一维的序列保守性特征,结构特征以及二维的共进化特征,然后采用2D的深度残差网络 (ResNet) 块对特征进行提取融合捕获上下文来预测残基对是否接触,协助蛋白质的从头折叠.该方法使用的ResNet网络相比前面提到的早期研究方法,学习到了更高阶 (high-order) 的残基对关系而且训练数据从单一到大量蛋白质家族上挖掘,因而精度有了明显的提升[74]. ...
Accurate de novo prediction of protein contact map by ultra-deep learning model
1
2017
... 早期的蛋白质折叠将蛋白质三维结构中的物理接触(contact)作为约束.通过分析蛋白质序列残基的共进化信息,将序列中残基的共变关系映射到蛋白质三维空间结构中的物理接触中.共进化指的是在蛋白质家族的进化演变中,由于环境以及自身进化的需要,某些残基发生突变后,为了维持某些主要的功能或者结构不变,其他残基随之发生共同变化 (co-evolution) 的现象.从蛋白质家族的多序列比对 (Multiple Sequence Alignment, MSA) 中统计不同位置上不同残基对共同出现的频率大小进而估计它们之间的相互作用,根据相互作用大小判断在空间结构上是否接触还是排斥.主要的估计方法有:稀疏逆协方差方差估计[67],互信息最大化[68],直接耦合分析 (Direct Coupling Analysis, DCA) [69].这样基于概率统计模型得到残基相互作用对估计量的方法,显而易见依赖于MSA的丰富程度并且难以达到满意的精度(主要是噪声以及信息的不足).但是由于结合了全局信息,相比“孤立”预测残基对的方法,还是有了很大的突破[70-72].随着人工智能技术的发展,解决手段就变得而更为丰富多样起来,预测精度也有了突破性的进展.2016年许锦波课题组提出的“RaptorX-Contact”方法首次将深度神经网络应用在蛋白质结构领域[73],在CASP12比赛中一举夺冠,证明了深度学习算法在该领域的可行性.该方法将残基对之间的相互作用关系看作图像问题,提取一维的序列保守性特征,结构特征以及二维的共进化特征,然后采用2D的深度残差网络 (ResNet) 块对特征进行提取融合捕获上下文来预测残基对是否接触,协助蛋白质的从头折叠.该方法使用的ResNet网络相比前面提到的早期研究方法,学习到了更高阶 (high-order) 的残基对关系而且训练数据从单一到大量蛋白质家族上挖掘,因而精度有了明显的提升[74]. ...
Distance-based protein folding powered by deep learning
1
2019
... 早期的蛋白质折叠将蛋白质三维结构中的物理接触(contact)作为约束.通过分析蛋白质序列残基的共进化信息,将序列中残基的共变关系映射到蛋白质三维空间结构中的物理接触中.共进化指的是在蛋白质家族的进化演变中,由于环境以及自身进化的需要,某些残基发生突变后,为了维持某些主要的功能或者结构不变,其他残基随之发生共同变化 (co-evolution) 的现象.从蛋白质家族的多序列比对 (Multiple Sequence Alignment, MSA) 中统计不同位置上不同残基对共同出现的频率大小进而估计它们之间的相互作用,根据相互作用大小判断在空间结构上是否接触还是排斥.主要的估计方法有:稀疏逆协方差方差估计[67],互信息最大化[68],直接耦合分析 (Direct Coupling Analysis, DCA) [69].这样基于概率统计模型得到残基相互作用对估计量的方法,显而易见依赖于MSA的丰富程度并且难以达到满意的精度(主要是噪声以及信息的不足).但是由于结合了全局信息,相比“孤立”预测残基对的方法,还是有了很大的突破[70-72].随着人工智能技术的发展,解决手段就变得而更为丰富多样起来,预测精度也有了突破性的进展.2016年许锦波课题组提出的“RaptorX-Contact”方法首次将深度神经网络应用在蛋白质结构领域[73],在CASP12比赛中一举夺冠,证明了深度学习算法在该领域的可行性.该方法将残基对之间的相互作用关系看作图像问题,提取一维的序列保守性特征,结构特征以及二维的共进化特征,然后采用2D的深度残差网络 (ResNet) 块对特征进行提取融合捕获上下文来预测残基对是否接触,协助蛋白质的从头折叠.该方法使用的ResNet网络相比前面提到的早期研究方法,学习到了更高阶 (high-order) 的残基对关系而且训练数据从单一到大量蛋白质家族上挖掘,因而精度有了明显的提升[74]. ...
Deep learning extends de novo protein modelling coverage of genomes using iteratively predicted structural constraints
1
2019
... 除了上面提到的接触约束,CASP13上DeepMind提出的AlphaFold1,则将这一约束扩展到了残基间的距离约束.然后将离散化的距离预测值通过采样插值转化成可微的残基距离分布函数,进而通过直接优化该函数求解距离和角度的最优解,从而确定最终的蛋白质三维结构[15].AlphaFold1的成功不仅仅是预测精度的显著提高,更是作为一种信号:深度神经网络可以有效识别蛋白质序列中的信号以及共进化信息的模式,并将其转化到高精度的距离分布上.考虑到三维空间的特性,trRosetta相比AlphaFold1还引入了5个角度的预测值来表示残基间的相对方向进一步加强了残基间的约束,并且精度提高了6.5%[16].David T. Jones 组提出的DMPfold,预测的是相对残基间的距离,主链氢键以及扭转角[75].当学习到这些约束后,类似于RaptorX,输入到crystallography and NMR system (CNS) [76]中作为约束指导蛋白质从头折叠.在2022年的CASP15上,张阳组郑伟博士在已有的I-TASSER基础上提出的D-I-TASSER算法[77],将AttentionPotential以及DeepPotential[78]两个深度学习算法预测出的高准确度的氢键 (hydrogen-bond) 网络,接触图以及距离图等约束加入到I-TASSER 中采用的力场能量项中,然后通过蒙特卡洛模拟进行迭代的片段组装装配最终的蛋白质结构构象,位列蛋白质单体单结构域比赛第一名. ...
