Synthetic Biology Journal ›› 2025, Vol. 6 ›› Issue (3): 603-616.DOI: 10.12211/2096-8280.2025-002
• Invited Review • Previous Articles Next Articles
Challenges and opportunities in text mining-based protein function annotation
ZHANG Chengxin1,2
- 1.CAS Key Laboratory of Quantitative Engineering Biology,Shenzhen Institute of Synthetic Biology,Shenzhen Institutes of Advanced Technology,Chinese Academy of Sciences,Shenzhen 518055,Guangdong,China
2.Department of Computational Medicine & Bioinformatics,University of Michigan,Ann Arbor 48109,Michigan,USA
-
Received:2025-01-02Revised:2025-03-04Online:2025-06-27Published:2025-06-30 -
Contact:ZHANG Chengxin
基于文本数据挖掘的蛋白功能预测:机遇与挑战
张成辛1,2
- 1.中国科学院深圳先进技术研究院,深圳合成生物学创新研究院,中国科学院定量工程生物学重点实验室,广东 深圳 518055
2.密歇根大学计算医学与生物信息学院,美国 密歇根州 安娜堡 48109
-
通讯作者:张成辛 -
作者简介:张成辛 (1991—),男,博士,研究员。研究方向为蛋白质与RNA的结构与功能预测。E-mail:cx.zhang2@siat.ac.cn
CLC Number:
Cite this article
ZHANG Chengxin. Challenges and opportunities in text mining-based protein function annotation[J]. Synthetic Biology Journal, 2025, 6(3): 603-616.
张成辛. 基于文本数据挖掘的蛋白功能预测:机遇与挑战[J]. 合成生物学, 2025, 6(3): 603-616.
share this article
Add to citation manager EndNote|Ris|BibTeX
URL: https://synbioj.cip.com.cn/EN/10.12211/2096-8280.2025-002
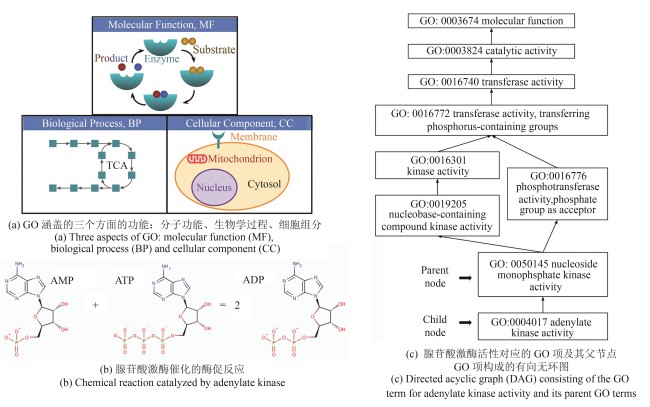
Fig. 1 Illustration of Gene Ontology (GO)
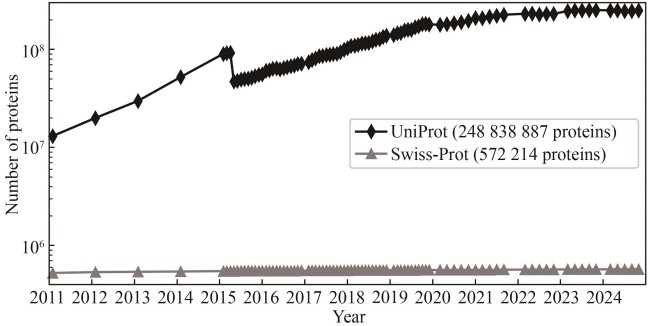
Fig. 2 Accumulation of protein entries in the UniProt and Swiss-Prot databases within the past 14 years(The drop in the number of UniProt proteins in 2015 was caused by the removal of redundant microbial proteins, i.e., if two proteins are from different strains or isolates of the same species are almost identical, only one protein is kept.)
