Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product
1
1987
... 对遗传信息进行编辑和对基因表达进行调控是研究各种遗传信息的具体功能、揭示基因独特生理机制的重要方法.进入新世纪后,来源于原核生物的簇状规则间隔短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR)/CRISPR相关蛋白(CRISPR-associated protein, Cas protein)系统蓬勃发展,成为应用于基因组工程中的重要技术.早在1987年,研究人员就在大肠杆菌基因组上发现了这一段特殊的序列[1].2002年,Jansen等[2]通过大量观察创造了CRISPR缩写代替了之前发现的重复片段.随后,Brouns等[3]揭示了该系统对DNA的靶向作用及原理,从而推测出该系统具有作为基因编辑工具的潜力.2013年,张锋团队[4]首次实现CRISPR/Cas9系统在哺乳动物细胞中的多基因编辑,开启了基因编辑技术的新篇章.依据Cas蛋白的结构和进化关系,目前CRISPR/Cas系统共分六种类型,其中Ⅰ、Ⅲ、Ⅳ型系统的Cas蛋白为多亚基,Ⅱ、Ⅴ、Ⅵ型系统的Cas蛋白为单亚基[5-6].由于单亚基CRISPR/Cas系统机制简单且易操作,目前研究成熟和应用广泛的大多为单亚基系统,如Ⅱ型的CRISPR/Cas9系统、Ⅴ型的CRISPR/Cas12a(Cpf1)系统和Ⅵ型的CRISPR/Cas13a(C2c2)系统等.其中CRISPR/Cas9系统凭借其操作简便、精准编辑、多重编辑等优势,与传统的DNA修饰工具如大范围核酸酶(meganucleases, MGNs)[7]、锌指核酸酶(zinc finger nucleases, ZFNs)[8]、转录激活因子样效应蛋白核酸酶(transcription activator-like effector nucleases, TALENs)[9]等区分开来,使其在基础科学到应用科学领域均得到了极大的发展与应用. ...
Identification of genes that are associated with DNA repeats in prokaryotes
1
2002
... 对遗传信息进行编辑和对基因表达进行调控是研究各种遗传信息的具体功能、揭示基因独特生理机制的重要方法.进入新世纪后,来源于原核生物的簇状规则间隔短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR)/CRISPR相关蛋白(CRISPR-associated protein, Cas protein)系统蓬勃发展,成为应用于基因组工程中的重要技术.早在1987年,研究人员就在大肠杆菌基因组上发现了这一段特殊的序列[1].2002年,Jansen等[2]通过大量观察创造了CRISPR缩写代替了之前发现的重复片段.随后,Brouns等[3]揭示了该系统对DNA的靶向作用及原理,从而推测出该系统具有作为基因编辑工具的潜力.2013年,张锋团队[4]首次实现CRISPR/Cas9系统在哺乳动物细胞中的多基因编辑,开启了基因编辑技术的新篇章.依据Cas蛋白的结构和进化关系,目前CRISPR/Cas系统共分六种类型,其中Ⅰ、Ⅲ、Ⅳ型系统的Cas蛋白为多亚基,Ⅱ、Ⅴ、Ⅵ型系统的Cas蛋白为单亚基[5-6].由于单亚基CRISPR/Cas系统机制简单且易操作,目前研究成熟和应用广泛的大多为单亚基系统,如Ⅱ型的CRISPR/Cas9系统、Ⅴ型的CRISPR/Cas12a(Cpf1)系统和Ⅵ型的CRISPR/Cas13a(C2c2)系统等.其中CRISPR/Cas9系统凭借其操作简便、精准编辑、多重编辑等优势,与传统的DNA修饰工具如大范围核酸酶(meganucleases, MGNs)[7]、锌指核酸酶(zinc finger nucleases, ZFNs)[8]、转录激活因子样效应蛋白核酸酶(transcription activator-like effector nucleases, TALENs)[9]等区分开来,使其在基础科学到应用科学领域均得到了极大的发展与应用. ...
Small CRISPR RNAs guide antiviral defense in prokaryotes
1
2008
... 对遗传信息进行编辑和对基因表达进行调控是研究各种遗传信息的具体功能、揭示基因独特生理机制的重要方法.进入新世纪后,来源于原核生物的簇状规则间隔短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR)/CRISPR相关蛋白(CRISPR-associated protein, Cas protein)系统蓬勃发展,成为应用于基因组工程中的重要技术.早在1987年,研究人员就在大肠杆菌基因组上发现了这一段特殊的序列[1].2002年,Jansen等[2]通过大量观察创造了CRISPR缩写代替了之前发现的重复片段.随后,Brouns等[3]揭示了该系统对DNA的靶向作用及原理,从而推测出该系统具有作为基因编辑工具的潜力.2013年,张锋团队[4]首次实现CRISPR/Cas9系统在哺乳动物细胞中的多基因编辑,开启了基因编辑技术的新篇章.依据Cas蛋白的结构和进化关系,目前CRISPR/Cas系统共分六种类型,其中Ⅰ、Ⅲ、Ⅳ型系统的Cas蛋白为多亚基,Ⅱ、Ⅴ、Ⅵ型系统的Cas蛋白为单亚基[5-6].由于单亚基CRISPR/Cas系统机制简单且易操作,目前研究成熟和应用广泛的大多为单亚基系统,如Ⅱ型的CRISPR/Cas9系统、Ⅴ型的CRISPR/Cas12a(Cpf1)系统和Ⅵ型的CRISPR/Cas13a(C2c2)系统等.其中CRISPR/Cas9系统凭借其操作简便、精准编辑、多重编辑等优势,与传统的DNA修饰工具如大范围核酸酶(meganucleases, MGNs)[7]、锌指核酸酶(zinc finger nucleases, ZFNs)[8]、转录激活因子样效应蛋白核酸酶(transcription activator-like effector nucleases, TALENs)[9]等区分开来,使其在基础科学到应用科学领域均得到了极大的发展与应用. ...
Multiplex genome engineering using CRISPR/Cas systems
7
2013
... 对遗传信息进行编辑和对基因表达进行调控是研究各种遗传信息的具体功能、揭示基因独特生理机制的重要方法.进入新世纪后,来源于原核生物的簇状规则间隔短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR)/CRISPR相关蛋白(CRISPR-associated protein, Cas protein)系统蓬勃发展,成为应用于基因组工程中的重要技术.早在1987年,研究人员就在大肠杆菌基因组上发现了这一段特殊的序列[1].2002年,Jansen等[2]通过大量观察创造了CRISPR缩写代替了之前发现的重复片段.随后,Brouns等[3]揭示了该系统对DNA的靶向作用及原理,从而推测出该系统具有作为基因编辑工具的潜力.2013年,张锋团队[4]首次实现CRISPR/Cas9系统在哺乳动物细胞中的多基因编辑,开启了基因编辑技术的新篇章.依据Cas蛋白的结构和进化关系,目前CRISPR/Cas系统共分六种类型,其中Ⅰ、Ⅲ、Ⅳ型系统的Cas蛋白为多亚基,Ⅱ、Ⅴ、Ⅵ型系统的Cas蛋白为单亚基[5-6].由于单亚基CRISPR/Cas系统机制简单且易操作,目前研究成熟和应用广泛的大多为单亚基系统,如Ⅱ型的CRISPR/Cas9系统、Ⅴ型的CRISPR/Cas12a(Cpf1)系统和Ⅵ型的CRISPR/Cas13a(C2c2)系统等.其中CRISPR/Cas9系统凭借其操作简便、精准编辑、多重编辑等优势,与传统的DNA修饰工具如大范围核酸酶(meganucleases, MGNs)[7]、锌指核酸酶(zinc finger nucleases, ZFNs)[8]、转录激活因子样效应蛋白核酸酶(transcription activator-like effector nucleases, TALENs)[9]等区分开来,使其在基础科学到应用科学领域均得到了极大的发展与应用. ...
... CRISPR系统利用单链向导RNA(single guide RNA,sgRNA或gRNA)靶向目的位点,随后Cas9等核酸酶切断目的区域的双链核酸,诱导细胞内源DNA损伤修复机制工作,最终借助该机制达到编辑遗传信息的目的[10].CRISPR/Cas9系统在发挥基因敲除、基因插入、单碱基编辑等主要编辑手段时主要依靠的是细胞内源的非同源末端连接修复机制(non-homologous end joining,NHEJ)、微同源介导末端连接机制(microhomology-mediated end joining,MMEJ)或同源重组修复机制(homology directed repair,HDR)[11].NHEJ系统进行双链断裂(double-strand breaks, DSBs)修复时,首先Ku异源二聚体识别并结合至两断裂末端,随后由DNA聚合酶和核酸外切酶对该处进行加工处理,最后进行末端连接形成双链.在对末端进行修饰时,由于外切酶和聚合酶结合顺序的不确定性则造成修复位点碱基缺失和插入突变等不同情况,致使目的基因失去活性[12].也有报道将Cas9蛋白靶向目的基因或染色体上两处位点,则会造成目的基因内或染色体上整段序列的缺失[4,13-15].MMEJ在修复双链断裂时的断裂末端比对过程中使用的微同源序列易导致原始断裂侧翼缺失,进而造成染色体异常如缺失、易位、倒位和其他复杂的重排.Cas9蛋白造成DNA双链断裂后,在损伤序列的同源供体DNA如姐妹染色单体存在的情况下,HDR则以同源DNA序列为模板进行无错修复.若人为地提供一段含有外源基因或替换了特定位置核苷酸的同源供体DNA(donor),则可利用HDR实现精准的基因插入或单碱基编辑(图1)[16].随着CRISPR/Cas9技术的普及和发展,其基本的基因编辑能力和性能已不能满足在基因组工程和各个领域中的应用.例如,除了受限于CRISPR/Cas9系统本身的PAM(protospacer adjacent motif)依赖性和脱靶效应,该系统在应用于代谢工程中时,还被其编辑靶点的数量和效率所制约[17],而应用在人类疾病基因疗法时还需进一步提高该系统的保真性、安全性等因素[18].因此,自张锋团队首次应用CRISPR/Cas9系统在哺乳动物细胞中实现多重编辑后,国内外的科研人员围绕该系统的多元化应用展开了研究,如通过优化Cas9蛋白拓宽该系统的识别范围并提高其靶向特异性[19-21]、通过融合效应结构域的方式使该系统获得动态可控性或进行转录调控[22-24]、为了进行表观基因组编辑或在非HDR介导的细胞中进行单碱基编辑将功能缺失的Cas9蛋白与甲基转移酶或脱氨酶融合等[25-26].2019年,哈佛大学David R. Liu团队开发的先导编辑(prime editing,PE)作为基于gRNA工程的多功能精准基因编辑工具,可以在不引入靶基因双链断裂及同源片段的条件下介导靶向的插入、缺失及所有碱基编辑,大大拓展了CRISPR/Cas9系统多功能基因组编辑的范围和能力[27].多重CRISPR技术是通过同时引入多个gRNA或多种直系同源Cas9蛋白,从而实现基因组上多靶点的同时靶向或多功能正交编辑.该技术不仅极大提高了基因编辑和转录调控的范围和效率,更促进了强大的生物工程应用.因此,如今越来越多的CRISPR/Cas9工具不仅可以实现定点改造,还可以实现多靶点的同时改造.发展趋势证明了基于RNA靶向性的CRISPR/Cas9技术凭借简单可编程性和广泛适用性具有强大优势,同时凸显出CRISPR/Cas9系统与TALENs、ZFNs等相比的优势[28]. ...