Version 1.2 of the crystallography and NMR system
1
2007
... 除了上面提到的接触约束,CASP13上DeepMind提出的AlphaFold1,则将这一约束扩展到了残基间的距离约束.然后将离散化的距离预测值通过采样插值转化成可微的残基距离分布函数,进而通过直接优化该函数求解距离和角度的最优解,从而确定最终的蛋白质三维结构[15].AlphaFold1的成功不仅仅是预测精度的显著提高,更是作为一种信号:深度神经网络可以有效识别蛋白质序列中的信号以及共进化信息的模式,并将其转化到高精度的距离分布上.考虑到三维空间的特性,trRosetta相比AlphaFold1还引入了5个角度的预测值来表示残基间的相对方向进一步加强了残基间的约束,并且精度提高了6.5%[16].David T. Jones 组提出的DMPfold,预测的是相对残基间的距离,主链氢键以及扭转角[75].当学习到这些约束后,类似于RaptorX,输入到crystallography and NMR system (CNS) [76]中作为约束指导蛋白质从头折叠.在2022年的CASP15上,张阳组郑伟博士在已有的I-TASSER基础上提出的D-I-TASSER算法[77],将AttentionPotential以及DeepPotential[78]两个深度学习算法预测出的高准确度的氢键 (hydrogen-bond) 网络,接触图以及距离图等约束加入到I-TASSER 中采用的力场能量项中,然后通过蒙特卡洛模拟进行迭代的片段组装装配最终的蛋白质结构构象,位列蛋白质单体单结构域比赛第一名. ...
Integrating deep neural network models with I-TASSER for accurate protein structure prediction
1
... 除了上面提到的接触约束,CASP13上DeepMind提出的AlphaFold1,则将这一约束扩展到了残基间的距离约束.然后将离散化的距离预测值通过采样插值转化成可微的残基距离分布函数,进而通过直接优化该函数求解距离和角度的最优解,从而确定最终的蛋白质三维结构[15].AlphaFold1的成功不仅仅是预测精度的显著提高,更是作为一种信号:深度神经网络可以有效识别蛋白质序列中的信号以及共进化信息的模式,并将其转化到高精度的距离分布上.考虑到三维空间的特性,trRosetta相比AlphaFold1还引入了5个角度的预测值来表示残基间的相对方向进一步加强了残基间的约束,并且精度提高了6.5%[16].David T. Jones 组提出的DMPfold,预测的是相对残基间的距离,主链氢键以及扭转角[75].当学习到这些约束后,类似于RaptorX,输入到crystallography and NMR system (CNS) [76]中作为约束指导蛋白质从头折叠.在2022年的CASP15上,张阳组郑伟博士在已有的I-TASSER基础上提出的D-I-TASSER算法[77],将AttentionPotential以及DeepPotential[78]两个深度学习算法预测出的高准确度的氢键 (hydrogen-bond) 网络,接触图以及距离图等约束加入到I-TASSER 中采用的力场能量项中,然后通过蒙特卡洛模拟进行迭代的片段组装装配最终的蛋白质结构构象,位列蛋白质单体单结构域比赛第一名. ...
Deep learning geometrical potential for high-accuracy ab initio protein structure prediction
1
2022
... 除了上面提到的接触约束,CASP13上DeepMind提出的AlphaFold1,则将这一约束扩展到了残基间的距离约束.然后将离散化的距离预测值通过采样插值转化成可微的残基距离分布函数,进而通过直接优化该函数求解距离和角度的最优解,从而确定最终的蛋白质三维结构[15].AlphaFold1的成功不仅仅是预测精度的显著提高,更是作为一种信号:深度神经网络可以有效识别蛋白质序列中的信号以及共进化信息的模式,并将其转化到高精度的距离分布上.考虑到三维空间的特性,trRosetta相比AlphaFold1还引入了5个角度的预测值来表示残基间的相对方向进一步加强了残基间的约束,并且精度提高了6.5%[16].David T. Jones 组提出的DMPfold,预测的是相对残基间的距离,主链氢键以及扭转角[75].当学习到这些约束后,类似于RaptorX,输入到crystallography and NMR system (CNS) [76]中作为约束指导蛋白质从头折叠.在2022年的CASP15上,张阳组郑伟博士在已有的I-TASSER基础上提出的D-I-TASSER算法[77],将AttentionPotential以及DeepPotential[78]两个深度学习算法预测出的高准确度的氢键 (hydrogen-bond) 网络,接触图以及距离图等约束加入到I-TASSER 中采用的力场能量项中,然后通过蒙特卡洛模拟进行迭代的片段组装装配最终的蛋白质结构构象,位列蛋白质单体单结构域比赛第一名. ...