| 证据编码 | 详细解释 |
|---|---|
| Inferred from Experiment (EXP) | 实验验证的生物功能 |
| Inferred from Direct Assay(IDA) | 生物化学或细胞生物学实验验证的生物功能 |
| Inferred from Physical Interaction(IPI) | 实验验证的蛋白-蛋白、蛋白-核酸或蛋白-小分子配体相互作用 |
| Inferred from Mutant Phenotype(IMP) | 根据同一个基因的两个等位基因的功能差异推测的生物功能 |
| Inferred from Genetic Interaction(IGI) | 涉及两个或以上的基因的序列改变或者表达量改变的实验验证的生物功能 |
| Inferred from Expression Pattern(IEP) | 根据基因表达的位置或者基因表达时间推测的生物过程 |
| Inferred from High Throughput Experiment(HTP) | 高通量实验验证的生物功能 |
| Inferred from High Throughput Direct Assay(HDA) | 高通量生物化学实验或高通量细胞生物学实验验证的生物功能 |
| Inferred from High Throughput Mutant Phenotype(HMP) | 根据高通量实验中一个基因的两个等位基因的功能差异推测的生物功能 |
| Inferred from Hight Throughput Genetic Interaction(HGI) | 涉及两个或以上的基因的序列改变或者表达量改变的高通量实验验证的生物功能 |
| Inferred from High Throughput Expression Pattern(HEP) | 根据高通量实验中基因表达的位置或者基因表达时间推测的生物过程 |
| Inferred from Sequence or Structural Similarity (ISS) | 根据序列分析或者结构相似性预测并经过人工审核的生物功能 |
| Inferred from Sequence Orthology(ISO) | 根据直系同源关系预测并经过人工审核的生物功能 |
| Inferred from Sequence Alignment(ISA) | 根据序列比对预测的生物功能;功能预测与序列比对本身都经过人工审核 |
| Inferred from Sequence Model(ISM) | 基于隐马尔科夫模型(如Pfam)等蛋白家族的统计模型预测并经过人工审核的生物功能 |
| Inferred from Genomic Context(IGC) | 根据目标基因在基因组上邻近的其他基因元件预测并经过人工审核的生物功能 |
| Inferred from Reviewed Computational Analysis(RCA) | 根据大规模实验数据(如酵母双杂交、质谱、基因芯片)预测或者结合多种类型的数据预测并经过人工审核的生物功能 |
| Inferred from Biological Aspect of Ancestor(IBA) | 根据系统发生树中的先祖基因的功能推测的后代基因的生物功能 |
| Inferred from Biological Aspect of Descendant(IBD) | 根据系统发生树中的后代基因的功能推测的先祖基因的生物功能 |
| Inferred from Key Residues(IKR) | 根据关键氨基酸残基缺失推测的生物功能缺失 |
| Inferred from Rapid Divergence(IRD) | 根据后代基因与先祖基因在进化上的快速分歧推断的生物功能缺失 |
| Traceable Author Statement(TAS) | 根据综述文献或者实验文献的介绍或讨论章节中的引用文献总结的生物功能 |
| Non-traceable Author Statement(NAS) | 根据文献中没有明确实验依据或引用支持的文字描述总结的生物功能 |
| Inferred by Curator(IC) | 根据蛋白的已有功能注释推测的相关生物功能;例如,根据一个真核蛋白的已知功能“RNA聚合酶Ⅱ活性”推测该蛋白应具有功能注释“细胞核” |
| Inferred from Electronic Annotation(IEA) | 无人工审核的计算预测得到的生物功能 |
Table 1 Evidence codes used for Gene Ontology annotation
| 证据编码 | 详细解释 |
|---|---|
| Inferred from Experiment (EXP) | 实验验证的生物功能 |
| Inferred from Direct Assay(IDA) | 生物化学或细胞生物学实验验证的生物功能 |
| Inferred from Physical Interaction(IPI) | 实验验证的蛋白-蛋白、蛋白-核酸或蛋白-小分子配体相互作用 |
| Inferred from Mutant Phenotype(IMP) | 根据同一个基因的两个等位基因的功能差异推测的生物功能 |
| Inferred from Genetic Interaction(IGI) | 涉及两个或以上的基因的序列改变或者表达量改变的实验验证的生物功能 |
| Inferred from Expression Pattern(IEP) | 根据基因表达的位置或者基因表达时间推测的生物过程 |
| Inferred from High Throughput Experiment(HTP) | 高通量实验验证的生物功能 |