... 酿脓链球菌(Streptococcus pyogenes)Cas9(SpCas9)是第一种被应用于哺乳动物细胞基因组编辑的Cas9蛋白[4,39-40],目前仍是CRISPR系统在各应用领域中最常用的Cas9蛋白.在发挥靶序列切割的功能时,SpCas9、CRISPR RNA(crRNA)、反式激活CRISPR RNA(trans-activating crRNA, tracrRNA)结合形成核糖核蛋白复合物(ribonucleoprotein,RNP)共同参与这一过程.在天然CRISPR/Cas9系统中,crRNA作为识别模块,其上的间隔区段能够识别与之互补的特异性靶序列(常见20个核苷酸长度),而靶序列下游必须有三个碱基的PAM序列NGG(N代表A、G、C和T中的任一碱基);tracrRNA和crRNA上的核苷酸链有能互补配对的部分,配对后促使RNP形成以及激活Cas9蛋白[41-42].在目前通用的CRISPR/Cas9系统中,crRNA与tracrRNA被融合成一条sgRNA发挥相同的作用[39].也有研究发现截短gRNA的间隔区段(如14 nt的间隔区段)能够使RNP保持识别和结合靶序列的能力,但无法引入DSB[43];而当gRNA的间隔区段长度在16~20 nt时均可有效进行基因编辑,且17~20 nt的gRNA编辑效率无显著性差异[44-45].最后,位于Cas9蛋白上的HNH和RuvC结构域发挥核酸内切酶活性,分别切断靶向链和非靶向链,最终使靶DNA形成平末端的双链断裂,实现“打靶”[39,42]. ...
... 除了开发Cas9的突变体以弥补和拓展CRISPR/Cas9系统的不足与应用范围外,对Cas9 直系同源蛋白进行挖掘和开发也能够使该系统识别范围得到极大扩展.如来自嗜热链球菌(Streptococcus thermophilus)的StCas9能识别NNGAAW(其中W代表A或T)的PAM序列[4,35],来自脑膜炎奈瑟菌(Neisseria meningitidis)的NmCas9识别NNNNGATT序列[36,60-61],来自金黄色葡萄球菌(Staphylococcus aureus)的SaCas9识别NNGRRT序列(其中R代表A或G)[37],这些SpCas9同源蛋白或其组合应用均已在细菌和哺乳动物细胞中得到了开发.此外,SaCas9在肽链长度较小的条件下(1053个氨基酸,SpCas9含有1368个氨基酸)拥有与SpCas9相当的编辑效率,这使其在大小受限制的传递载体如腺相关病毒(adeno-associated virus,AAV)中得到广泛应用.来自曲状杆菌(Campylobacter jejuni)的CjCas9多肽链上仅含984个氨基酸,能识别PAM序列NNNVRYM,已被报道可以通过AAV递送至小鼠肌纤维和视网膜色素上皮细胞(retinal pigment epithelium,RPE)中并成功进行基因编辑[38].工程化的新型Cas9突变体、gRNA与Cas9直系同源的不断发掘为CRISPR/Cas9系统在各种背景下的应用提供了源源不断的新方法和工具,也证明了该系统作为第三代“基因剪刀”目前仍然处于高速发展阶段.该系统中存在的PAM依赖性、脱靶率较高等问题都需要利用进化工程或计算机辅助理性设计等方式对Cas9蛋白进行进一步探索和改造才能得到解决,使其能够在更多的场景下最大程度地发挥自身高灵活性、高精准性、易编程性的优势. ...
... 目前,多gRNA的表达策略主要有三种(图3):①每个不同gRNA表达盒通过Pol Ⅲ启动子单独启动表达,表达盒之间串联在一起[4];②多个不同gRNA在一个表达盒内由Pol Ⅱ或Pol Ⅲ启动表达为一条转录本,然后再通过mRNA上gRNA之间的tRNA序列切割加工机制、核酶自切割能力或Csy4核酸酶切割释放为成熟的gRNA[11];③一条转录本内的多个gRNA也可以在CRISPR系统自然的自我加工活性的基础上被切割释放为成熟gRNA.2013年,Cong等[4]依据CRISPR基因位点的间隔区排列的自然结构,首次将Pol Ⅲ驱动的表达盒串联实现两个靶基因间隔区的多表达并成功编辑.2018年,Ferreira等[113]首次在酿酒酵母(Saccharomyces cerevisiae)中利用铜绿假单胞菌(Pseudomonas aeruginosa)来源的核酸内切酶Csy4处理单转录本中的多个gRNA,并最终获得高达96%四基因同时敲除效率,并借助CRISPRi系统对3个靶基因进行了稳定的扰动,证明了使用Csy4可以快速有效地进行多重基因组编辑和基因调控.基于此,McCarty等[114]开发的酵母模块化克隆多gRNA快速组装策略通过两步PCR反应及一步Golden Gate反应可在两天内实现单阵列12个gRNA的组装,每个gRNA两侧都有Csy4识别位点从而将转录本加工为成熟的gRNA.同年,刘子鹤团队[115]基于tRNA转录本切割机制构建了一种gRNA-tRNA阵列多gRNA表达系统(GTR-CRISPR系统)并成功在酿酒酵母中以80%的效率同时敲除8个基因.通过使用更快速的Lightning GTR-CRISPR系统,该团队成功在10天中敲除了8个目的基因,简化了酿酒酵母的脂质代谢网络,最终获得了游离脂肪酸30倍的提升效果.该系统的开发在极大地提高了CRISPR/Cas9系统用于酿酒酵母基因组多重编辑效率的同时也揭示该系统在多重应用领域蕴藏的巨大潜力. ...
... [4]依据CRISPR基因位点的间隔区排列的自然结构,首次将Pol Ⅲ驱动的表达盒串联实现两个靶基因间隔区的多表达并成功编辑.2018年,Ferreira等[113]首次在酿酒酵母(Saccharomyces cerevisiae)中利用铜绿假单胞菌(Pseudomonas aeruginosa)来源的核酸内切酶Csy4处理单转录本中的多个gRNA,并最终获得高达96%四基因同时敲除效率,并借助CRISPRi系统对3个靶基因进行了稳定的扰动,证明了使用Csy4可以快速有效地进行多重基因组编辑和基因调控.基于此,McCarty等[114]开发的酵母模块化克隆多gRNA快速组装策略通过两步PCR反应及一步Golden Gate反应可在两天内实现单阵列12个gRNA的组装,每个gRNA两侧都有Csy4识别位点从而将转录本加工为成熟的gRNA.同年,刘子鹤团队[115]基于tRNA转录本切割机制构建了一种gRNA-tRNA阵列多gRNA表达系统(GTR-CRISPR系统)并成功在酿酒酵母中以80%的效率同时敲除8个基因.通过使用更快速的Lightning GTR-CRISPR系统,该团队成功在10天中敲除了8个目的基因,简化了酿酒酵母的脂质代谢网络,最终获得了游离脂肪酸30倍的提升效果.该系统的开发在极大地提高了CRISPR/Cas9系统用于酿酒酵母基因组多重编辑效率的同时也揭示该系统在多重应用领域蕴藏的巨大潜力. ...
... Research on the applications of the multiplexed CRISPR/Cas9 system
Table 3| 宿主细胞 | 多重CRISPR策略 | 靶向目标数量 | 应用/概念验证 | 参考文献 |
|---|
| 哺乳动物细胞 | 2个间隔区通过天然CRISPR序列自我加工 | 2个靶标(1个基因) | 对1个靶基因敲除,效率为1.6% | [4] |
| 酿酒酵母 | Casy4核酸酶加工 | 3个基因 | 同时对3个不同启动子进行激活,使得3个报告基因荧光强度提升了2倍 | [113] |
| 酿酒酵母 | 基于Csy4核酸酶加工的多gRNA快速组装 | 12个靶标(3个基因) | 通过12个gRNA靶向使得3个报告基因荧光强度分别被抑制了92%、81%和95% | [114] |
| 酿酒酵母 | 基于tRNA转录本切割机制的多gRNA表达系统 | 8个基因 | 通过两轮8个基因的敲除获得了30倍的游离脂肪酸产量 | [115] |
| 酿酒酵母 | 多个gRNA表达盒通过pol Ⅲ启动子启动 | 3个基因 | 将适配子与gRNA融合进行多基因调控,获得了不同的紫罗兰素生物合成产物 | [118] |
| 酿酒酵母 | 基于Csy4核酸酶加工的多gRNA快速组装策略及多Cas9正交组合调控 | 3个基因 | 通过组合调控使两个报告基因表达分别被抑制和激活5倍,同时以95%的效率敲除第三个基因 | [119] |
| 酿酒酵母 | 通过质粒表达多个gRNA表达盒以及截短gRNA用于调控 | 3个基因 | 通过对三个基因分别进行编辑、激活和抑制获得了α-檀香烯2.66倍的产量 | [43] |
| 马克思克鲁维酵母(Kluyveromyces marxianus) | 基于tRNA转录本切割机制的多gRNA表达系统 | 6个靶标(4个基因) | 通过对4个基因进行调控,使乙酸乙酯的产量提升了3.8倍 | [120] |
| 大肠杆菌 | 多个gRNA表达盒通过质粒表达 | 3个基因 | 通过对3个基因进行组合调控得到了2.3倍苹果酸产量菌株 | [121] |
| 大肠杆菌 | 多个gRNA表达盒通过质粒表达 | 3个基因 | 对苹果酸合成途径基因进行组合调控后获得了2.3倍苹果酸产量菌株 | [121] |
| 枯草芽孢杆菌(Bacillus subtilis) | 多个gRNA表达盒整合至基因组 | 3个基因 | 通过多基因动态组合调控提高了N-乙酰葡糖胺产量 | [122] |
| 天蓝色链霉菌(Streptomyces coelicolor) | 多个gRNA表达盒与dCas9在质粒上表达 | 4个基因 | 通过对4个靶基因同时进行抑制,使其mRNA表达量降为对照的2%~32% | [123] |
2015年,Zalatan等[118]通过多sgRNA表达盒策略及sgRNA工程将适配体与识别区结合,将配体与转录因子融合表达后同时对3个目标基因进行激活或抑制调控,最终在酿酒酵母中获得了不同的紫罗兰素生物合成途径产物.Lobs等[120]在对一株马克思克鲁维酵母(Kluyveromyces marxianus)中乙酸酯的主要代谢途径和前体物质进行鉴定后,利用CRISPRi系统对ACO2b、SDH2、RIP1和MSS51四个基因进行多重调控,最终获得了乙酸乙酯3.8倍的提升.2017年,Lian等[119]在表征了多种同源Cas9蛋白的基因敲除、转录激活和转录抑制效率后,优化并组合三种同源蛋白最终构建了一套完全正交的三功能CRISPR代谢调控系统.由于正交的Cas9同源蛋白所识别的PAM序列不同,发挥不同功能的同源Cas9之间互不干扰,使得两个被调控报告基因的表达强度均被激活和抑制了5倍,并使第三个报告基因的敲除效率达到了95%.该系统通过一步组合调控使得改造的酿酒酵母中胡萝卜素产量提升了3倍,同时使表面葡聚糖酶展示量提高了2.5倍.原核生物具有相对较低的代谢网络复杂性,同时其无核膜包被的拟核能够实现转录与翻译同时进行,这使得原核生物(如大肠杆菌)成为多基因操作及表达调控的模式生物[122-123].2018年,Gao等[121]通过多重CRISPRi系统对苹果酸生物合成途径中的基因进行组合调控后得到了2.3倍的苹果酸产量菌株. ...