End-to-end differentiable learning of protein structure
1
2019
... 2019年Mohammed AlQuraishi提出RGN方法首次尝试使用深度学习算法端到端的从蛋白质序列直接预测最终的3D坐标[79],而不是通过前面介绍的“两步式”方法.其主要思想是将每个残基作为一个可微基元,然后从两个方向:N端到C端,C端到N端,预测在已有的所有残基的局部结构下,当前残基加入后的空间结构,从而将整个蛋白质残基序列串联起来,得到最终蛋白质结构.这个过程中,考虑了当前残基与相邻残基之间的相互作用关系,并实现了“多个尺寸”的蛋白质表示学习.实验证明相比CASP11,CASP12上排名第一的Server组来说,该方法在对于具有新折叠的自由建模中,表现优异.但是该方法输入是蛋白质序列独热 (one-hot) 编码以及位置保守性特异矩阵 (position-specific scoring matrices,PSSM) ,然后通过LSTM去实现序列的编码框架,预测出每个残基的扭转角参数.PSSM相比前面提到的MSA中提取的共进化信息,并不包含残基对间的相互作用,只着重单个残基在单个位置上的进化保守性.因此,导致该方法:1)依赖PSSM矩阵的特征准确性;2)忽略残基对间的相互作用(MSA中共进化信息不是线性的,成本高,且不适合RGN的循环方法).而之后在CASP14比赛上,DeepMind提出AlphaFold2[14],完全抛弃了AlphaFold1传统的“两步式”思路,通过图推理的方式直接实现了“端到端”(end-to-end) 的蛋白质结构预测方法,转变了结合人工智能研究蛋白质结构研究新范式.因此,由该方法引发的“AI蛋白质折叠”被《MIT Technology Review》评为“全球十大突破性技术”.AlphaFold2主要由神经网络EvoFormer和结构模块两部分组成.EvoFormer中序列信息和从MSA中抽取的进化特征之间进行信息交换,直接推理出在空间和进化关系中残基对的配对表征.结构模块则用于将得到的特征转化为三维坐标结构.AlphaFold2的优势在于信息流之间的注意力机制,包括从MSA中学习到配对特征表示与序列上每个残基的特征表示之间的相互信息交流(基于注意力机制),通过几何空间约束形成的具有共残基的相互作用残基对之间的信息交流(三角注意力机制).得到更新后的配对残基特征以及单残基特征后,通过结构模块不断迭代更新坐标系预测当前残基和相邻残基之间肽键的角度和距离偏移,最终得到整个蛋白质的全局笛卡尔系坐标.平均自由建模精度(GDT打分)达到80以上,而在CASAP13(AlphaFold出现)之前,这一个值最高是40左右. ...
Language models of protein sequences at the scale of evolution enable accurate structure prediction
3
2022
... 对于AlphaFold2来说,尽管其预测精度在CASP14上表现惊人,但是后续研究者陆续发现其高度依赖共进化信息以及模板信息,而且对于一条蛋白质在CPU上进行搜索需要大概30分钟[80].因此,从2022年起,陆续有工作直接从已有序列出发,不再显式利用共进化信息,通过大规模语言预训练任务(一般采用的模型框架是Transformer)在海量蛋白质序列数据库中学习残基的表示以及残基对的表示关系,直接输入到AlphaFold2的结构模块中,输出蛋白质结构的3D坐标[80-83].这些方法相比基于共进化的方法(AlphaFold2)来说最显著的优势是时间上提升了一个数量级,对于宏基因数量组的蛋白质结构从时间尺度上成为可能.Meta-FAIR提出的ESMFold[80],不仅推理速度比AlphaFold2快,同时对于低复杂度序列的推理精度与AlphaFold2相当.除此之外,还有Ratul Chowdhury提出的RGN2[83],华深智药提出的OmegaFold[82], 上海天壤科技开发的TRFold方法,山东大学杨建益团队提出的trRosettaX-Single[81]等方法.上述方法基本思路差别不大,各个团队在模型框架上存在一些技巧的差别.例如,trRosettaX-Single采用了知识蒸馏的思想,利用基于进化的模型作为“老师”去指导仅基于序列的“学生”模型去获得一个比较理想的结果.这些方法预测一个蛋白根据计算资源和长度的不同,计算时间基本在毫秒到秒级,同时不依赖于共进化信息.这种优势对于缺少同源信息的酶设计改造来说,是非常有必要的. ...
... [80-83].这些方法相比基于共进化的方法(AlphaFold2)来说最显著的优势是时间上提升了一个数量级,对于宏基因数量组的蛋白质结构从时间尺度上成为可能.Meta-FAIR提出的ESMFold[80],不仅推理速度比AlphaFold2快,同时对于低复杂度序列的推理精度与AlphaFold2相当.除此之外,还有Ratul Chowdhury提出的RGN2[83],华深智药提出的OmegaFold[82], 上海天壤科技开发的TRFold方法,山东大学杨建益团队提出的trRosettaX-Single[81]等方法.上述方法基本思路差别不大,各个团队在模型框架上存在一些技巧的差别.例如,trRosettaX-Single采用了知识蒸馏的思想,利用基于进化的模型作为“老师”去指导仅基于序列的“学生”模型去获得一个比较理想的结果.这些方法预测一个蛋白根据计算资源和长度的不同,计算时间基本在毫秒到秒级,同时不依赖于共进化信息.这种优势对于缺少同源信息的酶设计改造来说,是非常有必要的. ...
... [80],不仅推理速度比AlphaFold2快,同时对于低复杂度序列的推理精度与AlphaFold2相当.除此之外,还有Ratul Chowdhury提出的RGN2[83],华深智药提出的OmegaFold[82], 上海天壤科技开发的TRFold方法,山东大学杨建益团队提出的trRosettaX-Single[81]等方法.上述方法基本思路差别不大,各个团队在模型框架上存在一些技巧的差别.例如,trRosettaX-Single采用了知识蒸馏的思想,利用基于进化的模型作为“老师”去指导仅基于序列的“学生”模型去获得一个比较理想的结果.这些方法预测一个蛋白根据计算资源和长度的不同,计算时间基本在毫秒到秒级,同时不依赖于共进化信息.这种优势对于缺少同源信息的酶设计改造来说,是非常有必要的. ...