| Inferred from High Throughput Direct Assay(HDA) | 高通量生物化学实验或高通量细胞生物学实验验证的生物功能 |
| Inferred from High Throughput Mutant Phenotype(HMP) | 根据高通量实验中一个基因的两个等位基因的功能差异推测的生物功能 |
| Inferred from Hight Throughput Genetic Interaction(HGI) | 涉及两个或以上的基因的序列改变或者表达量改变的高通量实验验证的生物功能 |
| Inferred from High Throughput Expression Pattern(HEP) | 根据高通量实验中基因表达的位置或者基因表达时间推测的生物过程 |
| Inferred from Sequence or Structural Similarity (ISS) | 根据序列分析或者结构相似性预测并经过人工审核的生物功能 |
| Inferred from Sequence Orthology(ISO) | 根据直系同源关系预测并经过人工审核的生物功能 |
| Inferred from Sequence Alignment(ISA) | 根据序列比对预测的生物功能;功能预测与序列比对本身都经过人工审核 |
| Inferred from Sequence Model(ISM) | 基于隐马尔科夫模型(如Pfam)等蛋白家族的统计模型预测并经过人工审核的生物功能 |
| Inferred from Genomic Context(IGC) | 根据目标基因在基因组上邻近的其他基因元件预测并经过人工审核的生物功能 |
| Inferred from Reviewed Computational Analysis(RCA) | 根据大规模实验数据(如酵母双杂交、质谱、基因芯片)预测或者结合多种类型的数据预测并经过人工审核的生物功能 |
| Inferred from Biological Aspect of Ancestor(IBA) | 根据系统发生树中的先祖基因的功能推测的后代基因的生物功能 |
| Inferred from Biological Aspect of Descendant(IBD) | 根据系统发生树中的后代基因的功能推测的先祖基因的生物功能 |
| Inferred from Key Residues(IKR) | 根据关键氨基酸残基缺失推测的生物功能缺失 |
| Inferred from Rapid Divergence(IRD) | 根据后代基因与先祖基因在进化上的快速分歧推断的生物功能缺失 |
| Traceable Author Statement(TAS) | 根据综述文献或者实验文献的介绍或讨论章节中的引用文献总结的生物功能 |
| Non-traceable Author Statement(NAS) | 根据文献中没有明确实验依据或引用支持的文字描述总结的生物功能 |
| Inferred by Curator(IC) | 根据蛋白的已有功能注释推测的相关生物功能;例如,根据一个真核蛋白的已知功能“RNA聚合酶Ⅱ活性”推测该蛋白应具有功能注释“细胞核” |
| Inferred from Electronic Annotation(IEA) | 无人工审核的计算预测得到的生物功能 |
| 方法 | 功能预测的信息来源(特征) | 机器学习模型 |
|---|---|---|
| GOtcha、Blast2GO、BAR+ | BLASTp搜索得到的同源序列 | 无 |
| ConFunc、PFP、GoFDR | PSI-BLAST搜索得到的同源序列 | 无 |
| HFSP | MMseqs2搜索得到的同源序列 | 无 |
| ProFunc | BLASTp搜索得到的同源序列、SSM与Jess结构搜索得到的相似结构 | 无 |
| COFACTOR | BLASTp与PSI-BLAST搜索得到的同源序列、TM-align结构搜索得到的相似结构、蛋白-蛋白互作 | 无 |
| MetaGO | BLASTp与PSI-BLAST搜索得到的同源序列、TM-align结构搜索得到的相似结构、蛋白-蛋白互作 | 逻辑回归 |
| StarFunc | BLASTp搜索得到的同源序列、Foldseek与TM-align结构搜索得到的相似结构、Pfam蛋白结构域家族、蛋白-蛋白互作、目标蛋白序列(ESM蛋白语言模型提取的特征) | 逻辑回归、全连接神经网络、随机森林 |
| DeepFRI、Struct2Go | 三维结构提取的残基接触图、目标蛋白序列(独热编码) | 图卷积神经网络 |
| TALE-cmap | 三维结构提取的残基接触图、多序列比对(ESM-MSA蛋白语言模型提取的特征) | Transformer |
| CLEAN-Contact | 三维结构提取的残基接触图、目标蛋白序列(ESM蛋白语言模型提取的特征) | 卷积神经网络 |
| MS-kNN | 同源序列、基因表达谱、蛋白-蛋白互作 | k-最近邻 |
| INGA | BLASTp搜索得到的同源序列、蛋白-蛋白互作、Pfam蛋白结构域家族 | 无 |
| GOLabeler | BLASTp搜索得到的同源序列、InterPro蛋白结构域家族、目标蛋白序列(连续三个氨基酸残基序列片段的频率、ProFET程序提取的序列特征) | 逻辑回归、梯度增强树 |
| NetGO | BLASTp搜索得到的同源序列、InterPro蛋白结构域家族、蛋白-蛋白互作、目标蛋白序列(连续三个氨基酸残基序列片段的频率、ProFET程序提取的序列特征) | 逻辑回归、梯度增强树 |