CRISPR-Cas systems and RNA-guided interference
1
2013
... 对遗传信息进行编辑和对基因表达进行调控是研究各种遗传信息的具体功能、揭示基因独特生理机制的重要方法.进入新世纪后,来源于原核生物的簇状规则间隔短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR)/CRISPR相关蛋白(CRISPR-associated protein, Cas protein)系统蓬勃发展,成为应用于基因组工程中的重要技术.早在1987年,研究人员就在大肠杆菌基因组上发现了这一段特殊的序列[1].2002年,Jansen等[2]通过大量观察创造了CRISPR缩写代替了之前发现的重复片段.随后,Brouns等[3]揭示了该系统对DNA的靶向作用及原理,从而推测出该系统具有作为基因编辑工具的潜力.2013年,张锋团队[4]首次实现CRISPR/Cas9系统在哺乳动物细胞中的多基因编辑,开启了基因编辑技术的新篇章.依据Cas蛋白的结构和进化关系,目前CRISPR/Cas系统共分六种类型,其中Ⅰ、Ⅲ、Ⅳ型系统的Cas蛋白为多亚基,Ⅱ、Ⅴ、Ⅵ型系统的Cas蛋白为单亚基[5-6].由于单亚基CRISPR/Cas系统机制简单且易操作,目前研究成熟和应用广泛的大多为单亚基系统,如Ⅱ型的CRISPR/Cas9系统、Ⅴ型的CRISPR/Cas12a(Cpf1)系统和Ⅵ型的CRISPR/Cas13a(C2c2)系统等.其中CRISPR/Cas9系统凭借其操作简便、精准编辑、多重编辑等优势,与传统的DNA修饰工具如大范围核酸酶(meganucleases, MGNs)[7]、锌指核酸酶(zinc finger nucleases, ZFNs)[8]、转录激活因子样效应蛋白核酸酶(transcription activator-like effector nucleases, TALENs)[9]等区分开来,使其在基础科学到应用科学领域均得到了极大的发展与应用. ...
Discovery and functional characterization of diverse class 2 CRISPR-cas systems
1
2015
... 对遗传信息进行编辑和对基因表达进行调控是研究各种遗传信息的具体功能、揭示基因独特生理机制的重要方法.进入新世纪后,来源于原核生物的簇状规则间隔短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR)/CRISPR相关蛋白(CRISPR-associated protein, Cas protein)系统蓬勃发展,成为应用于基因组工程中的重要技术.早在1987年,研究人员就在大肠杆菌基因组上发现了这一段特殊的序列[1].2002年,Jansen等[2]通过大量观察创造了CRISPR缩写代替了之前发现的重复片段.随后,Brouns等[3]揭示了该系统对DNA的靶向作用及原理,从而推测出该系统具有作为基因编辑工具的潜力.2013年,张锋团队[4]首次实现CRISPR/Cas9系统在哺乳动物细胞中的多基因编辑,开启了基因编辑技术的新篇章.依据Cas蛋白的结构和进化关系,目前CRISPR/Cas系统共分六种类型,其中Ⅰ、Ⅲ、Ⅳ型系统的Cas蛋白为多亚基,Ⅱ、Ⅴ、Ⅵ型系统的Cas蛋白为单亚基[5-6].由于单亚基CRISPR/Cas系统机制简单且易操作,目前研究成熟和应用广泛的大多为单亚基系统,如Ⅱ型的CRISPR/Cas9系统、Ⅴ型的CRISPR/Cas12a(Cpf1)系统和Ⅵ型的CRISPR/Cas13a(C2c2)系统等.其中CRISPR/Cas9系统凭借其操作简便、精准编辑、多重编辑等优势,与传统的DNA修饰工具如大范围核酸酶(meganucleases, MGNs)[7]、锌指核酸酶(zinc finger nucleases, ZFNs)[8]、转录激活因子样效应蛋白核酸酶(transcription activator-like effector nucleases, TALENs)[9]等区分开来,使其在基础科学到应用科学领域均得到了极大的发展与应用. ...
Origins of programmable nucleases for genome engineering
1
2016
... 对遗传信息进行编辑和对基因表达进行调控是研究各种遗传信息的具体功能、揭示基因独特生理机制的重要方法.进入新世纪后,来源于原核生物的簇状规则间隔短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR)/CRISPR相关蛋白(CRISPR-associated protein, Cas protein)系统蓬勃发展,成为应用于基因组工程中的重要技术.早在1987年,研究人员就在大肠杆菌基因组上发现了这一段特殊的序列[1].2002年,Jansen等[2]通过大量观察创造了CRISPR缩写代替了之前发现的重复片段.随后,Brouns等[3]揭示了该系统对DNA的靶向作用及原理,从而推测出该系统具有作为基因编辑工具的潜力.2013年,张锋团队[4]首次实现CRISPR/Cas9系统在哺乳动物细胞中的多基因编辑,开启了基因编辑技术的新篇章.依据Cas蛋白的结构和进化关系,目前CRISPR/Cas系统共分六种类型,其中Ⅰ、Ⅲ、Ⅳ型系统的Cas蛋白为多亚基,Ⅱ、Ⅴ、Ⅵ型系统的Cas蛋白为单亚基[5-6].由于单亚基CRISPR/Cas系统机制简单且易操作,目前研究成熟和应用广泛的大多为单亚基系统,如Ⅱ型的CRISPR/Cas9系统、Ⅴ型的CRISPR/Cas12a(Cpf1)系统和Ⅵ型的CRISPR/Cas13a(C2c2)系统等.其中CRISPR/Cas9系统凭借其操作简便、精准编辑、多重编辑等优势,与传统的DNA修饰工具如大范围核酸酶(meganucleases, MGNs)[7]、锌指核酸酶(zinc finger nucleases, ZFNs)[8]、转录激活因子样效应蛋白核酸酶(transcription activator-like effector nucleases, TALENs)[9]等区分开来,使其在基础科学到应用科学领域均得到了极大的发展与应用. ...
Gene targeting using zinc finger nucleases
1
2005
... 对遗传信息进行编辑和对基因表达进行调控是研究各种遗传信息的具体功能、揭示基因独特生理机制的重要方法.进入新世纪后,来源于原核生物的簇状规则间隔短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR)/CRISPR相关蛋白(CRISPR-associated protein, Cas protein)系统蓬勃发展,成为应用于基因组工程中的重要技术.早在1987年,研究人员就在大肠杆菌基因组上发现了这一段特殊的序列[1].2002年,Jansen等[2]通过大量观察创造了CRISPR缩写代替了之前发现的重复片段.随后,Brouns等[3]揭示了该系统对DNA的靶向作用及原理,从而推测出该系统具有作为基因编辑工具的潜力.2013年,张锋团队[4]首次实现CRISPR/Cas9系统在哺乳动物细胞中的多基因编辑,开启了基因编辑技术的新篇章.依据Cas蛋白的结构和进化关系,目前CRISPR/Cas系统共分六种类型,其中Ⅰ、Ⅲ、Ⅳ型系统的Cas蛋白为多亚基,Ⅱ、Ⅴ、Ⅵ型系统的Cas蛋白为单亚基[5-6].由于单亚基CRISPR/Cas系统机制简单且易操作,目前研究成熟和应用广泛的大多为单亚基系统,如Ⅱ型的CRISPR/Cas9系统、Ⅴ型的CRISPR/Cas12a(Cpf1)系统和Ⅵ型的CRISPR/Cas13a(C2c2)系统等.其中CRISPR/Cas9系统凭借其操作简便、精准编辑、多重编辑等优势,与传统的DNA修饰工具如大范围核酸酶(meganucleases, MGNs)[7]、锌指核酸酶(zinc finger nucleases, ZFNs)[8]、转录激活因子样效应蛋白核酸酶(transcription activator-like effector nucleases, TALENs)[9]等区分开来,使其在基础科学到应用科学领域均得到了极大的发展与应用. ...
TALENs: a widely applicable technology for targeted genome editing
2
2013
... 对遗传信息进行编辑和对基因表达进行调控是研究各种遗传信息的具体功能、揭示基因独特生理机制的重要方法.进入新世纪后,来源于原核生物的簇状规则间隔短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR)/CRISPR相关蛋白(CRISPR-associated protein, Cas protein)系统蓬勃发展,成为应用于基因组工程中的重要技术.早在1987年,研究人员就在大肠杆菌基因组上发现了这一段特殊的序列[1].2002年,Jansen等[2]通过大量观察创造了CRISPR缩写代替了之前发现的重复片段.随后,Brouns等[3]揭示了该系统对DNA的靶向作用及原理,从而推测出该系统具有作为基因编辑工具的潜力.2013年,张锋团队[4]首次实现CRISPR/Cas9系统在哺乳动物细胞中的多基因编辑,开启了基因编辑技术的新篇章.依据Cas蛋白的结构和进化关系,目前CRISPR/Cas系统共分六种类型,其中Ⅰ、Ⅲ、Ⅳ型系统的Cas蛋白为多亚基,Ⅱ、Ⅴ、Ⅵ型系统的Cas蛋白为单亚基[5-6].由于单亚基CRISPR/Cas系统机制简单且易操作,目前研究成熟和应用广泛的大多为单亚基系统,如Ⅱ型的CRISPR/Cas9系统、Ⅴ型的CRISPR/Cas12a(Cpf1)系统和Ⅵ型的CRISPR/Cas13a(C2c2)系统等.其中CRISPR/Cas9系统凭借其操作简便、精准编辑、多重编辑等优势,与传统的DNA修饰工具如大范围核酸酶(meganucleases, MGNs)[7]、锌指核酸酶(zinc finger nucleases, ZFNs)[8]、转录激活因子样效应蛋白核酸酶(transcription activator-like effector nucleases, TALENs)[9]等区分开来,使其在基础科学到应用科学领域均得到了极大的发展与应用. ...
... 转录因子通过结合或识别基因上游特定区域,进而调节RNA聚合酶的招募,最终使靶基因表达上调或下调[79-80].其不仅可以与启动子区域结合,多个转录因子间也可结合形成转录因子复合体进而对靶基因进行更复杂、精细的调控,促进或抑制DNA上遗传信息向RNA转录的过程.在基因组范围上对靶基因进行调控是探究基因功能、重塑代谢调控网络的重要方法[62].自20世纪末以来,研究人员就已经将自然存在的或工程化后的DNA靶向蛋白如四环素阻遏蛋白[81-82]、Gal4蛋白[83]、锌指蛋白[84]以及TALE蛋白[85-87]等与转录因子融合进行靶基因调控.但由于依赖蛋白质靶向 DNA的特性,靶向不同的DNA序列时需要工程化设计不同的蛋白质氨基酸序列,使得这些蛋白普遍可编程性较差且分子庞大,因此开发用于基因组范围的转录调控蛋白文库相当耗时且价格昂贵[9].CRISPR基因调控系统包括CRISPR激活系统(CRISPR activation system,CRISPRa)和CRISPR干扰系统(CRISPR interference system,CRISPRi),该系统将转录激活或抑制因子与dCas9蛋白融合后,借助CRISPR系统精准的靶向性和灵活的可编程性将融合蛋白靶向至目的基因从而对该基因的转录进行激活或干扰(图2)[62,81-88].不同于其他DNA靶向蛋白,CRISPR激活系统的dCas9-effector融合蛋白利用gRNA与靶序列的碱基互补配对进行精确定位并最终实现目的基因调控,同时gRNA的易编程性和dCas9蛋白较小的体量使该系统在基因组范围上进行调控成为了可能. ...