Single-sequence protein structure prediction using supervised transformer protein language models
1
2022
... 对于AlphaFold2来说,尽管其预测精度在CASP14上表现惊人,但是后续研究者陆续发现其高度依赖共进化信息以及模板信息,而且对于一条蛋白质在CPU上进行搜索需要大概30分钟[80].因此,从2022年起,陆续有工作直接从已有序列出发,不再显式利用共进化信息,通过大规模语言预训练任务(一般采用的模型框架是Transformer)在海量蛋白质序列数据库中学习残基的表示以及残基对的表示关系,直接输入到AlphaFold2的结构模块中,输出蛋白质结构的3D坐标[80-83].这些方法相比基于共进化的方法(AlphaFold2)来说最显著的优势是时间上提升了一个数量级,对于宏基因数量组的蛋白质结构从时间尺度上成为可能.Meta-FAIR提出的ESMFold[80],不仅推理速度比AlphaFold2快,同时对于低复杂度序列的推理精度与AlphaFold2相当.除此之外,还有Ratul Chowdhury提出的RGN2[83],华深智药提出的OmegaFold[82], 上海天壤科技开发的TRFold方法,山东大学杨建益团队提出的trRosettaX-Single[81]等方法.上述方法基本思路差别不大,各个团队在模型框架上存在一些技巧的差别.例如,trRosettaX-Single采用了知识蒸馏的思想,利用基于进化的模型作为“老师”去指导仅基于序列的“学生”模型去获得一个比较理想的结果.这些方法预测一个蛋白根据计算资源和长度的不同,计算时间基本在毫秒到秒级,同时不依赖于共进化信息.这种优势对于缺少同源信息的酶设计改造来说,是非常有必要的. ...
High-resolution de novo structure prediction from primary sequence
1
2022
... 对于AlphaFold2来说,尽管其预测精度在CASP14上表现惊人,但是后续研究者陆续发现其高度依赖共进化信息以及模板信息,而且对于一条蛋白质在CPU上进行搜索需要大概30分钟[80].因此,从2022年起,陆续有工作直接从已有序列出发,不再显式利用共进化信息,通过大规模语言预训练任务(一般采用的模型框架是Transformer)在海量蛋白质序列数据库中学习残基的表示以及残基对的表示关系,直接输入到AlphaFold2的结构模块中,输出蛋白质结构的3D坐标[80-83].这些方法相比基于共进化的方法(AlphaFold2)来说最显著的优势是时间上提升了一个数量级,对于宏基因数量组的蛋白质结构从时间尺度上成为可能.Meta-FAIR提出的ESMFold[80],不仅推理速度比AlphaFold2快,同时对于低复杂度序列的推理精度与AlphaFold2相当.除此之外,还有Ratul Chowdhury提出的RGN2[83],华深智药提出的OmegaFold[82], 上海天壤科技开发的TRFold方法,山东大学杨建益团队提出的trRosettaX-Single[81]等方法.上述方法基本思路差别不大,各个团队在模型框架上存在一些技巧的差别.例如,trRosettaX-Single采用了知识蒸馏的思想,利用基于进化的模型作为“老师”去指导仅基于序列的“学生”模型去获得一个比较理想的结果.这些方法预测一个蛋白根据计算资源和长度的不同,计算时间基本在毫秒到秒级,同时不依赖于共进化信息.这种优势对于缺少同源信息的酶设计改造来说,是非常有必要的. ...
Single-sequence protein structure prediction using a language model and deep learning
2
2022
... 对于AlphaFold2来说,尽管其预测精度在CASP14上表现惊人,但是后续研究者陆续发现其高度依赖共进化信息以及模板信息,而且对于一条蛋白质在CPU上进行搜索需要大概30分钟[80].因此,从2022年起,陆续有工作直接从已有序列出发,不再显式利用共进化信息,通过大规模语言预训练任务(一般采用的模型框架是Transformer)在海量蛋白质序列数据库中学习残基的表示以及残基对的表示关系,直接输入到AlphaFold2的结构模块中,输出蛋白质结构的3D坐标[80-83].这些方法相比基于共进化的方法(AlphaFold2)来说最显著的优势是时间上提升了一个数量级,对于宏基因数量组的蛋白质结构从时间尺度上成为可能.Meta-FAIR提出的ESMFold[80],不仅推理速度比AlphaFold2快,同时对于低复杂度序列的推理精度与AlphaFold2相当.除此之外,还有Ratul Chowdhury提出的RGN2[83],华深智药提出的OmegaFold[82], 上海天壤科技开发的TRFold方法,山东大学杨建益团队提出的trRosettaX-Single[81]等方法.上述方法基本思路差别不大,各个团队在模型框架上存在一些技巧的差别.例如,trRosettaX-Single采用了知识蒸馏的思想,利用基于进化的模型作为“老师”去指导仅基于序列的“学生”模型去获得一个比较理想的结果.这些方法预测一个蛋白根据计算资源和长度的不同,计算时间基本在毫秒到秒级,同时不依赖于共进化信息.这种优势对于缺少同源信息的酶设计改造来说,是非常有必要的. ...
... [83],华深智药提出的OmegaFold[82], 上海天壤科技开发的TRFold方法,山东大学杨建益团队提出的trRosettaX-Single[81]等方法.上述方法基本思路差别不大,各个团队在模型框架上存在一些技巧的差别.例如,trRosettaX-Single采用了知识蒸馏的思想,利用基于进化的模型作为“老师”去指导仅基于序列的“学生”模型去获得一个比较理想的结果.这些方法预测一个蛋白根据计算资源和长度的不同,计算时间基本在毫秒到秒级,同时不依赖于共进化信息.这种优势对于缺少同源信息的酶设计改造来说,是非常有必要的. ...