| NetGO2.0 | BLASTp搜索得到的同源序列、InterPro蛋白结构域家族、蛋白-蛋白互作、目标蛋白序列(连续三个氨基酸残基序列片段的频率、独热编码)、PubMed摘要 | 逻辑回归、双向长短期记忆神经网络、梯度增强树 |
| DeepGO、DeepGOplus、ProteInfer、DeepEC、ECPICK | 目标蛋白序列(独热编码) | 卷积神经网络 |
| ATGO+ | BLASTp搜索得到的同源序列、目标蛋白序列(ESM蛋白语言模型提取的特征) | 全连接神经网络 |
| InterLabelGO+ | DIAMOND搜索得到的同源序列、目标蛋白序列(ESM蛋白语言模型提取的特征) | 全连接神经网络 |
| DeepGO-SE | 目标蛋白序列(ESM蛋白语言模型提取的特征)、蛋白-蛋白互作 | 全连接神经网络、图注意力网络 |
| DeepECtransformer | DIAMOND搜索得到的同源序列、目标蛋白序列(ESM蛋白语言模型提取的特征) | 注意力网络 |
| CLEAN | 目标蛋白序列(ESM蛋白语言模型提取的特征) | 全连接神经网络 |
Table 2 Existing methods for protein function prediction
| 方法 | 功能预测的信息来源(特征) | 机器学习模型 |
|---|---|---|
| GOtcha、Blast2GO、BAR+ | BLASTp搜索得到的同源序列 | 无 |
| ConFunc、PFP、GoFDR | PSI-BLAST搜索得到的同源序列 | 无 |
| HFSP | MMseqs2搜索得到的同源序列 | 无 |
| ProFunc | BLASTp搜索得到的同源序列、SSM与Jess结构搜索得到的相似结构 | 无 |
| COFACTOR | BLASTp与PSI-BLAST搜索得到的同源序列、TM-align结构搜索得到的相似结构、蛋白-蛋白互作 | 无 |
| MetaGO | BLASTp与PSI-BLAST搜索得到的同源序列、TM-align结构搜索得到的相似结构、蛋白-蛋白互作 | 逻辑回归 |
| StarFunc | BLASTp搜索得到的同源序列、Foldseek与TM-align结构搜索得到的相似结构、Pfam蛋白结构域家族、蛋白-蛋白互作、目标蛋白序列(ESM蛋白语言模型提取的特征) | 逻辑回归、全连接神经网络、随机森林 |
| DeepFRI、Struct2Go | 三维结构提取的残基接触图、目标蛋白序列(独热编码) | 图卷积神经网络 |
| TALE-cmap | 三维结构提取的残基接触图、多序列比对(ESM-MSA蛋白语言模型提取的特征) | Transformer |
| CLEAN-Contact | 三维结构提取的残基接触图、目标蛋白序列(ESM蛋白语言模型提取的特征) | 卷积神经网络 |
| MS-kNN | 同源序列、基因表达谱、蛋白-蛋白互作 | k-最近邻 |
| INGA | BLASTp搜索得到的同源序列、蛋白-蛋白互作、Pfam蛋白结构域家族 | 无 |
| GOLabeler | BLASTp搜索得到的同源序列、InterPro蛋白结构域家族、目标蛋白序列(连续三个氨基酸残基序列片段的频率、ProFET程序提取的序列特征) | 逻辑回归、梯度增强树 |
| NetGO | BLASTp搜索得到的同源序列、InterPro蛋白结构域家族、蛋白-蛋白互作、目标蛋白序列(连续三个氨基酸残基序列片段的频率、ProFET程序提取的序列特征) | 逻辑回归、梯度增强树 |
| NetGO2.0 | BLASTp搜索得到的同源序列、InterPro蛋白结构域家族、蛋白-蛋白互作、目标蛋白序列(连续三个氨基酸残基序列片段的频率、独热编码)、PubMed摘要 | 逻辑回归、双向长短期记忆神经网络、梯度增强树 |
| DeepGO、DeepGOplus、ProteInfer、DeepEC、ECPICK | 目标蛋白序列(独热编码) | 卷积神经网络 |
| ATGO+ | BLASTp搜索得到的同源序列、目标蛋白序列(ESM蛋白语言模型提取的特征) | 全连接神经网络 |
| InterLabelGO+ | DIAMOND搜索得到的同源序列、目标蛋白序列(ESM蛋白语言模型提取的特征) | 全连接神经网络 |
| DeepGO-SE | 目标蛋白序列(ESM蛋白语言模型提取的特征)、蛋白-蛋白互作 | 全连接神经网络、图注意力网络 |
| DeepECtransformer | DIAMOND搜索得到的同源序列、目标蛋白序列(ESM蛋白语言模型提取的特征) | 注意力网络 |
| CLEAN | 目标蛋白序列(ESM蛋白语言模型提取的特征) | 全连接神经网络 |
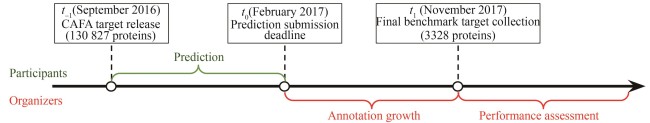
Fig. 3 Timeline of CAFA3
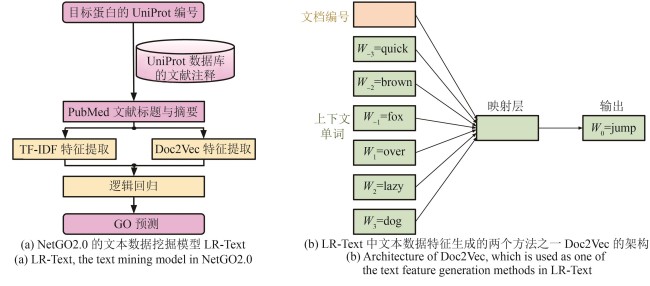
Fig. 4 Text mining-based protein GO term prediction in NetGO2.0(In this example, the Doc2Vec neural network model is trained to predict the masked word “jump” given its context in the sentence “The quick brown fox ___ over the lazy dog.” The word “the” is excluded from the input sentence as it does not have meaningful information.)