DNA double-strand break repair: all's well that ends well
1
2006
... CRISPR系统利用单链向导RNA(single guide RNA,sgRNA或gRNA)靶向目的位点,随后Cas9等核酸酶切断目的区域的双链核酸,诱导细胞内源DNA损伤修复机制工作,最终借助该机制达到编辑遗传信息的目的[10].CRISPR/Cas9系统在发挥基因敲除、基因插入、单碱基编辑等主要编辑手段时主要依靠的是细胞内源的非同源末端连接修复机制(non-homologous end joining,NHEJ)、微同源介导末端连接机制(microhomology-mediated end joining,MMEJ)或同源重组修复机制(homology directed repair,HDR)[11].NHEJ系统进行双链断裂(double-strand breaks, DSBs)修复时,首先Ku异源二聚体识别并结合至两断裂末端,随后由DNA聚合酶和核酸外切酶对该处进行加工处理,最后进行末端连接形成双链.在对末端进行修饰时,由于外切酶和聚合酶结合顺序的不确定性则造成修复位点碱基缺失和插入突变等不同情况,致使目的基因失去活性[12].也有报道将Cas9蛋白靶向目的基因或染色体上两处位点,则会造成目的基因内或染色体上整段序列的缺失[4,13-15].MMEJ在修复双链断裂时的断裂末端比对过程中使用的微同源序列易导致原始断裂侧翼缺失,进而造成染色体异常如缺失、易位、倒位和其他复杂的重排.Cas9蛋白造成DNA双链断裂后,在损伤序列的同源供体DNA如姐妹染色单体存在的情况下,HDR则以同源DNA序列为模板进行无错修复.若人为地提供一段含有外源基因或替换了特定位置核苷酸的同源供体DNA(donor),则可利用HDR实现精准的基因插入或单碱基编辑(图1)[16].随着CRISPR/Cas9技术的普及和发展,其基本的基因编辑能力和性能已不能满足在基因组工程和各个领域中的应用.例如,除了受限于CRISPR/Cas9系统本身的PAM(protospacer adjacent motif)依赖性和脱靶效应,该系统在应用于代谢工程中时,还被其编辑靶点的数量和效率所制约[17],而应用在人类疾病基因疗法时还需进一步提高该系统的保真性、安全性等因素[18].因此,自张锋团队首次应用CRISPR/Cas9系统在哺乳动物细胞中实现多重编辑后,国内外的科研人员围绕该系统的多元化应用展开了研究,如通过优化Cas9蛋白拓宽该系统的识别范围并提高其靶向特异性[19-21]、通过融合效应结构域的方式使该系统获得动态可控性或进行转录调控[22-24]、为了进行表观基因组编辑或在非HDR介导的细胞中进行单碱基编辑将功能缺失的Cas9蛋白与甲基转移酶或脱氨酶融合等[25-26].2019年,哈佛大学David R. Liu团队开发的先导编辑(prime editing,PE)作为基于gRNA工程的多功能精准基因编辑工具,可以在不引入靶基因双链断裂及同源片段的条件下介导靶向的插入、缺失及所有碱基编辑,大大拓展了CRISPR/Cas9系统多功能基因组编辑的范围和能力[27].多重CRISPR技术是通过同时引入多个gRNA或多种直系同源Cas9蛋白,从而实现基因组上多靶点的同时靶向或多功能正交编辑.该技术不仅极大提高了基因编辑和转录调控的范围和效率,更促进了强大的生物工程应用.因此,如今越来越多的CRISPR/Cas9工具不仅可以实现定点改造,还可以实现多靶点的同时改造.发展趋势证明了基于RNA靶向性的CRISPR/Cas9技术凭借简单可编程性和广泛适用性具有强大优势,同时凸显出CRISPR/Cas9系统与TALENs、ZFNs等相比的优势[28]. ...
Genome engineering for crop improvement and future agriculture
2
2021
... CRISPR系统利用单链向导RNA(single guide RNA,sgRNA或gRNA)靶向目的位点,随后Cas9等核酸酶切断目的区域的双链核酸,诱导细胞内源DNA损伤修复机制工作,最终借助该机制达到编辑遗传信息的目的[10].CRISPR/Cas9系统在发挥基因敲除、基因插入、单碱基编辑等主要编辑手段时主要依靠的是细胞内源的非同源末端连接修复机制(non-homologous end joining,NHEJ)、微同源介导末端连接机制(microhomology-mediated end joining,MMEJ)或同源重组修复机制(homology directed repair,HDR)[11].NHEJ系统进行双链断裂(double-strand breaks, DSBs)修复时,首先Ku异源二聚体识别并结合至两断裂末端,随后由DNA聚合酶和核酸外切酶对该处进行加工处理,最后进行末端连接形成双链.在对末端进行修饰时,由于外切酶和聚合酶结合顺序的不确定性则造成修复位点碱基缺失和插入突变等不同情况,致使目的基因失去活性[12].也有报道将Cas9蛋白靶向目的基因或染色体上两处位点,则会造成目的基因内或染色体上整段序列的缺失[4,13-15].MMEJ在修复双链断裂时的断裂末端比对过程中使用的微同源序列易导致原始断裂侧翼缺失,进而造成染色体异常如缺失、易位、倒位和其他复杂的重排.Cas9蛋白造成DNA双链断裂后,在损伤序列的同源供体DNA如姐妹染色单体存在的情况下,HDR则以同源DNA序列为模板进行无错修复.若人为地提供一段含有外源基因或替换了特定位置核苷酸的同源供体DNA(donor),则可利用HDR实现精准的基因插入或单碱基编辑(图1)[16].随着CRISPR/Cas9技术的普及和发展,其基本的基因编辑能力和性能已不能满足在基因组工程和各个领域中的应用.例如,除了受限于CRISPR/Cas9系统本身的PAM(protospacer adjacent motif)依赖性和脱靶效应,该系统在应用于代谢工程中时,还被其编辑靶点的数量和效率所制约[17],而应用在人类疾病基因疗法时还需进一步提高该系统的保真性、安全性等因素[18].因此,自张锋团队首次应用CRISPR/Cas9系统在哺乳动物细胞中实现多重编辑后,国内外的科研人员围绕该系统的多元化应用展开了研究,如通过优化Cas9蛋白拓宽该系统的识别范围并提高其靶向特异性[19-21]、通过融合效应结构域的方式使该系统获得动态可控性或进行转录调控[22-24]、为了进行表观基因组编辑或在非HDR介导的细胞中进行单碱基编辑将功能缺失的Cas9蛋白与甲基转移酶或脱氨酶融合等[25-26].2019年,哈佛大学David R. Liu团队开发的先导编辑(prime editing,PE)作为基于gRNA工程的多功能精准基因编辑工具,可以在不引入靶基因双链断裂及同源片段的条件下介导靶向的插入、缺失及所有碱基编辑,大大拓展了CRISPR/Cas9系统多功能基因组编辑的范围和能力[27].多重CRISPR技术是通过同时引入多个gRNA或多种直系同源Cas9蛋白,从而实现基因组上多靶点的同时靶向或多功能正交编辑.该技术不仅极大提高了基因编辑和转录调控的范围和效率,更促进了强大的生物工程应用.因此,如今越来越多的CRISPR/Cas9工具不仅可以实现定点改造,还可以实现多靶点的同时改造.发展趋势证明了基于RNA靶向性的CRISPR/Cas9技术凭借简单可编程性和广泛适用性具有强大优势,同时凸显出CRISPR/Cas9系统与TALENs、ZFNs等相比的优势[28]. ...
... 目前,多gRNA的表达策略主要有三种(图3):①每个不同gRNA表达盒通过Pol Ⅲ启动子单独启动表达,表达盒之间串联在一起[4];②多个不同gRNA在一个表达盒内由Pol Ⅱ或Pol Ⅲ启动表达为一条转录本,然后再通过mRNA上gRNA之间的tRNA序列切割加工机制、核酶自切割能力或Csy4核酸酶切割释放为成熟的gRNA[11];③一条转录本内的多个gRNA也可以在CRISPR系统自然的自我加工活性的基础上被切割释放为成熟gRNA.2013年,Cong等[4]依据CRISPR基因位点的间隔区排列的自然结构,首次将Pol Ⅲ驱动的表达盒串联实现两个靶基因间隔区的多表达并成功编辑.2018年,Ferreira等[113]首次在酿酒酵母(Saccharomyces cerevisiae)中利用铜绿假单胞菌(Pseudomonas aeruginosa)来源的核酸内切酶Csy4处理单转录本中的多个gRNA,并最终获得高达96%四基因同时敲除效率,并借助CRISPRi系统对3个靶基因进行了稳定的扰动,证明了使用Csy4可以快速有效地进行多重基因组编辑和基因调控.基于此,McCarty等[114]开发的酵母模块化克隆多gRNA快速组装策略通过两步PCR反应及一步Golden Gate反应可在两天内实现单阵列12个gRNA的组装,每个gRNA两侧都有Csy4识别位点从而将转录本加工为成熟的gRNA.同年,刘子鹤团队[115]基于tRNA转录本切割机制构建了一种gRNA-tRNA阵列多gRNA表达系统(GTR-CRISPR系统)并成功在酿酒酵母中以80%的效率同时敲除8个基因.通过使用更快速的Lightning GTR-CRISPR系统,该团队成功在10天中敲除了8个目的基因,简化了酿酒酵母的脂质代谢网络,最终获得了游离脂肪酸30倍的提升效果.该系统的开发在极大地提高了CRISPR/Cas9系统用于酿酒酵母基因组多重编辑效率的同时也揭示该系统在多重应用领域蕴藏的巨大潜力. ...
Non-homologous DNA end joining and alternative pathways to double-strand break repair
1
2017
... CRISPR系统利用单链向导RNA(single guide RNA,sgRNA或gRNA)靶向目的位点,随后Cas9等核酸酶切断目的区域的双链核酸,诱导细胞内源DNA损伤修复机制工作,最终借助该机制达到编辑遗传信息的目的[10].CRISPR/Cas9系统在发挥基因敲除、基因插入、单碱基编辑等主要编辑手段时主要依靠的是细胞内源的非同源末端连接修复机制(non-homologous end joining,NHEJ)、微同源介导末端连接机制(microhomology-mediated end joining,MMEJ)或同源重组修复机制(homology directed repair,HDR)[11].NHEJ系统进行双链断裂(double-strand breaks, DSBs)修复时,首先Ku异源二聚体识别并结合至两断裂末端,随后由DNA聚合酶和核酸外切酶对该处进行加工处理,最后进行末端连接形成双链.在对末端进行修饰时,由于外切酶和聚合酶结合顺序的不确定性则造成修复位点碱基缺失和插入突变等不同情况,致使目的基因失去活性[12].也有报道将Cas9蛋白靶向目的基因或染色体上两处位点,则会造成目的基因内或染色体上整段序列的缺失[4,13-15].MMEJ在修复双链断裂时的断裂末端比对过程中使用的微同源序列易导致原始断裂侧翼缺失,进而造成染色体异常如缺失、易位、倒位和其他复杂的重排.Cas9蛋白造成DNA双链断裂后,在损伤序列的同源供体DNA如姐妹染色单体存在的情况下,HDR则以同源DNA序列为模板进行无错修复.若人为地提供一段含有外源基因或替换了特定位置核苷酸的同源供体DNA(donor),则可利用HDR实现精准的基因插入或单碱基编辑(图1)[16].随着CRISPR/Cas9技术的普及和发展,其基本的基因编辑能力和性能已不能满足在基因组工程和各个领域中的应用.例如,除了受限于CRISPR/Cas9系统本身的PAM(protospacer adjacent motif)依赖性和脱靶效应,该系统在应用于代谢工程中时,还被其编辑靶点的数量和效率所制约[17],而应用在人类疾病基因疗法时还需进一步提高该系统的保真性、安全性等因素[18].因此,自张锋团队首次应用CRISPR/Cas9系统在哺乳动物细胞中实现多重编辑后,国内外的科研人员围绕该系统的多元化应用展开了研究,如通过优化Cas9蛋白拓宽该系统的识别范围并提高其靶向特异性[19-21]、通过融合效应结构域的方式使该系统获得动态可控性或进行转录调控[22-24]、为了进行表观基因组编辑或在非HDR介导的细胞中进行单碱基编辑将功能缺失的Cas9蛋白与甲基转移酶或脱氨酶融合等[25-26].2019年,哈佛大学David R. Liu团队开发的先导编辑(prime editing,PE)作为基于gRNA工程的多功能精准基因编辑工具,可以在不引入靶基因双链断裂及同源片段的条件下介导靶向的插入、缺失及所有碱基编辑,大大拓展了CRISPR/Cas9系统多功能基因组编辑的范围和能力[27].多重CRISPR技术是通过同时引入多个gRNA或多种直系同源Cas9蛋白,从而实现基因组上多靶点的同时靶向或多功能正交编辑.该技术不仅极大提高了基因编辑和转录调控的范围和效率,更促进了强大的生物工程应用.因此,如今越来越多的CRISPR/Cas9工具不仅可以实现定点改造,还可以实现多靶点的同时改造.发展趋势证明了基于RNA靶向性的CRISPR/Cas9技术凭借简单可编程性和广泛适用性具有强大优势,同时凸显出CRISPR/Cas9系统与TALENs、ZFNs等相比的优势[28]. ...
Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity
1
2013
... CRISPR系统利用单链向导RNA(single guide RNA,sgRNA或gRNA)靶向目的位点,随后Cas9等核酸酶切断目的区域的双链核酸,诱导细胞内源DNA损伤修复机制工作,最终借助该机制达到编辑遗传信息的目的[10].CRISPR/Cas9系统在发挥基因敲除、基因插入、单碱基编辑等主要编辑手段时主要依靠的是细胞内源的非同源末端连接修复机制(non-homologous end joining,NHEJ)、微同源介导末端连接机制(microhomology-mediated end joining,MMEJ)或同源重组修复机制(homology directed repair,HDR)[11].NHEJ系统进行双链断裂(double-strand breaks, DSBs)修复时,首先Ku异源二聚体识别并结合至两断裂末端,随后由DNA聚合酶和核酸外切酶对该处进行加工处理,最后进行末端连接形成双链.在对末端进行修饰时,由于外切酶和聚合酶结合顺序的不确定性则造成修复位点碱基缺失和插入突变等不同情况,致使目的基因失去活性[12].也有报道将Cas9蛋白靶向目的基因或染色体上两处位点,则会造成目的基因内或染色体上整段序列的缺失[4,13-15].MMEJ在修复双链断裂时的断裂末端比对过程中使用的微同源序列易导致原始断裂侧翼缺失,进而造成染色体异常如缺失、易位、倒位和其他复杂的重排.Cas9蛋白造成DNA双链断裂后,在损伤序列的同源供体DNA如姐妹染色单体存在的情况下,HDR则以同源DNA序列为模板进行无错修复.若人为地提供一段含有外源基因或替换了特定位置核苷酸的同源供体DNA(donor),则可利用HDR实现精准的基因插入或单碱基编辑(图1)[16].随着CRISPR/Cas9技术的普及和发展,其基本的基因编辑能力和性能已不能满足在基因组工程和各个领域中的应用.例如,除了受限于CRISPR/Cas9系统本身的PAM(protospacer adjacent motif)依赖性和脱靶效应,该系统在应用于代谢工程中时,还被其编辑靶点的数量和效率所制约[17],而应用在人类疾病基因疗法时还需进一步提高该系统的保真性、安全性等因素[18].因此,自张锋团队首次应用CRISPR/Cas9系统在哺乳动物细胞中实现多重编辑后,国内外的科研人员围绕该系统的多元化应用展开了研究,如通过优化Cas9蛋白拓宽该系统的识别范围并提高其靶向特异性[19-21]、通过融合效应结构域的方式使该系统获得动态可控性或进行转录调控[22-24]、为了进行表观基因组编辑或在非HDR介导的细胞中进行单碱基编辑将功能缺失的Cas9蛋白与甲基转移酶或脱氨酶融合等[25-26].2019年,哈佛大学David R. Liu团队开发的先导编辑(prime editing,PE)作为基于gRNA工程的多功能精准基因编辑工具,可以在不引入靶基因双链断裂及同源片段的条件下介导靶向的插入、缺失及所有碱基编辑,大大拓展了CRISPR/Cas9系统多功能基因组编辑的范围和能力[27].多重CRISPR技术是通过同时引入多个gRNA或多种直系同源Cas9蛋白,从而实现基因组上多靶点的同时靶向或多功能正交编辑.该技术不仅极大提高了基因编辑和转录调控的范围和效率,更促进了强大的生物工程应用.因此,如今越来越多的CRISPR/Cas9工具不仅可以实现定点改造,还可以实现多靶点的同时改造.发展趋势证明了基于RNA靶向性的CRISPR/Cas9技术凭借简单可编程性和广泛适用性具有强大优势,同时凸显出CRISPR/Cas9系统与TALENs、ZFNs等相比的优势[28]. ...
Genome engineering using the CRISPR-Cas9 system
0
2013
Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR-Cas9 library
1
2016
... CRISPR系统利用单链向导RNA(single guide RNA,sgRNA或gRNA)靶向目的位点,随后Cas9等核酸酶切断目的区域的双链核酸,诱导细胞内源DNA损伤修复机制工作,最终借助该机制达到编辑遗传信息的目的[10].CRISPR/Cas9系统在发挥基因敲除、基因插入、单碱基编辑等主要编辑手段时主要依靠的是细胞内源的非同源末端连接修复机制(non-homologous end joining,NHEJ)、微同源介导末端连接机制(microhomology-mediated end joining,MMEJ)或同源重组修复机制(homology directed repair,HDR)[11].NHEJ系统进行双链断裂(double-strand breaks, DSBs)修复时,首先Ku异源二聚体识别并结合至两断裂末端,随后由DNA聚合酶和核酸外切酶对该处进行加工处理,最后进行末端连接形成双链.在对末端进行修饰时,由于外切酶和聚合酶结合顺序的不确定性则造成修复位点碱基缺失和插入突变等不同情况,致使目的基因失去活性[12].也有报道将Cas9蛋白靶向目的基因或染色体上两处位点,则会造成目的基因内或染色体上整段序列的缺失[4,13-15].MMEJ在修复双链断裂时的断裂末端比对过程中使用的微同源序列易导致原始断裂侧翼缺失,进而造成染色体异常如缺失、易位、倒位和其他复杂的重排.Cas9蛋白造成DNA双链断裂后,在损伤序列的同源供体DNA如姐妹染色单体存在的情况下,HDR则以同源DNA序列为模板进行无错修复.若人为地提供一段含有外源基因或替换了特定位置核苷酸的同源供体DNA(donor),则可利用HDR实现精准的基因插入或单碱基编辑(图1)[16].随着CRISPR/Cas9技术的普及和发展,其基本的基因编辑能力和性能已不能满足在基因组工程和各个领域中的应用.例如,除了受限于CRISPR/Cas9系统本身的PAM(protospacer adjacent motif)依赖性和脱靶效应,该系统在应用于代谢工程中时,还被其编辑靶点的数量和效率所制约[17],而应用在人类疾病基因疗法时还需进一步提高该系统的保真性、安全性等因素[18].因此,自张锋团队首次应用CRISPR/Cas9系统在哺乳动物细胞中实现多重编辑后,国内外的科研人员围绕该系统的多元化应用展开了研究,如通过优化Cas9蛋白拓宽该系统的识别范围并提高其靶向特异性[19-21]、通过融合效应结构域的方式使该系统获得动态可控性或进行转录调控[22-24]、为了进行表观基因组编辑或在非HDR介导的细胞中进行单碱基编辑将功能缺失的Cas9蛋白与甲基转移酶或脱氨酶融合等[25-26].2019年,哈佛大学David R. Liu团队开发的先导编辑(prime editing,PE)作为基于gRNA工程的多功能精准基因编辑工具,可以在不引入靶基因双链断裂及同源片段的条件下介导靶向的插入、缺失及所有碱基编辑,大大拓展了CRISPR/Cas9系统多功能基因组编辑的范围和能力[27].多重CRISPR技术是通过同时引入多个gRNA或多种直系同源Cas9蛋白,从而实现基因组上多靶点的同时靶向或多功能正交编辑.该技术不仅极大提高了基因编辑和转录调控的范围和效率,更促进了强大的生物工程应用.因此,如今越来越多的CRISPR/Cas9工具不仅可以实现定点改造,还可以实现多靶点的同时改造.发展趋势证明了基于RNA靶向性的CRISPR/Cas9技术凭借简单可编程性和广泛适用性具有强大优势,同时凸显出CRISPR/Cas9系统与TALENs、ZFNs等相比的优势[28]. ...
Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9
2
2016
... CRISPR系统利用单链向导RNA(single guide RNA,sgRNA或gRNA)靶向目的位点,随后Cas9等核酸酶切断目的区域的双链核酸,诱导细胞内源DNA损伤修复机制工作,最终借助该机制达到编辑遗传信息的目的[10].CRISPR/Cas9系统在发挥基因敲除、基因插入、单碱基编辑等主要编辑手段时主要依靠的是细胞内源的非同源末端连接修复机制(non-homologous end joining,NHEJ)、微同源介导末端连接机制(microhomology-mediated end joining,MMEJ)或同源重组修复机制(homology directed repair,HDR)[11].NHEJ系统进行双链断裂(double-strand breaks, DSBs)修复时,首先Ku异源二聚体识别并结合至两断裂末端,随后由DNA聚合酶和核酸外切酶对该处进行加工处理,最后进行末端连接形成双链.在对末端进行修饰时,由于外切酶和聚合酶结合顺序的不确定性则造成修复位点碱基缺失和插入突变等不同情况,致使目的基因失去活性[12].也有报道将Cas9蛋白靶向目的基因或染色体上两处位点,则会造成目的基因内或染色体上整段序列的缺失[4,13-15].MMEJ在修复双链断裂时的断裂末端比对过程中使用的微同源序列易导致原始断裂侧翼缺失,进而造成染色体异常如缺失、易位、倒位和其他复杂的重排.Cas9蛋白造成DNA双链断裂后,在损伤序列的同源供体DNA如姐妹染色单体存在的情况下,HDR则以同源DNA序列为模板进行无错修复.若人为地提供一段含有外源基因或替换了特定位置核苷酸的同源供体DNA(donor),则可利用HDR实现精准的基因插入或单碱基编辑(图1)[16].随着CRISPR/Cas9技术的普及和发展,其基本的基因编辑能力和性能已不能满足在基因组工程和各个领域中的应用.例如,除了受限于CRISPR/Cas9系统本身的PAM(protospacer adjacent motif)依赖性和脱靶效应,该系统在应用于代谢工程中时,还被其编辑靶点的数量和效率所制约[17],而应用在人类疾病基因疗法时还需进一步提高该系统的保真性、安全性等因素[18].因此,自张锋团队首次应用CRISPR/Cas9系统在哺乳动物细胞中实现多重编辑后,国内外的科研人员围绕该系统的多元化应用展开了研究,如通过优化Cas9蛋白拓宽该系统的识别范围并提高其靶向特异性[19-21]、通过融合效应结构域的方式使该系统获得动态可控性或进行转录调控[22-24]、为了进行表观基因组编辑或在非HDR介导的细胞中进行单碱基编辑将功能缺失的Cas9蛋白与甲基转移酶或脱氨酶融合等[25-26].2019年,哈佛大学David R. Liu团队开发的先导编辑(prime editing,PE)作为基于gRNA工程的多功能精准基因编辑工具,可以在不引入靶基因双链断裂及同源片段的条件下介导靶向的插入、缺失及所有碱基编辑,大大拓展了CRISPR/Cas9系统多功能基因组编辑的范围和能力[27].多重CRISPR技术是通过同时引入多个gRNA或多种直系同源Cas9蛋白,从而实现基因组上多靶点的同时靶向或多功能正交编辑.该技术不仅极大提高了基因编辑和转录调控的范围和效率,更促进了强大的生物工程应用.因此,如今越来越多的CRISPR/Cas9工具不仅可以实现定点改造,还可以实现多靶点的同时改造.发展趋势证明了基于RNA靶向性的CRISPR/Cas9技术凭借简单可编程性和广泛适用性具有强大优势,同时凸显出CRISPR/Cas9系统与TALENs、ZFNs等相比的优势[28]. ...
... 单点突变是人类遗传病中最常见的基因变异,且该变异通常会造成严重的后果.如镰刀形红细胞贫血症患者由于其血红蛋白基因中一对碱基对发生突变使得血红蛋白β亚基的N端第6个氨基酸发生改变,最终使血红蛋白结构发生改变并失去氧气输送功能[104].因此,利用CRISPR/Ca9系统的靶向性进行高效的单核苷酸转换是建立遗传病模型、开发基因纠正疗法的重要手段,同时促进了基础生物学研究和合成生物学的发展.通过共递送一段含有待编辑碱基的同源DNA,CRIPSR/Cas9系统可在HDR活性高的细胞中以较高的效率将目的碱基替换为任意碱基[16],而在HDR活性较低的细胞如大多数细菌和有丝分裂后期细胞中,该策略则无法正常发挥作用.此外,诱导HDR时Cas9对靶序列产生的双链切割带来的脱靶诱变风险以及HDR进行DNA修复时对细胞活力产生的不利影响均会使CRIPSR/Cas9系统在遗传病的基因疗法等应用场景上受到限制[105-106]. ...