Accurate prediction of protein structures and interactions using a three-track neural network
2
2021
... 第一部分中我们提到,对于酶的改造和设计这两个应用场景,设计新酶的折叠能力是至关重要的.不论是在给定结构还是在给定功能约束下,设计的新酶如果不能正常折叠或者折叠后偏离预设结构,则减弱甚至丧失给定的功能.因此在设计过程中结合设计后新酶的折叠状态,相比不考虑再去实验验证筛选(几千几万条),在时间和实验成本上都占有优势.然而,折叠后的构象,实际上就是蛋白质结构预测的目标.结合第二部分中对蛋白质结构预测工具的梳理,可以看到蛋白质结构预测在人工智能强大的拟合能力帮助下最近几年来获得了突破性的进展.许多蛋白质结构预测工具由于预测的高效快速被广泛应用,例如:trRosetta[16],RoseTTAFold[84]等.那么,从设计酶的“可折叠性”出发,探索将蛋白质结构预测工具与现有的酶设计改造方法相结合,将会是一条有效的酶智能设计改造思路,有助于探索更为广阔的蛋白质序列空间. ...
... 在介绍应用的时候,我们提出三种应用方式.这三种应用的前提均是认为AlphaFold2这类蛋白质结构预测工具学习到了蛋白质序列到结构的复杂关系,对蛋白质结构的全局以及局部结构预测的准确度是可信的.随着越来越多结构预测工具的开发,根据不同任务(无同源序列),数据(α螺旋结构比例较高)等,可以将AlphaFold2替换成其他的结构预测工具.例如上面提到的David Baker组提出的RFjoint[45]采用的就是他们组的另一个非常优秀的工具RoseTTAFold[84]. ...
Combinatorial assembly and design of enzymes
1
2023
... 考虑酶的“可折叠性”,最直观的解决办法是快速预测设计的新酶的结构,检验其是否具有给定结构.因此,第一种预测是将蛋白结构预测工具作为一个监督者,约束生成的序列具有折叠成给定结构的能力(如图1左上角)[41-45,85].这个思路实施起来的最大难点是从序列预测结构的精度限制.但是现在得益于结构预测的突破性进展,使的这种设计新酶成为可能.其基本思路是在设计序列的时候,加入一个辅助的“监督者”对于生成的序列是否可以折叠且具有给定的构象进行评分.根据得分对蛋白质序列通过基于梯度的,梯度自由的,或者神经网络构造的优化方法来更新序列.通过不断重复迭代这一过程,最终得到构象约束下的收敛序列.设计序列的时候一般遵从最小能量的原则.但是,我们不清楚给定的构象就一定是设计的这条序列折叠后的最低能量构象.因此结构预测做为“监督器”实际上去计算了在给定结构情况下蛋白质序列的最大联合概率. ...
Modeling alternate conformations with Alphafold2 via modification of the multiple sequence alignment
2
2021
... (1PLDDT (predicted local distance difference test): 描述每个残基预测的局部置信度,局部结构指标,0-100,越大越好2PAE (predicted aligned error): 估计氨基酸在每一个位置上的误差,成对计算指标3Disfavoured: 反应预测序列类型于当前结构微环境的“适配性”4TMscore (template modeling score): 衡量蛋白质结构间的相似度,全局结构指标,0-1,越大越好5RMSD (root-mean-square deviation): 表示两个蛋白结构对齐后的结构差异,一般认为小于2Å预测准确.越小越好(图中展示的代表性方法分别按照1,2,3三部分从上至下摘自文献[43,45],[42-43],[86-87])) ...
... 还有研究工作利用AlphaFold2研究不同的构象变化(图1左小角).AlphaFold2虽然在单体结构上训练,但是可被成功应用在多肽与蛋白质的复合物结构预测中[97].因此,合理推断AlphaFold2学习到了蛋白质在功能改变过程中构象的动态集合或者是由于突变导致的构象改变.有工作利用不同深度的MSA输入到AlphaFold2中去研究这种构象的异质性 (Conformational heterogeneity) [86].Guillem Casadevall提出了一种新的观点,将基于AlphaFold2的新模板策略结合分子动力学模拟,发现不同突变的色氨酸合酶的β亚基 (TrpB) 结构域的一些不同闭合模式[87]. ...
Estimating conformational heterogeneity of tryptophan synthase with a template-based Alphafold2 approach
2
2022
... (1PLDDT (predicted local distance difference test): 描述每个残基预测的局部置信度,局部结构指标,0-100,越大越好2PAE (predicted aligned error): 估计氨基酸在每一个位置上的误差,成对计算指标3Disfavoured: 反应预测序列类型于当前结构微环境的“适配性”4TMscore (template modeling score): 衡量蛋白质结构间的相似度,全局结构指标,0-1,越大越好5RMSD (root-mean-square deviation): 表示两个蛋白结构对齐后的结构差异,一般认为小于2Å预测准确.越小越好(图中展示的代表性方法分别按照1,2,3三部分从上至下摘自文献[43,45],[42-43],[86-87])) ...
... 还有研究工作利用AlphaFold2研究不同的构象变化(图1左小角).AlphaFold2虽然在单体结构上训练,但是可被成功应用在多肽与蛋白质的复合物结构预测中[97].因此,合理推断AlphaFold2学习到了蛋白质在功能改变过程中构象的动态集合或者是由于突变导致的构象改变.有工作利用不同深度的MSA输入到AlphaFold2中去研究这种构象的异质性 (Conformational heterogeneity) [86].Guillem Casadevall提出了一种新的观点,将基于AlphaFold2的新模板策略结合分子动力学模拟,发现不同突变的色氨酸合酶的β亚基 (TrpB) 结构域的一些不同闭合模式[87]. ...