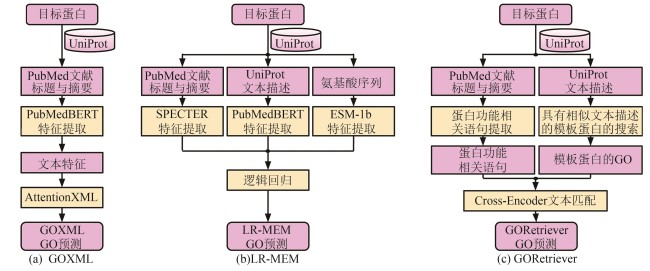
Fig. 5 Three text mining-based models for protein GO term prediction in GOCurator
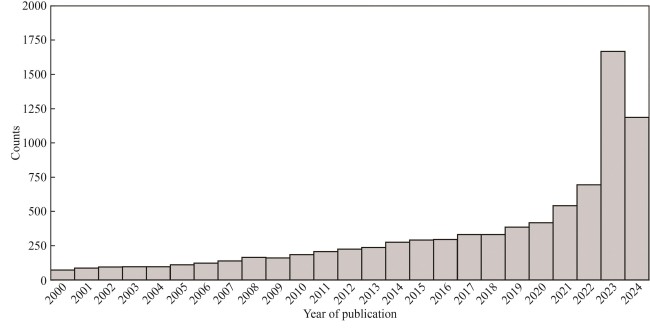
Fig. 6 Publication year for 8935 PubMed citations added to UniProt in 2024(549 papers published in or before year 1999 are not shown)
| 1 | ASHBURNER M, BALL C A, BLAKE J A, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium[J]. Nature Genetics, 2000, 25(1): 25-29. |
| 2 | International Union of Biochemistry, Nomenclature Committee. Enzyme nomenclature, 1978: recommendations of the Nomenclature Committee of the International Union of Biochemistry on the nomenclature and classification of enzymes[M]. New York: Academic Press, 1979. |
| 3 | GARGANO M A, MATENTZOGLU N, COLEMAN B, et al. The human phenotype ontology in 2024: phenotypes around the world[J]. Nucleic Acids Research, 2024, 52(D1): D1333-D1346. |
| 4 | The UniProt Consortium. UniProt: the universal protein knowledgebase in 2025[J]. Nucleic Acids Research, 2025, 53(D1): D609–D617. |
| 5 | HUNTLEY R P, SAWFORD T, MUTOWO-MEULLENET P, et al. The GOA database: gene Ontology annotation updates for 2015[J]. Nucleic Acids Research, 2015, 43(Database issue): D1057-D1063. |
| 6 | FELDMANN P, EICHER E N, LEEVERS S J, et al. Control of growth and differentiation by Drosophila RasGAP, a homolog of p120 ras-GTPase-activating protein[J]. Molecular and Cellular Biology, 1999, 19(3): 1928-1937. |
| 7 | GAUDET P, LIVSTONE M S, LEWIS S E, et al. Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium[J]. Briefings in Bioinformatics, 2011, 12(5): 449-462. |
| 8 | WEI X Q, ZHANG C X, FREDDOLINO P L, et al. Detecting Gene Ontology misannotations using taxon-specific rate ratio comparisons[J]. Bioinformatics, 2020, 36(16): 4383-4388. |
| 9 | MARTIN D M A, BERRIMAN M, BARTON G J. GOtcha: a new method for prediction of protein function assessed by the annotation of seven genomes[J]. BMC Bioinformatics, 2004, 5: 178. |
| 10 | CONESA A, GÖTZ S. Blast2GO: a comprehensive suite for functional analysis in plant genomics[J]. International Journal of Plant Genomics, 2008, 2008(1): 619832. |
| 11 | PIOVESAN D, MARTELLI P L, FARISELLI P, et al. BAR-PLUS: the Bologna Annotation Resource Plus for functional and structural annotation of protein sequences[J]. Nucleic Acids Research, 2011, 39(Web Server issue): W197-W202. |
| 12 | ALTSCHUL S F, MADDEN T L, SCHÄFFER A A, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs[J]. Nucleic Acids Research, 1997, 25(17): 3389-3402. |
| 13 | WASS M N, STERNBERG M J E. ConFunc: functional annotation in the twilight zone[J]. Bioinformatics, 2008, 24(6): 798-806. |
| 14 | HAWKINS T, CHITALE M, LUBAN S, et al. PFP Automated prediction of gene ontology functional annotations with confidence scores using protein sequence data[J]. Proteins: Structure, Function, and Bioinformatics, 2009, 74(3): 566-582. |
| 15 | GONG Q T, NING W, TIAN W D. GoFDR: a sequence alignment based method for predicting protein functions[J]. Methods, 2016, 93: 3-14. |
| 16 | MAHLICH Y, STEINEGGER M, ROST B, et al. HFSP: high speed homology-driven function annotation of proteins[J]. Bioinformatics, 2018, 34(13): i304-i312. |
| 17 | STEINEGGER M, SÖDING J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets[J]. Nature Biotechnology, 2017, 35(11): 1026-1028. |
| 18 | KULMANOV M, HOEHNDORF R. DeepGOPlus: improved protein function prediction from sequence[J]. Bioinformatics, 2020, 36(2): 422-429. |
| 19 | KULMANOV M, HOEHNDORF R. DeepGOZero: improving protein function prediction from sequence and zero-shot learning based on ontology axioms[J]. Bioinformatics, 2022, 38(S1): i238-i245. |
| 20 | YUAN Q M, XIE J J, XIE J C, et al. Fast and accurate protein function prediction from sequence through pretrained language model and homology-based label diffusion[J]. Briefings in Bioinformatics, 2023, 24(3): bbad117. |
| 21 | BUCHFINK B, REUTER K, DROST H G. Sensitive protein alignments at tree-of-life scale using DIAMOND[J]. Nature Methods, 2021, 18(4): 366-368. |