A Cas9-based toolkit to program gene expression in Saccharomyces cerevisiae
1
2016
... CRISPR系统利用单链向导RNA(single guide RNA,sgRNA或gRNA)靶向目的位点,随后Cas9等核酸酶切断目的区域的双链核酸,诱导细胞内源DNA损伤修复机制工作,最终借助该机制达到编辑遗传信息的目的[10].CRISPR/Cas9系统在发挥基因敲除、基因插入、单碱基编辑等主要编辑手段时主要依靠的是细胞内源的非同源末端连接修复机制(non-homologous end joining,NHEJ)、微同源介导末端连接机制(microhomology-mediated end joining,MMEJ)或同源重组修复机制(homology directed repair,HDR)[11].NHEJ系统进行双链断裂(double-strand breaks, DSBs)修复时,首先Ku异源二聚体识别并结合至两断裂末端,随后由DNA聚合酶和核酸外切酶对该处进行加工处理,最后进行末端连接形成双链.在对末端进行修饰时,由于外切酶和聚合酶结合顺序的不确定性则造成修复位点碱基缺失和插入突变等不同情况,致使目的基因失去活性[12].也有报道将Cas9蛋白靶向目的基因或染色体上两处位点,则会造成目的基因内或染色体上整段序列的缺失[4,13-15].MMEJ在修复双链断裂时的断裂末端比对过程中使用的微同源序列易导致原始断裂侧翼缺失,进而造成染色体异常如缺失、易位、倒位和其他复杂的重排.Cas9蛋白造成DNA双链断裂后,在损伤序列的同源供体DNA如姐妹染色单体存在的情况下,HDR则以同源DNA序列为模板进行无错修复.若人为地提供一段含有外源基因或替换了特定位置核苷酸的同源供体DNA(donor),则可利用HDR实现精准的基因插入或单碱基编辑(图1)[16].随着CRISPR/Cas9技术的普及和发展,其基本的基因编辑能力和性能已不能满足在基因组工程和各个领域中的应用.例如,除了受限于CRISPR/Cas9系统本身的PAM(protospacer adjacent motif)依赖性和脱靶效应,该系统在应用于代谢工程中时,还被其编辑靶点的数量和效率所制约[17],而应用在人类疾病基因疗法时还需进一步提高该系统的保真性、安全性等因素[18].因此,自张锋团队首次应用CRISPR/Cas9系统在哺乳动物细胞中实现多重编辑后,国内外的科研人员围绕该系统的多元化应用展开了研究,如通过优化Cas9蛋白拓宽该系统的识别范围并提高其靶向特异性[19-21]、通过融合效应结构域的方式使该系统获得动态可控性或进行转录调控[22-24]、为了进行表观基因组编辑或在非HDR介导的细胞中进行单碱基编辑将功能缺失的Cas9蛋白与甲基转移酶或脱氨酶融合等[25-26].2019年,哈佛大学David R. Liu团队开发的先导编辑(prime editing,PE)作为基于gRNA工程的多功能精准基因编辑工具,可以在不引入靶基因双链断裂及同源片段的条件下介导靶向的插入、缺失及所有碱基编辑,大大拓展了CRISPR/Cas9系统多功能基因组编辑的范围和能力[27].多重CRISPR技术是通过同时引入多个gRNA或多种直系同源Cas9蛋白,从而实现基因组上多靶点的同时靶向或多功能正交编辑.该技术不仅极大提高了基因编辑和转录调控的范围和效率,更促进了强大的生物工程应用.因此,如今越来越多的CRISPR/Cas9工具不仅可以实现定点改造,还可以实现多靶点的同时改造.发展趋势证明了基于RNA靶向性的CRISPR/Cas9技术凭借简单可编程性和广泛适用性具有强大优势,同时凸显出CRISPR/Cas9系统与TALENs、ZFNs等相比的优势[28]. ...
In situ gene therapy via AAV-CRISPR-Cas9-mediated targeted gene regulation
1
2018
... CRISPR系统利用单链向导RNA(single guide RNA,sgRNA或gRNA)靶向目的位点,随后Cas9等核酸酶切断目的区域的双链核酸,诱导细胞内源DNA损伤修复机制工作,最终借助该机制达到编辑遗传信息的目的[10].CRISPR/Cas9系统在发挥基因敲除、基因插入、单碱基编辑等主要编辑手段时主要依靠的是细胞内源的非同源末端连接修复机制(non-homologous end joining,NHEJ)、微同源介导末端连接机制(microhomology-mediated end joining,MMEJ)或同源重组修复机制(homology directed repair,HDR)[11].NHEJ系统进行双链断裂(double-strand breaks, DSBs)修复时,首先Ku异源二聚体识别并结合至两断裂末端,随后由DNA聚合酶和核酸外切酶对该处进行加工处理,最后进行末端连接形成双链.在对末端进行修饰时,由于外切酶和聚合酶结合顺序的不确定性则造成修复位点碱基缺失和插入突变等不同情况,致使目的基因失去活性[12].也有报道将Cas9蛋白靶向目的基因或染色体上两处位点,则会造成目的基因内或染色体上整段序列的缺失[4,13-15].MMEJ在修复双链断裂时的断裂末端比对过程中使用的微同源序列易导致原始断裂侧翼缺失,进而造成染色体异常如缺失、易位、倒位和其他复杂的重排.Cas9蛋白造成DNA双链断裂后,在损伤序列的同源供体DNA如姐妹染色单体存在的情况下,HDR则以同源DNA序列为模板进行无错修复.若人为地提供一段含有外源基因或替换了特定位置核苷酸的同源供体DNA(donor),则可利用HDR实现精准的基因插入或单碱基编辑(图1)[16].随着CRISPR/Cas9技术的普及和发展,其基本的基因编辑能力和性能已不能满足在基因组工程和各个领域中的应用.例如,除了受限于CRISPR/Cas9系统本身的PAM(protospacer adjacent motif)依赖性和脱靶效应,该系统在应用于代谢工程中时,还被其编辑靶点的数量和效率所制约[17],而应用在人类疾病基因疗法时还需进一步提高该系统的保真性、安全性等因素[18].因此,自张锋团队首次应用CRISPR/Cas9系统在哺乳动物细胞中实现多重编辑后,国内外的科研人员围绕该系统的多元化应用展开了研究,如通过优化Cas9蛋白拓宽该系统的识别范围并提高其靶向特异性[19-21]、通过融合效应结构域的方式使该系统获得动态可控性或进行转录调控[22-24]、为了进行表观基因组编辑或在非HDR介导的细胞中进行单碱基编辑将功能缺失的Cas9蛋白与甲基转移酶或脱氨酶融合等[25-26].2019年,哈佛大学David R. Liu团队开发的先导编辑(prime editing,PE)作为基于gRNA工程的多功能精准基因编辑工具,可以在不引入靶基因双链断裂及同源片段的条件下介导靶向的插入、缺失及所有碱基编辑,大大拓展了CRISPR/Cas9系统多功能基因组编辑的范围和能力[27].多重CRISPR技术是通过同时引入多个gRNA或多种直系同源Cas9蛋白,从而实现基因组上多靶点的同时靶向或多功能正交编辑.该技术不仅极大提高了基因编辑和转录调控的范围和效率,更促进了强大的生物工程应用.因此,如今越来越多的CRISPR/Cas9工具不仅可以实现定点改造,还可以实现多靶点的同时改造.发展趋势证明了基于RNA靶向性的CRISPR/Cas9技术凭借简单可编程性和广泛适用性具有强大优势,同时凸显出CRISPR/Cas9系统与TALENs、ZFNs等相比的优势[28]. ...
Engineered CRISPR-Cas9 nucleases with altered PAM specificities
5
2015
... CRISPR系统利用单链向导RNA(single guide RNA,sgRNA或gRNA)靶向目的位点,随后Cas9等核酸酶切断目的区域的双链核酸,诱导细胞内源DNA损伤修复机制工作,最终借助该机制达到编辑遗传信息的目的[10].CRISPR/Cas9系统在发挥基因敲除、基因插入、单碱基编辑等主要编辑手段时主要依靠的是细胞内源的非同源末端连接修复机制(non-homologous end joining,NHEJ)、微同源介导末端连接机制(microhomology-mediated end joining,MMEJ)或同源重组修复机制(homology directed repair,HDR)[11].NHEJ系统进行双链断裂(double-strand breaks, DSBs)修复时,首先Ku异源二聚体识别并结合至两断裂末端,随后由DNA聚合酶和核酸外切酶对该处进行加工处理,最后进行末端连接形成双链.在对末端进行修饰时,由于外切酶和聚合酶结合顺序的不确定性则造成修复位点碱基缺失和插入突变等不同情况,致使目的基因失去活性[12].也有报道将Cas9蛋白靶向目的基因或染色体上两处位点,则会造成目的基因内或染色体上整段序列的缺失[4,13-15].MMEJ在修复双链断裂时的断裂末端比对过程中使用的微同源序列易导致原始断裂侧翼缺失,进而造成染色体异常如缺失、易位、倒位和其他复杂的重排.Cas9蛋白造成DNA双链断裂后,在损伤序列的同源供体DNA如姐妹染色单体存在的情况下,HDR则以同源DNA序列为模板进行无错修复.若人为地提供一段含有外源基因或替换了特定位置核苷酸的同源供体DNA(donor),则可利用HDR实现精准的基因插入或单碱基编辑(图1)[16].随着CRISPR/Cas9技术的普及和发展,其基本的基因编辑能力和性能已不能满足在基因组工程和各个领域中的应用.例如,除了受限于CRISPR/Cas9系统本身的PAM(protospacer adjacent motif)依赖性和脱靶效应,该系统在应用于代谢工程中时,还被其编辑靶点的数量和效率所制约[17],而应用在人类疾病基因疗法时还需进一步提高该系统的保真性、安全性等因素[18].因此,自张锋团队首次应用CRISPR/Cas9系统在哺乳动物细胞中实现多重编辑后,国内外的科研人员围绕该系统的多元化应用展开了研究,如通过优化Cas9蛋白拓宽该系统的识别范围并提高其靶向特异性[19-21]、通过融合效应结构域的方式使该系统获得动态可控性或进行转录调控[22-24]、为了进行表观基因组编辑或在非HDR介导的细胞中进行单碱基编辑将功能缺失的Cas9蛋白与甲基转移酶或脱氨酶融合等[25-26].2019年,哈佛大学David R. Liu团队开发的先导编辑(prime editing,PE)作为基于gRNA工程的多功能精准基因编辑工具,可以在不引入靶基因双链断裂及同源片段的条件下介导靶向的插入、缺失及所有碱基编辑,大大拓展了CRISPR/Cas9系统多功能基因组编辑的范围和能力[27].多重CRISPR技术是通过同时引入多个gRNA或多种直系同源Cas9蛋白,从而实现基因组上多靶点的同时靶向或多功能正交编辑.该技术不仅极大提高了基因编辑和转录调控的范围和效率,更促进了强大的生物工程应用.因此,如今越来越多的CRISPR/Cas9工具不仅可以实现定点改造,还可以实现多靶点的同时改造.发展趋势证明了基于RNA靶向性的CRISPR/Cas9技术凭借简单可编程性和广泛适用性具有强大优势,同时凸显出CRISPR/Cas9系统与TALENs、ZFNs等相比的优势[28]. ...