Design in the DARK: learning deep generative models for de novo protein design
1
2022
... David T. Jones尝试将AlphaFold2引入固定骨架设计序列的过程中,以约束生成的序列能够折叠成给定的骨架,并且正交实验中也验证了分子动力学方法模拟的结构对AlphaFold2监督后的实验结构高度支持[41].其具体流程是:(1)生成初始蛋白序列.基于研究者之前提出的基于自回归的Transformer蛋白质序列生成模型[88]生成1000条初始序列.同时对于得到的序列用AlphaFold2预测其结构,并与要设计的骨架结构用TM-align[89]做结构比对.最后选择结构比对得分最高的那部分结构的序列为初始序列,不具有高结构置信度的序列则用丙氨酸填充.这样做的好处是保证初始的序列是可收敛的,否则可能序列太随机导致最后没办法折叠.(2)在序列空间中执行贪婪的半随机游走,逐步突变起始序列进行迭代的端到端的设计.这里面AlphaFold2的作用有两个,一个是预测序列结构,比较与要设计结构的距离直方图损失.根据损失是否减小来判断突变序列是否合理.另一个是确定该序列中哪一部分残基位点要被突变,修改.举例来说,从起始序列出发并通过AlphaFold2预测其结构以及每一个残基的pLDDT打分(衡量每个残基的局部结构合理性).这里,计算预测结构中的距离直方图并与要设计的骨架结构的直方图计算损失.同时,利用每个残基的pLDDT打分设置为序列位点是否要被采样的概率.得分较高代表此处残基是稳定的,反之则是下一次迭代序列设计采样的点.在下次迭代采样中,对于选定的采样位点进行饱和突变,直到距离直方图损失减小,才接受序列的突变采样.这样设置的好处是对于与要设计结构的高度匹配的序列不再改变,大量减少采样时间尽快收敛以及可能引起的负协同效应.作者在人工设计的Top7上进行测试,得到的序列结构不论是通过AlphaFold2,trRosetta还是基于片段从头折叠的方法,均被证实与要设计的骨架可能是同一种折叠.该工作应用AlphaFold2在初始序列设计上保证了与目标结构的局部高结构匹配度,同时在序列设计过程中利用AlphaFold2预测的结构与目标结构的距离直方图损失约束其设计序列保持全局结构相似性以及利用残基位点可信度增强局部残基结构稳定性.同年,S. Kashif Sadiq也在bioRxiv上提交AlphaDesign工作,基本思路也是利用AlphaFold2预测的结构与要设计的骨架结构的差异来限制调整序列的优化,采用的优化函数是基于进化的遗传算法来迭代生成序列[42].主要差别在于该方法利用预测结构的三维坐标信息差异构建目标函数优化而不仅仅是二维的配对距离直方图约束,可能在结构约束上更加有效.而且该方法扩展了可能的设计任务的范围,设计了一些长度在32到256的结构稳定的从头设计的具有不同折叠的单体蛋白,同源二聚体,异源二聚体,同源低聚物(三聚体到六聚体).Baker组提出的trDesign是第一个提出将结构预测工具trRosetta应用到蛋白质序列设计中的工作,考虑的也是二维距离直方图的损失来更新梯度,更新被表示为PSSM的序列[43].但是受限于trRosetta利用的是二维的结构约束,在正交验证中发现基于这种反向传播的方式设计序列不能很好的对三维结构进行编码.且上述三个工作均是基于给定骨架设计序列,限制了实际设计酶的应用需求.后来Baker组提出的“幻想” (hallucination) 的方法,不从给定骨架结构出发设计序列,而是考虑在这种目标结构缺失的条件下,是否能随机产生结构和序列.其实现是通过最大化设计序列的结构与随机背景序列的差异约束,从而约束该序列折叠后的结构具有一个典型的2维结构特性[44].实验中幻想了2000条序列,聚类后发现均可以在已有的PDB结构库中寻找到相似的折叠.实验验证的时候有62条是可溶表达的(实验验证了129条),且CD的圆二色谱和目标结构的二级结构分布吻合.相比传统设计验证的方法,仅仅129条实验验证且48%的成功率,极大的减少了人工验证的成本和时间.但是由于trRosetta精度有限以及二维结构约束的不足,在接下来的工作中将RoseTTAFold嵌入到具有给定motif的序列设计中[45].RoseTTAFold显示利用SE-3 Transformer预测三维结构坐标以及二维距离分布,大大提高了序列设计的准确性.在免疫相关蛋白中,成功设计出携带中和性抗体表位的蛋白以及与新冠病毒S突刺蛋白受体结合的ACE2类似物蛋白.后续提出的RFjoint,不再通过神经网络不断迭代推理以及反向传播来设计序列,而是将结构预测和序列设计两大任务结合起来,直接训练全新的模型[45].这样的好处是减少了反向推理时间大大减少了设计的时间成本. ...