| 22 | ZHANG C X, LYDIA FREDDOLINO P. A large-scale assessment of sequence database search tools for homology-based protein function prediction[EB/OL]. bioRxiv, 2023: 2023.11.14.567021. (2023-11-16)[2024-12-01]. . |
| 23 | ZHANG C X, FREDDOLINO L, ZHANG Y. COFACTOR: improved protein function prediction by combining structure, sequence and protein-protein interaction information[J]. Nucleic Acids Research, 2017, 45(W1): W291-W299. |
| 24 | ZHANG C X, ZHENG W, FREDDOLINO P L, et al. MetaGO: predicting gene ontology of non-homologous proteins through low-resolution protein structure prediction and protein-protein network mapping[J]. Journal of Molecular Biology, 2018, 430(15): 2256-2265. |
| 25 | ZHANG Y, SKOLNICK J. TM-align: a protein structure alignment algorithm based on the TM-score[J]. Nucleic Acids Research, 2005, 33(7): 2302-2309. |
| 26 | ZHANG C X, ZHANG X, FREDDOLINO L, et al. BioLiP2: an updated structure database for biologically relevant ligand-protein interactions[J]. Nucleic Acids Research, 2024, 52(D1): D404-D412. |
| 27 | LASKOWSKI R A. The ProFunc function prediction server[J]. Methods in Molecular Biology, 2017, 1611: 75-95. |
| 28 | KRISSINEL E, HENRICK K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions[J]. Acta Crystallographica Section D, Biological Crystallography, 2004, 60(Pt 12 Pt 1): 2256-2268. |
| 29 | BARKER J A, THORNTON J M. An algorithm for constraint-based structural template matching: application to 3D templates with statistical analysis[J]. Bioinformatics, 2003, 19(13): 1644-1649. |
| 30 | ZHANG C X, LIU Q C, FREDDOLINO L. StarFunc: fusing template-based and deep learning approaches for accurate protein function prediction[EB/OL]. bioRxiv, 2024: 2024.05.15.. |
| 31 | VAN KEMPEN M, KIM S S, TUMESCHEIT C, et al. Fast and accurate protein structure search with Foldseek[J]. Nature Biotechnology, 2024, 42(2): 243-246. |
| 32 | VARADI M, ANYANGO S, DESHPANDE M, et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models[J]. Nucleic Acids Research, 2022, 50(D1): D439-D444. |
| 33 | MISTRY J, CHUGURANSKY S, WILLIAMS L, et al. Pfam: the protein families database in 2021[J]. Nucleic Acids Research, 2021, 49(D1): D412-D419. |
| 34 | LIU Q C, ZHANG C X, FREDDOLINO L. InterLabelGO+: unraveling label correlations in protein function prediction[J]. Bioinformatics, 2024, 40(11): btae655. |
| 35 | GLIGORIJEVIĆ V, RENFREW P D, KOSCIOLEK T, et al. Structure-based protein function prediction using graph convolutional networks[J]. Nature Communications, 2021, 12(1): 3168. |
| 36 | MA W J, ZHANG S G, LI Z, et al. Enhancing protein function prediction performance by utilizing AlphaFold-predicted protein structures[J]. Journal of Chemical Information and Modeling, 2022, 62(17): 4008-4017. |
| 37 | QIU X Y, WU H, SHAO J Y. TALE-cmap: protein function prediction based on a TALE-based architecture and the structure information from contact map[J]. Computers in Biology and Medicine, 2022, 149: 105938. |
| 38 | YANG Y X, JERGER A, FENG S, et al. Improved enzyme functional annotation prediction using contrastive learning with structural inference[J]. Communications Biology, 2024, 7(1): 1690. |
| 39 | LAN L, DJURIC N, GUO Y H, et al. MS-kNN: protein function prediction by integrating multiple data sources[J]. BMC Bioinformatics, 2013, 14(): S8. |
| 40 | PIOVESAN D, TOSATTO S C E. INGA 2.0: improving protein function prediction for the dark proteome[J]. Nucleic Acids Research, 2019, 47(W1): W373-W378. |
| 41 | YOU R H, ZHANG Z H, XIONG Y, et al. GOLabeler: improving sequence-based large-scale protein function prediction by learning to rank[J]. Bioinformatics, 2018, 34(14): 2465-2473. |
| 42 | BLUM M, CHANG H Y, CHUGURANSKY S, et al. The InterPro protein families and domains database: 20 years on[J]. Nucleic Acids Research, 2021, 49(D1): D344-D354. |
| 43 | CHEN T Q, GUESTRIN C. XGBoost: a scalable tree boosting system[C/OL]//Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. San Francisco California USA. ACM, 2016: 785-794. (2016-08-13)[2024-12-01]. . |
| 44 | YOU R H, YAO S W, XIONG Y, et al. NetGO: improving large-scale protein function prediction with massive network information[J]. Nucleic Acids Research, 2019, 47(W1): W379-W387. |