... Major Cas9 variants and orthologues as well as their features
Table 1| 变体及直系同源 | 改造位点或方式 | PAM | 特性 | 参考文献 |
|---|
| SpCas9 | 原生酿脓链球菌Cas9 | NGG | 1368个氨基酸 | [29] |
| dCas9 | D10A,H840A | NGG | 核酸酶活性失活 | [29] |
| nCas9(D10A) | D10A | NGG | 切口酶,非靶向链切割活性失活 | [30] |
| nCas9(H840A) | H840A | NGG | 切口酶,靶向链切割活性失活 | [30] |
| SpCas9NG | R1335V,L1111R,D1135V,G1218R,E1219F,A1322R,T1337R | NG | PAM改变;通过Cas9理性设计获得 | [31] |
| VRERSpCas9 | D1135A,G1218R,R1335E,T1337R | NGCG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| VQRSpCas9 | D1135V,R1335Q,T1337R | NGAN,NGNG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| EQRSpCas9 | D1335E,R1335Q,T1337R | NGAG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| xCas9 | A262T,R324L,S409I,E480K,E543D,M694I,E1219V | NG,GAA,GAT | PAM改变;通过噬菌体辅助持续进化获得 | [20] |
| SpG | D1135L,S1136W,G1218K,E1219Q,R1335Q,T1337R | NGN | PAM改变;通过SpCas9理性设计获得 | [32] |
| SpRY | SpG,A61R,L1111R,A1322R,N1317R,R1333P | NRN,NYN | PAM改变;通过对SpG进一步理性设计获得 | [32] |
| evoCas9 | M495V, Y515N, K526E, R661Q | NGG | 保真性提高,编辑效率接近天然SpCas9 | [33] |
| fCas9 | SpdCas9与FokI融合 | NGG | 保真性提高;通过将FokI核酸酶与dCas9融合获得 | [34] |
| StCas9 | 原生嗜热链球菌Cas9 | NNAGAAW | 1121个氨基酸 | [35] |
| NmCas9 | 原生脑膜炎奈瑟菌Cas9 | NNNNGATT | 1082个氨基酸 | [36] |
| SaCas9 | 原生金黄色葡萄球菌Cas9 | NNGRRT | 1053个氨基酸;基因组编辑效率与SpCas9相当 | [37] |
| CjCas9 | 原生曲状杆菌Cas9 | NNNVRYM | 984个氨基酸 | [38] |
注:V代表A、C或G;R代表A或G;Y代表C或T;M代表A或C. ...
... [
19]
| EQRSpCas9 | D1335E,R1335Q,T1337R | NGAG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| xCas9 | A262T,R324L,S409I,E480K,E543D,M694I,E1219V | NG,GAA,GAT | PAM改变;通过噬菌体辅助持续进化获得 | [20] |
| SpG | D1135L,S1136W,G1218K,E1219Q,R1335Q,T1337R | NGN | PAM改变;通过SpCas9理性设计获得 | [32] |
| SpRY | SpG,A61R,L1111R,A1322R,N1317R,R1333P | NRN,NYN | PAM改变;通过对SpG进一步理性设计获得 | [32] |
| evoCas9 | M495V, Y515N, K526E, R661Q | NGG | 保真性提高,编辑效率接近天然SpCas9 | [33] |
| fCas9 | SpdCas9与FokI融合 | NGG | 保真性提高;通过将FokI核酸酶与dCas9融合获得 | [34] |
| StCas9 | 原生嗜热链球菌Cas9 | NNAGAAW | 1121个氨基酸 | [35] |
| NmCas9 | 原生脑膜炎奈瑟菌Cas9 | NNNNGATT | 1082个氨基酸 | [36] |
| SaCas9 | 原生金黄色葡萄球菌Cas9 | NNGRRT | 1053个氨基酸;基因组编辑效率与SpCas9相当 | [37] |
| CjCas9 | 原生曲状杆菌Cas9 | NNNVRYM | 984个氨基酸 | [38] |
注:V代表A、C或G;R代表A或G;Y代表C或T;M代表A或C. ...
... [
19]
| xCas9 | A262T,R324L,S409I,E480K,E543D,M694I,E1219V | NG,GAA,GAT | PAM改变;通过噬菌体辅助持续进化获得 | [20] |
| SpG | D1135L,S1136W,G1218K,E1219Q,R1335Q,T1337R | NGN | PAM改变;通过SpCas9理性设计获得 | [32] |
| SpRY | SpG,A61R,L1111R,A1322R,N1317R,R1333P | NRN,NYN | PAM改变;通过对SpG进一步理性设计获得 | [32] |
| evoCas9 | M495V, Y515N, K526E, R661Q | NGG | 保真性提高,编辑效率接近天然SpCas9 | [33] |
| fCas9 | SpdCas9与FokI融合 | NGG | 保真性提高;通过将FokI核酸酶与dCas9融合获得 | [34] |
| StCas9 | 原生嗜热链球菌Cas9 | NNAGAAW | 1121个氨基酸 | [35] |
| NmCas9 | 原生脑膜炎奈瑟菌Cas9 | NNNNGATT | 1082个氨基酸 | [36] |
| SaCas9 | 原生金黄色葡萄球菌Cas9 | NNGRRT | 1053个氨基酸;基因组编辑效率与SpCas9相当 | [37] |
| CjCas9 | 原生曲状杆菌Cas9 | NNNVRYM | 984个氨基酸 | [38] |
注:V代表A、C或G;R代表A或G;Y代表C或T;M代表A或C. ...
... 尽管CRISPR/Cas9系统已在多种生物和细胞的基因组上得到了应用[46-51],Cas9对于PAM序列NGG的严格识别、复杂基因组中gRNA潜在的脱靶效应以及该系统作为医疗手段使用时的保真性仍大大限制其在更广范围下的应用与开发.如在人类基因组DNA上平均每8个碱基对存在一个NGG位点[52],Fu等[53]在人类细胞中表征了CRISPR/Cas9系统的脱靶效应,结果显示该系统的脱靶率高达66%.因此,研究人员通过工程化Cas9蛋白或gRNA等方式,使CRISPR/Cas9系统的靶向范围、靶向特异性和保真性均得到了拓展和提高[19-20].2018年,张锋团队和Osamu Nureki团队[31]通过对SpCas9进行理性设计获得了能够识别NG PAM序列的突变体蛋白SpCas9-NG,并证明了其在人类细胞中的基因组编辑能力以及与胞苷脱氨酶融合后仍具有单碱基编辑能力.2020年,David R. Liu团队[54]利用噬菌体辅助持续进化(phage-assisted continuous evolution,PACE)方法获得了三种识别非G PAM序列的SpCas9突变体,从而进一步提高了CRISPR/Cas9系统的靶向范围.Walton等[32]通过改造与PAM序列直接作用的氨基酸位点获得了能稳定识别NRN PAM序列的突变体蛋白SpRY,同时其能以较低效率识别NYN PAM序列,使其在靶点识别上几乎不受PAM限制.Hu等[20]应用PACE方法获得一种能够识别NG、GAA及GAT等PAM序列的SpCas9突变体xCas9,同时其能以优于SpCas9的特异性靶向NGG,且对各种PAM的靶向能力均在人类细胞中的基因编辑、转录调控以及胞嘧啶与腺嘌呤核苷酸的碱基修饰中得到了应用.2014年,Guilinger等[34]将FokⅠ核酸酶与核酸酶活性失活的Cas9蛋白(catalytically inactive Cas9,dCas9)融合后得到了一种融合蛋白fCas9,只有两个fCas9单体同时结合在靶基因附近时才能切割目的基因,使其在人类细胞中进行基因编辑的特异性比野生型Cas9提高了140倍.多种研究已报道了将SpCas9或sgRNA工程化后,通过质粒载体携带并表达CRISPR/Cas9系统或通过RNP递送的方式均能够在基因组范围上降低CRISPR/Cas9系统的脱靶效应,并显著提高该系统的保真性[33,55-58].2018年,Casini等[33]筛选并表征了能够提高CRISPR/Cas9系统编辑效率的Cas9 REC3结构域突变库并组合了四种有利突变后,获得了一种保真性比野生型SpCas9提高了79倍的突变体蛋白evoCas9,同时编辑效率接近野生型SpCas9.除了对Cas9蛋白进行优化,对gRNA不断进行工程化改造也能使CRISPR/Cas9系统的性能得到显著提升.2014年,Fu等[44]证明了间隔区截短后的gRNA的脱靶率明显低于全长的gRNA(20 nt),最大降低幅度达5000倍.2019年,Kocak等[21]在工程化设计的sgRNA上的间隔区加入一“发夹”形RNA二级结构,该结构能够在发生脱靶编辑时在脱靶位点处提高gRNA或crRNA链入侵的热力学屏障而并不影响中靶效率,使CRISPR系统的特异性增加了两个数量级.2020年,Rose等[59]基于过短的gRNA(间隔区小于16 nt)与Cas9形成的RNP只能结合却不能切割靶位点特性设计的失活RNA(dead-RNAs,dRNAs)可保护多个脱靶位点,从而使得SpCas9的靶向特异性提高了40倍.相较于工程化改造的高特异性Cas9突变体来说,通过缩短gRNA的间隔区来提高靶向特异性的方法在设计和应用方面均更易用和灵活,预计将成为未来的研究热点方向. ...
Evolved Cas9 variants with broad PAM compatibility and high DNA specificity
3
2018
... Major Cas9 variants and orthologues as well as their features
Table 1| 变体及直系同源 | 改造位点或方式 | PAM | 特性 | 参考文献 |
|---|
| SpCas9 | 原生酿脓链球菌Cas9 | NGG | 1368个氨基酸 | [29] |
| dCas9 | D10A,H840A | NGG | 核酸酶活性失活 | [29] |
| nCas9(D10A) | D10A | NGG | 切口酶,非靶向链切割活性失活 | [30] |
| nCas9(H840A) | H840A | NGG | 切口酶,靶向链切割活性失活 | [30] |
| SpCas9NG | R1335V,L1111R,D1135V,G1218R,E1219F,A1322R,T1337R | NG | PAM改变;通过Cas9理性设计获得 | [31] |
| VRERSpCas9 | D1135A,G1218R,R1335E,T1337R | NGCG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| VQRSpCas9 | D1135V,R1335Q,T1337R | NGAN,NGNG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| EQRSpCas9 | D1335E,R1335Q,T1337R | NGAG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| xCas9 | A262T,R324L,S409I,E480K,E543D,M694I,E1219V | NG,GAA,GAT | PAM改变;通过噬菌体辅助持续进化获得 | [20] |
| SpG | D1135L,S1136W,G1218K,E1219Q,R1335Q,T1337R | NGN | PAM改变;通过SpCas9理性设计获得 | [32] |
| SpRY | SpG,A61R,L1111R,A1322R,N1317R,R1333P | NRN,NYN | PAM改变;通过对SpG进一步理性设计获得 | [32] |
| evoCas9 | M495V, Y515N, K526E, R661Q | NGG | 保真性提高,编辑效率接近天然SpCas9 | [33] |
| fCas9 | SpdCas9与FokI融合 | NGG | 保真性提高;通过将FokI核酸酶与dCas9融合获得 | [34] |
| StCas9 | 原生嗜热链球菌Cas9 | NNAGAAW | 1121个氨基酸 | [35] |
| NmCas9 | 原生脑膜炎奈瑟菌Cas9 | NNNNGATT | 1082个氨基酸 | [36] |
| SaCas9 | 原生金黄色葡萄球菌Cas9 | NNGRRT | 1053个氨基酸;基因组编辑效率与SpCas9相当 | [37] |
| CjCas9 | 原生曲状杆菌Cas9 | NNNVRYM | 984个氨基酸 | [38] |
注:V代表A、C或G;R代表A或G;Y代表C或T;M代表A或C. ...