TM-align: a protein structure alignment algorithm based on the TM-score
1
2005
... David T. Jones尝试将AlphaFold2引入固定骨架设计序列的过程中,以约束生成的序列能够折叠成给定的骨架,并且正交实验中也验证了分子动力学方法模拟的结构对AlphaFold2监督后的实验结构高度支持[41].其具体流程是:(1)生成初始蛋白序列.基于研究者之前提出的基于自回归的Transformer蛋白质序列生成模型[88]生成1000条初始序列.同时对于得到的序列用AlphaFold2预测其结构,并与要设计的骨架结构用TM-align[89]做结构比对.最后选择结构比对得分最高的那部分结构的序列为初始序列,不具有高结构置信度的序列则用丙氨酸填充.这样做的好处是保证初始的序列是可收敛的,否则可能序列太随机导致最后没办法折叠.(2)在序列空间中执行贪婪的半随机游走,逐步突变起始序列进行迭代的端到端的设计.这里面AlphaFold2的作用有两个,一个是预测序列结构,比较与要设计结构的距离直方图损失.根据损失是否减小来判断突变序列是否合理.另一个是确定该序列中哪一部分残基位点要被突变,修改.举例来说,从起始序列出发并通过AlphaFold2预测其结构以及每一个残基的pLDDT打分(衡量每个残基的局部结构合理性).这里,计算预测结构中的距离直方图并与要设计的骨架结构的直方图计算损失.同时,利用每个残基的pLDDT打分设置为序列位点是否要被采样的概率.得分较高代表此处残基是稳定的,反之则是下一次迭代序列设计采样的点.在下次迭代采样中,对于选定的采样位点进行饱和突变,直到距离直方图损失减小,才接受序列的突变采样.这样设置的好处是对于与要设计结构的高度匹配的序列不再改变,大量减少采样时间尽快收敛以及可能引起的负协同效应.作者在人工设计的Top7上进行测试,得到的序列结构不论是通过AlphaFold2,trRosetta还是基于片段从头折叠的方法,均被证实与要设计的骨架可能是同一种折叠.该工作应用AlphaFold2在初始序列设计上保证了与目标结构的局部高结构匹配度,同时在序列设计过程中利用AlphaFold2预测的结构与目标结构的距离直方图损失约束其设计序列保持全局结构相似性以及利用残基位点可信度增强局部残基结构稳定性.同年,S. Kashif Sadiq也在bioRxiv上提交AlphaDesign工作,基本思路也是利用AlphaFold2预测的结构与要设计的骨架结构的差异来限制调整序列的优化,采用的优化函数是基于进化的遗传算法来迭代生成序列[42].主要差别在于该方法利用预测结构的三维坐标信息差异构建目标函数优化而不仅仅是二维的配对距离直方图约束,可能在结构约束上更加有效.而且该方法扩展了可能的设计任务的范围,设计了一些长度在32到256的结构稳定的从头设计的具有不同折叠的单体蛋白,同源二聚体,异源二聚体,同源低聚物(三聚体到六聚体).Baker组提出的trDesign是第一个提出将结构预测工具trRosetta应用到蛋白质序列设计中的工作,考虑的也是二维距离直方图的损失来更新梯度,更新被表示为PSSM的序列[43].但是受限于trRosetta利用的是二维的结构约束,在正交验证中发现基于这种反向传播的方式设计序列不能很好的对三维结构进行编码.且上述三个工作均是基于给定骨架设计序列,限制了实际设计酶的应用需求.后来Baker组提出的“幻想” (hallucination) 的方法,不从给定骨架结构出发设计序列,而是考虑在这种目标结构缺失的条件下,是否能随机产生结构和序列.其实现是通过最大化设计序列的结构与随机背景序列的差异约束,从而约束该序列折叠后的结构具有一个典型的2维结构特性[44].实验中幻想了2000条序列,聚类后发现均可以在已有的PDB结构库中寻找到相似的折叠.实验验证的时候有62条是可溶表达的(实验验证了129条),且CD的圆二色谱和目标结构的二级结构分布吻合.相比传统设计验证的方法,仅仅129条实验验证且48%的成功率,极大的减少了人工验证的成本和时间.但是由于trRosetta精度有限以及二维结构约束的不足,在接下来的工作中将RoseTTAFold嵌入到具有给定motif的序列设计中[45].RoseTTAFold显示利用SE-3 Transformer预测三维结构坐标以及二维距离分布,大大提高了序列设计的准确性.在免疫相关蛋白中,成功设计出携带中和性抗体表位的蛋白以及与新冠病毒S突刺蛋白受体结合的ACE2类似物蛋白.后续提出的RFjoint,不再通过神经网络不断迭代推理以及反向传播来设计序列,而是将结构预测和序列设计两大任务结合起来,直接训练全新的模型[45].这样的好处是减少了反向推理时间大大减少了设计的时间成本. ...
Improving de novo protein binder design with deep learning
1
2022
... 在设计领域,有工作通过引入pAE等来自AlphaFold2的结构指标作为“筛选器”,为4个靶点受体蛋白设计了2万条伙伴(binder) 序列,并且做了相应的实验合成[90].最后发现基于pAE指标相比传统的Rosetta打分,筛选后的序列成功率在IL2RA以及LTK靶点上数量差异分别达到了8倍,30倍.这一数量变化证明了利用结构预测工具做“筛选器”的有效性. ...
Structure prediction and analysis of hepatitis E virus non-structural proteins from the replication and transcription machinery by AlphaFold2
1
2022
... 结构预测工具还可以作为一种辅助的结构分析,从预测的结构上分析其背后存在的催化机理,结合特异性等(图1右下角).通过分析突变体结构(AlphaFold2预测)与底物结合的复合物结构,来检验突变策略是否合适[91-94].Martin Bartas则利用AlphaFold2预测成功的蛋白质结构库,通过结构相似寻找具有Zα 结构域(高结构保守,与Z-DNA/Z-RNA结合形成)的蛋白结构[95].这种蛋白在相关文献报导中仅有8个,但是由于AlphaFold2对蛋白结构库的丰富,发现了185个推定可能有该结构域的蛋白质结构.Fengjiao Xin课题组利用AlphaFold2预测出酶序列的高精确度的合理结构,从结构角度上分析其催化性能,在与底物结合口袋附近的位点上进行合理突变,发现了高催化效率和或底物偏好性扩大的的突变体[96]. ...