| 45 | YAO S W, YOU R H, WANG S J, et al. NetGO 2.0: improving large-scale protein function prediction with massive sequence, text, domain, family and network information[J]. Nucleic Acids Research, 2021, 49(W1): W469-W475. |
| 46 | KULMANOV M, KHAN M A, HOEHNDORF R, et al. DeepGO: predicting protein functions from sequence and interactions using a deep ontology-aware classifier[J]. Bioinformatics, 2018, 34(4): 660-668. |
| 47 | SANDERSON T, BILESCHI M L, BELANGER D, et al. ProteInfer, deep neural networks for protein functional inference[J]. eLife, 2023, 12: e80942. |
| 48 | RYU J Y, KIM H U, LEE S Y. Deep learning enables high-quality and high-throughput prediction of enzyme commission numbers[J]. Proceedings of the National Academy of Sciences of the United States of America, 2019, 116(28): 13996-14001. |
| 49 | HAN S R, PARK M, KOSARAJU S, et al. Evidential deep learning for trustworthy prediction of enzyme commission number[J]. Briefings in Bioinformatics, 2023, 25(1): bbad401. |
| 50 | ZHU Y H, ZHANG C X, YU D J, et al. Integrating unsupervised language model with triplet neural networks for protein gene ontology prediction[J]. PLoS Computational Biology, 2022, 18(12): e1010793. |
| 51 | KULMANOV M, GUZMÁN-VEGA F J, ROGGLI P D, et al. DeepGO-SE: protein function prediction as Approximate Semantic Entailment[EB/OL]. bioRxiv, 2023: 2023.09.26.559473. (2023-09-28)[2024-12-01]. . |
| 52 | KIM G B, KIM J Y, LEE J A, et al. Functional annotation of enzyme-encoding genes using deep learning with transformer layers[J]. Nature Communications, 2023, 14(1): 7370. |
| 53 | YU T H, CUI H Y, LI J C, et al. Enzyme function prediction using contrastive learning[J]. Science, 2023, 379(6639): 1358-1363. |
| 54 | LIN Z M, AKIN H, RAO R, et al. Evolutionary-scale prediction of atomic-level protein structure with a language model[J]. Science, 2023, 379(6637): 1123-1130. |
| 55 | ELNAGGAR A, HEINZINGER M, DALLAGO C, et al. ProtTrans: toward understanding the language of life through self-supervised learning[J]. IEEE Transactions on Pattern Analysis and Machine Intelligence, 2022, 44(10): 7112-7127. |
| 56 | VASWANI A, SHAZEER N, PARMAR N, et al. Attention is all you need[C/OL]//Advances in Neural Information Processing Systems 30 (NIPS 2017), 2017[2024-12-01]. . |
| 57 | RADIVOJAC P, CLARK W T, ORON T R, et al. A large-scale evaluation of computational protein function prediction[J]. Nature Methods, 2013, 10(3): 221-227. |
| 58 | ZHOU N H, JIANG Y X, BERGQUIST T R, et al. The CAFA challenge reports improved protein function prediction and new functional annotations for hundreds of genes through experimental screens[J]. Genome Biology, 2019, 20(1): 244. |
| 59 | JIANG Y X, ORON T R, CLARK W T, et al. An expanded evaluation of protein function prediction methods shows an improvement in accuracy[J]. Genome Biology, 2016, 17(1): 184. |
| 60 | YAN H Y, WANG S J, LIU H C, et al. GORetriever: reranking protein-description-based GO candidates by literature-driven deep information retrieval for protein function annotation[J]. Bioinformatics, 2024, 40(S2): ii53-ii61. |
| 61 | CHUA Z M, RAJESH A, SINHA S, et al. PROTGOAT: improved automated protein function predictions using Protein Language Models[EB/OL]. bioRxiv, 2024: 2024.04. 01.587572 .(2024-04-02)[2024-12-01]. . |
| 62 | COZZETTO D, BUCHAN D W, BRYSON K, et al. Protein function prediction by massive integration of evolutionary analyses and multiple data sources[J]. BMC Bioinformatics, 2013, 14(3): S1. |
| 63 | YOU R H, HUANG X D, ZHU S F. DeepText2GO: improving large-scale protein function prediction with deep semantic text representation[J]. Methods, 2018, 145: 82-90. |
| 64 | LE Q, MIKOLOV T. Distributed representations of sentences and documents; proceedings of the international conference on machine learning[C/OL]//Proceedings of the 31st International Conference on Machine Learning, PMLR, 2014, 32(2): 1188-1196 [2024-12-04]. . |
