|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Challenges and opportunities in text mining-based protein function annotation
Synthetic Biology Journal
2025, 6 (3):
603-616.
DOI: 10.12211/2096-8280.2025-002
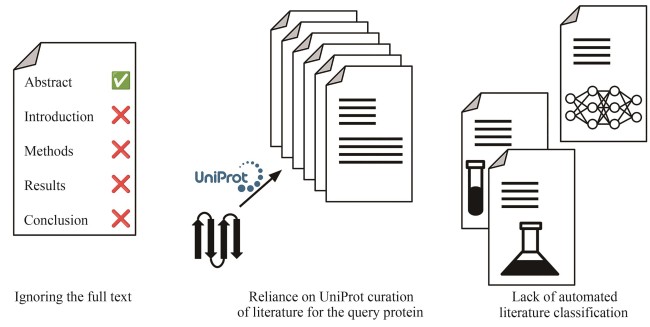
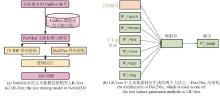
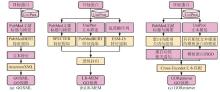
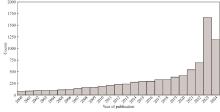
Understanding the biological function of proteins is crucial for advancing quantitative synthetic biology. Except for a small number of model organisms, most species contain many proteins whose functions have not been experimentally verified, necessitating the development of accurate, automated protein function annotation methods. Recent progress in protein bioinformatics, particularly in predicting protein structures and functions, has been driven significantly by the application of artificial intelligence (AI) algorithms, with a notable emphasis on deep learning models. For instance, the top-ranked methods in recent Critical Assessment of Function Annotation (CAFA) challenge have used deep learning models, primarily large language models, to perform text mining-based protein function annotation. These methods either predict Gene Ontology (GO) terms directly from text features extracted from scientific literatures or from template proteins with databases. Despite the extensive work in developing increasingly powerful deep learning models for text mining-based protein function annotation, several major challenges have been overlooked when parsing scientific literature data. This manuscript reviews existing methods and challenges in protein function annotation. First, many text mining-based protein function predictors rely exclusively on PubMed abstracts collected by UniProt curators for the query protein, ignoring literatures that have not been reviewed by biocurators. Consequently, protein functions predicted by text mining might overlap with those from manual curation of the UniProt Gene Ontology Annotation. Second, nearly all methods only parse PubMed abstracts, ignoring the more informative full-text documents often available in the PubMed Central and Europe PMC repositories. Third, few studies have been proposed to automatically differentiate between different categories of literatures, such as low and high throughput experiments, and computational predictions. This manuscript also proposes promising approaches to enhance text mining-based protein function annotation using the latest development in AI, which is expected to contribute to the development of next-generation text mining tools for more accurate function annotation.
Table 2
Existing methods for protein function prediction
Other Images/Table from this Article
|