Directed evolution engineering to improve activity of glucose dehydrogenase by increasing pocket hydrophobicity
0
2022
Analysis of insertions and extensions in the functional evolution of the ribonucleotide reductase family
0
2022
Eliminating host-guest incompatibility via enzyme mining enables the high-temperature production of N-acetylglucosamine
1
2023
... 结构预测工具还可以作为一种辅助的结构分析,从预测的结构上分析其背后存在的催化机理,结合特异性等(图1右下角).通过分析突变体结构(AlphaFold2预测)与底物结合的复合物结构,来检验突变策略是否合适[91-94].Martin Bartas则利用AlphaFold2预测成功的蛋白质结构库,通过结构相似寻找具有Zα 结构域(高结构保守,与Z-DNA/Z-RNA结合形成)的蛋白结构[95].这种蛋白在相关文献报导中仅有8个,但是由于AlphaFold2对蛋白结构库的丰富,发现了185个推定可能有该结构域的蛋白质结构.Fengjiao Xin课题组利用AlphaFold2预测出酶序列的高精确度的合理结构,从结构角度上分析其催化性能,在与底物结合口袋附近的位点上进行合理突变,发现了高催化效率和或底物偏好性扩大的的突变体[96]. ...
Searching for new Z-DNA/Z-RNA binding proteins based on structural similarity to experimentally validated zα domain
1
2022
... 结构预测工具还可以作为一种辅助的结构分析,从预测的结构上分析其背后存在的催化机理,结合特异性等(图1右下角).通过分析突变体结构(AlphaFold2预测)与底物结合的复合物结构,来检验突变策略是否合适[91-94].Martin Bartas则利用AlphaFold2预测成功的蛋白质结构库,通过结构相似寻找具有Zα 结构域(高结构保守,与Z-DNA/Z-RNA结合形成)的蛋白结构[95].这种蛋白在相关文献报导中仅有8个,但是由于AlphaFold2对蛋白结构库的丰富,发现了185个推定可能有该结构域的蛋白质结构.Fengjiao Xin课题组利用AlphaFold2预测出酶序列的高精确度的合理结构,从结构角度上分析其催化性能,在与底物结合口袋附近的位点上进行合理突变,发现了高催化效率和或底物偏好性扩大的的突变体[96]. ...
Engineering the active site pocket to enhance the catalytic efficiency of a novel feruloyl esterase derived from human intestinal bacteria Dorea formicigenerans
1
2022
... 结构预测工具还可以作为一种辅助的结构分析,从预测的结构上分析其背后存在的催化机理,结合特异性等(图1右下角).通过分析突变体结构(AlphaFold2预测)与底物结合的复合物结构,来检验突变策略是否合适[91-94].Martin Bartas则利用AlphaFold2预测成功的蛋白质结构库,通过结构相似寻找具有Zα 结构域(高结构保守,与Z-DNA/Z-RNA结合形成)的蛋白结构[95].这种蛋白在相关文献报导中仅有8个,但是由于AlphaFold2对蛋白结构库的丰富,发现了185个推定可能有该结构域的蛋白质结构.Fengjiao Xin课题组利用AlphaFold2预测出酶序列的高精确度的合理结构,从结构角度上分析其催化性能,在与底物结合口袋附近的位点上进行合理突变,发现了高催化效率和或底物偏好性扩大的的突变体[96]. ...
Harnessing protein folding neural networks for peptide-protein docking
1
2022
... 还有研究工作利用AlphaFold2研究不同的构象变化(图1左小角).AlphaFold2虽然在单体结构上训练,但是可被成功应用在多肽与蛋白质的复合物结构预测中[97].因此,合理推断AlphaFold2学习到了蛋白质在功能改变过程中构象的动态集合或者是由于突变导致的构象改变.有工作利用不同深度的MSA输入到AlphaFold2中去研究这种构象的异质性 (Conformational heterogeneity) [86].Guillem Casadevall提出了一种新的观点,将基于AlphaFold2的新模板策略结合分子动力学模拟,发现不同突变的色氨酸合酶的β亚基 (TrpB) 结构域的一些不同闭合模式[87]. ...
Learning deep representations of enzyme thermal adaptation
1
2022
... 第二个难点是对于深度学习模型来说,从海量数据中挖掘模式是合适的.但是现有的状况是酶的相关数据量小,没有统一的标准格式,冗余的.当然这也与特定学科有关系.很多研究工作利用迁移学习来解决数据量小的问题,比如DeepET在大的蛋白质序列-最佳生长温度 (OGT) 数据集上训练模型,然后迁移到预测酶的最佳催化温度和蛋白质的熔融温度[98].或者利用自然语言处理 (NLP) 中广泛使用的大规模语言预训练模型学习序列的表示,然后小数据集上微调,进行一些功能预测[21,26]. ...



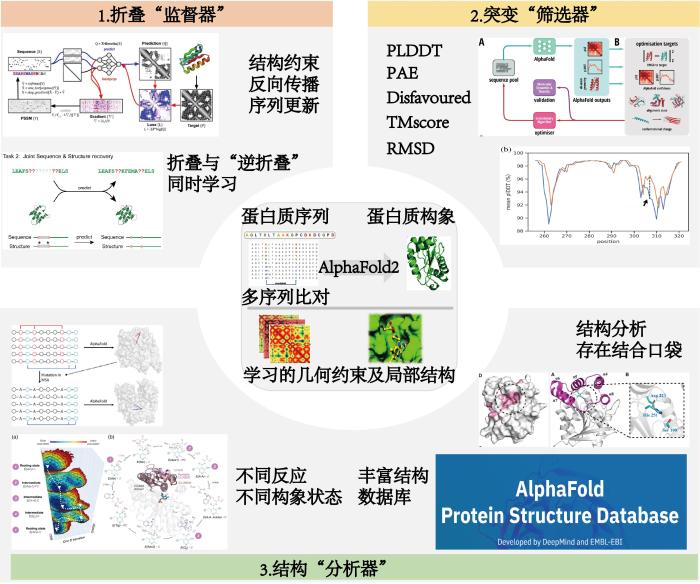


{kind=link}
{kind=link}