| 65 | GU Y, TINN R, CHENG H, et al. Domain-specific language model pretraining for biomedical natural language processing[J]. ACM Transactions on Computing for Healthcare, 2021, 3(1): 1-23. |
| 66 | COHAN A, FELDMAN S, BELTAGY I, et al. SPECTER: document-level representation learning using citation-informed transformers[EB/OL]. arXiv, 2020: 200407180. (2020-05-20)[2024-12-01]. . |
| 67 | REIMERS N, GUREVYCH I. Sentence-BERT: Sentence Embeddings using Siamese BERT-Networks[EB/OLJ]. arXiv, 2019: 190810084. (2019-08-27)[2024-12-01]. . |
| 68 | WU J S, YIN Q, ZHANG C X, et al. Function prediction for G protein-coupled receptors through text mining and induction matrix completion[J]. ACS Omega, 2019, 4(2): 3045-3054. |
| 69 | BADAL V D, KUNDROTAS P J, VAKSER I A. Text mining for protein docking[J]. PLoS Computational Biology, 2015, 11(12): e1004630. |
| 70 | KAFKAS Ş, HOEHNDORF R. Ontology based text mining of gene-phenotype associations: application to candidate gene prediction[J]. Database, 2019, 2019: baz019. |
| 71 | CZARNECKI J, NOBELI I, SMITH A M, et al. A text-mining system for extracting metabolic reactions from full-text articles[J]. BMC Bioinformatics, 2012, 13: 172. |
| 72 | VERSPOOR K M, COHN J D, RAVIKUMAR K E, et al. Text mining improves prediction of protein functional sites[J]. PLoS One, 2012, 7(2): e32171. |
| 73 | WEI X Q, ZOU S, XIE Z H, et al. EDIL3 deficiency ameliorates adverse cardiac remodelling by neutrophil extracellular traps (NET)-mediated macrophage polarization[J]. Cardiovascular Research, 2022, 118(9): 2179-2195. |
| 74 | PAFILIS E, BUTTIGIEG P L, FERRELL B, et al. EXTRACT: interactive extraction of environment metadata and term suggestion for metagenomic sample annotation[J]. Database, 2016, 2016: baw005. |
| 75 | WEI C H, KAO H Y, LU Z Y. PubTator: a web-based text mining tool for assisting biocuration[J]. Nucleic Acids Research, 2013, 41(Web Server issue): W518-W522. |
| 76 | WEBER L, SÄNGER M, MÜNCHMEYER J, et al. HunFlair: an easy-to-use tool for state-of-the-art biomedical named entity recognition[J]. Bioinformatics, 2021, 37(17): 2792-2794. |
| 77 | GIORGI J M, BADER G D. Towards reliable named entity recognition in the biomedical domain[J]. Bioinformatics, 2020, 36(1): 280-286. |
| 78 | FURRER L, JANCSO A, COLIC N, et al. OGER++: hybrid multi-type entity recognition[J]. Journal of Cheminformatics, 2019, 11(1): 7. |
| [1] | LI Mingchen, ZHONG Bozitao, YU Yuanxi, JIANG Fan, ZHANG Liang, TAN Yang, YU Huiqun, FAN Guisheng, HONG Liang. DeepSeek model analysis and its applications in AI-assistant protein engineering [J]. Synthetic Biology Journal, 2025, 6(3): 636-650. |
| [2] | ZHENG Yikun, ZHENG Jie, HU Guopeng. Research on the application of optogenetic tools in learning and memory [J]. Synthetic Biology Journal, 2025, 6(1): 87-104. |
| [3] | DONG Ying, MA Mengdan, HUANG Weiren. Progress in the miniaturization of CRISPR-Cas systems [J]. Synthetic Biology Journal, 2025, 6(1): 105-117. |
| [4] | ZHU Jingyong, LI Junxiang, LI Xuhui, ZHANG Jin, WU Wenjing. Advances in applications of deep learning for predicting sequence-based protein interactions [J]. Synthetic Biology Journal, 2024, 5(1): 88-106. |
| [5] | CHEN Zhihang, JI Menglin, QI Yifei. Research progress of artificial intelligence in desiging protein structures [J]. Synthetic Biology Journal, 2023, 4(3): 464-487. |
| [6] | SONG Yidong, YUAN Qianmu, YANG Yuedong. Application of deep learning in protein function prediction [J]. Synthetic Biology Journal, 2023, 4(3): 488-506. |
| [7] | LIU Renmei, LI Leshi, YANG Xiaoyan, CHEN Xianjun, YANG Yi. Technologies for precise spatiotemporal control of post-transcriptional RNA metabolism [J]. Synthetic Biology Journal, 2023, 4(1): 141-164. |
| [8] | LYU Jingwei, DENG Zixin, ZHANG Qi, DING Wei. Identification of RiPPs precursor peptides and cleavage sites based on deep learning [J]. Synthetic Biology Journal, 2022, 3(6): 1262-1276. |
| [9] | WANG Ye, WANG Haochen, YAN Minghao, HU Guanhua, WANG Xiaowo. Design of biomolecular sequences by artificial intelligence [J]. Synthetic Biology Journal, 2021, 2(1): 1-14. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||