... 尽管CRISPR/Cas9系统已在多种生物和细胞的基因组上得到了应用[46-51],Cas9对于PAM序列NGG的严格识别、复杂基因组中gRNA潜在的脱靶效应以及该系统作为医疗手段使用时的保真性仍大大限制其在更广范围下的应用与开发.如在人类基因组DNA上平均每8个碱基对存在一个NGG位点[52],Fu等[53]在人类细胞中表征了CRISPR/Cas9系统的脱靶效应,结果显示该系统的脱靶率高达66%.因此,研究人员通过工程化Cas9蛋白或gRNA等方式,使CRISPR/Cas9系统的靶向范围、靶向特异性和保真性均得到了拓展和提高[19-20].2018年,张锋团队和Osamu Nureki团队[31]通过对SpCas9进行理性设计获得了能够识别NG PAM序列的突变体蛋白SpCas9-NG,并证明了其在人类细胞中的基因组编辑能力以及与胞苷脱氨酶融合后仍具有单碱基编辑能力.2020年,David R. Liu团队[54]利用噬菌体辅助持续进化(phage-assisted continuous evolution,PACE)方法获得了三种识别非G PAM序列的SpCas9突变体,从而进一步提高了CRISPR/Cas9系统的靶向范围.Walton等[32]通过改造与PAM序列直接作用的氨基酸位点获得了能稳定识别NRN PAM序列的突变体蛋白SpRY,同时其能以较低效率识别NYN PAM序列,使其在靶点识别上几乎不受PAM限制.Hu等[20]应用PACE方法获得一种能够识别NG、GAA及GAT等PAM序列的SpCas9突变体xCas9,同时其能以优于SpCas9的特异性靶向NGG,且对各种PAM的靶向能力均在人类细胞中的基因编辑、转录调控以及胞嘧啶与腺嘌呤核苷酸的碱基修饰中得到了应用.2014年,Guilinger等[34]将FokⅠ核酸酶与核酸酶活性失活的Cas9蛋白(catalytically inactive Cas9,dCas9)融合后得到了一种融合蛋白fCas9,只有两个fCas9单体同时结合在靶基因附近时才能切割目的基因,使其在人类细胞中进行基因编辑的特异性比野生型Cas9提高了140倍.多种研究已报道了将SpCas9或sgRNA工程化后,通过质粒载体携带并表达CRISPR/Cas9系统或通过RNP递送的方式均能够在基因组范围上降低CRISPR/Cas9系统的脱靶效应,并显著提高该系统的保真性[33,55-58].2018年,Casini等[33]筛选并表征了能够提高CRISPR/Cas9系统编辑效率的Cas9 REC3结构域突变库并组合了四种有利突变后,获得了一种保真性比野生型SpCas9提高了79倍的突变体蛋白evoCas9,同时编辑效率接近野生型SpCas9.除了对Cas9蛋白进行优化,对gRNA不断进行工程化改造也能使CRISPR/Cas9系统的性能得到显著提升.2014年,Fu等[44]证明了间隔区截短后的gRNA的脱靶率明显低于全长的gRNA(20 nt),最大降低幅度达5000倍.2019年,Kocak等[21]在工程化设计的sgRNA上的间隔区加入一“发夹”形RNA二级结构,该结构能够在发生脱靶编辑时在脱靶位点处提高gRNA或crRNA链入侵的热力学屏障而并不影响中靶效率,使CRISPR系统的特异性增加了两个数量级.2020年,Rose等[59]基于过短的gRNA(间隔区小于16 nt)与Cas9形成的RNP只能结合却不能切割靶位点特性设计的失活RNA(dead-RNAs,dRNAs)可保护多个脱靶位点,从而使得SpCas9的靶向特异性提高了40倍.相较于工程化改造的高特异性Cas9突变体来说,通过缩短gRNA的间隔区来提高靶向特异性的方法在设计和应用方面均更易用和灵活,预计将成为未来的研究热点方向. ...
... [20]应用PACE方法获得一种能够识别NG、GAA及GAT等PAM序列的SpCas9突变体xCas9,同时其能以优于SpCas9的特异性靶向NGG,且对各种PAM的靶向能力均在人类细胞中的基因编辑、转录调控以及胞嘧啶与腺嘌呤核苷酸的碱基修饰中得到了应用.2014年,Guilinger等[34]将FokⅠ核酸酶与核酸酶活性失活的Cas9蛋白(catalytically inactive Cas9,dCas9)融合后得到了一种融合蛋白fCas9,只有两个fCas9单体同时结合在靶基因附近时才能切割目的基因,使其在人类细胞中进行基因编辑的特异性比野生型Cas9提高了140倍.多种研究已报道了将SpCas9或sgRNA工程化后,通过质粒载体携带并表达CRISPR/Cas9系统或通过RNP递送的方式均能够在基因组范围上降低CRISPR/Cas9系统的脱靶效应,并显著提高该系统的保真性[33,55-58].2018年,Casini等[33]筛选并表征了能够提高CRISPR/Cas9系统编辑效率的Cas9 REC3结构域突变库并组合了四种有利突变后,获得了一种保真性比野生型SpCas9提高了79倍的突变体蛋白evoCas9,同时编辑效率接近野生型SpCas9.除了对Cas9蛋白进行优化,对gRNA不断进行工程化改造也能使CRISPR/Cas9系统的性能得到显著提升.2014年,Fu等[44]证明了间隔区截短后的gRNA的脱靶率明显低于全长的gRNA(20 nt),最大降低幅度达5000倍.2019年,Kocak等[21]在工程化设计的sgRNA上的间隔区加入一“发夹”形RNA二级结构,该结构能够在发生脱靶编辑时在脱靶位点处提高gRNA或crRNA链入侵的热力学屏障而并不影响中靶效率,使CRISPR系统的特异性增加了两个数量级.2020年,Rose等[59]基于过短的gRNA(间隔区小于16 nt)与Cas9形成的RNP只能结合却不能切割靶位点特性设计的失活RNA(dead-RNAs,dRNAs)可保护多个脱靶位点,从而使得SpCas9的靶向特异性提高了40倍.相较于工程化改造的高特异性Cas9突变体来说,通过缩短gRNA的间隔区来提高靶向特异性的方法在设计和应用方面均更易用和灵活,预计将成为未来的研究热点方向. ...
Increasing the specificity of CRISPR systems with engineered RNA secondary structures
2
2019
... CRISPR系统利用单链向导RNA(single guide RNA,sgRNA或gRNA)靶向目的位点,随后Cas9等核酸酶切断目的区域的双链核酸,诱导细胞内源DNA损伤修复机制工作,最终借助该机制达到编辑遗传信息的目的[10].CRISPR/Cas9系统在发挥基因敲除、基因插入、单碱基编辑等主要编辑手段时主要依靠的是细胞内源的非同源末端连接修复机制(non-homologous end joining,NHEJ)、微同源介导末端连接机制(microhomology-mediated end joining,MMEJ)或同源重组修复机制(homology directed repair,HDR)[11].NHEJ系统进行双链断裂(double-strand breaks, DSBs)修复时,首先Ku异源二聚体识别并结合至两断裂末端,随后由DNA聚合酶和核酸外切酶对该处进行加工处理,最后进行末端连接形成双链.在对末端进行修饰时,由于外切酶和聚合酶结合顺序的不确定性则造成修复位点碱基缺失和插入突变等不同情况,致使目的基因失去活性[12].也有报道将Cas9蛋白靶向目的基因或染色体上两处位点,则会造成目的基因内或染色体上整段序列的缺失[4,13-15].MMEJ在修复双链断裂时的断裂末端比对过程中使用的微同源序列易导致原始断裂侧翼缺失,进而造成染色体异常如缺失、易位、倒位和其他复杂的重排.Cas9蛋白造成DNA双链断裂后,在损伤序列的同源供体DNA如姐妹染色单体存在的情况下,HDR则以同源DNA序列为模板进行无错修复.若人为地提供一段含有外源基因或替换了特定位置核苷酸的同源供体DNA(donor),则可利用HDR实现精准的基因插入或单碱基编辑(图1)[16].随着CRISPR/Cas9技术的普及和发展,其基本的基因编辑能力和性能已不能满足在基因组工程和各个领域中的应用.例如,除了受限于CRISPR/Cas9系统本身的PAM(protospacer adjacent motif)依赖性和脱靶效应,该系统在应用于代谢工程中时,还被其编辑靶点的数量和效率所制约[17],而应用在人类疾病基因疗法时还需进一步提高该系统的保真性、安全性等因素[18].因此,自张锋团队首次应用CRISPR/Cas9系统在哺乳动物细胞中实现多重编辑后,国内外的科研人员围绕该系统的多元化应用展开了研究,如通过优化Cas9蛋白拓宽该系统的识别范围并提高其靶向特异性[19-21]、通过融合效应结构域的方式使该系统获得动态可控性或进行转录调控[22-24]、为了进行表观基因组编辑或在非HDR介导的细胞中进行单碱基编辑将功能缺失的Cas9蛋白与甲基转移酶或脱氨酶融合等[25-26].2019年,哈佛大学David R. Liu团队开发的先导编辑(prime editing,PE)作为基于gRNA工程的多功能精准基因编辑工具,可以在不引入靶基因双链断裂及同源片段的条件下介导靶向的插入、缺失及所有碱基编辑,大大拓展了CRISPR/Cas9系统多功能基因组编辑的范围和能力[27].多重CRISPR技术是通过同时引入多个gRNA或多种直系同源Cas9蛋白,从而实现基因组上多靶点的同时靶向或多功能正交编辑.该技术不仅极大提高了基因编辑和转录调控的范围和效率,更促进了强大的生物工程应用.因此,如今越来越多的CRISPR/Cas9工具不仅可以实现定点改造,还可以实现多靶点的同时改造.发展趋势证明了基于RNA靶向性的CRISPR/Cas9技术凭借简单可编程性和广泛适用性具有强大优势,同时凸显出CRISPR/Cas9系统与TALENs、ZFNs等相比的优势[28]. ...
... 尽管CRISPR/Cas9系统已在多种生物和细胞的基因组上得到了应用[46-51],Cas9对于PAM序列NGG的严格识别、复杂基因组中gRNA潜在的脱靶效应以及该系统作为医疗手段使用时的保真性仍大大限制其在更广范围下的应用与开发.如在人类基因组DNA上平均每8个碱基对存在一个NGG位点[52],Fu等[53]在人类细胞中表征了CRISPR/Cas9系统的脱靶效应,结果显示该系统的脱靶率高达66%.因此,研究人员通过工程化Cas9蛋白或gRNA等方式,使CRISPR/Cas9系统的靶向范围、靶向特异性和保真性均得到了拓展和提高[19-20].2018年,张锋团队和Osamu Nureki团队[31]通过对SpCas9进行理性设计获得了能够识别NG PAM序列的突变体蛋白SpCas9-NG,并证明了其在人类细胞中的基因组编辑能力以及与胞苷脱氨酶融合后仍具有单碱基编辑能力.2020年,David R. Liu团队[54]利用噬菌体辅助持续进化(phage-assisted continuous evolution,PACE)方法获得了三种识别非G PAM序列的SpCas9突变体,从而进一步提高了CRISPR/Cas9系统的靶向范围.Walton等[32]通过改造与PAM序列直接作用的氨基酸位点获得了能稳定识别NRN PAM序列的突变体蛋白SpRY,同时其能以较低效率识别NYN PAM序列,使其在靶点识别上几乎不受PAM限制.Hu等[20]应用PACE方法获得一种能够识别NG、GAA及GAT等PAM序列的SpCas9突变体xCas9,同时其能以优于SpCas9的特异性靶向NGG,且对各种PAM的靶向能力均在人类细胞中的基因编辑、转录调控以及胞嘧啶与腺嘌呤核苷酸的碱基修饰中得到了应用.2014年,Guilinger等[34]将FokⅠ核酸酶与核酸酶活性失活的Cas9蛋白(catalytically inactive Cas9,dCas9)融合后得到了一种融合蛋白fCas9,只有两个fCas9单体同时结合在靶基因附近时才能切割目的基因,使其在人类细胞中进行基因编辑的特异性比野生型Cas9提高了140倍.多种研究已报道了将SpCas9或sgRNA工程化后,通过质粒载体携带并表达CRISPR/Cas9系统或通过RNP递送的方式均能够在基因组范围上降低CRISPR/Cas9系统的脱靶效应,并显著提高该系统的保真性[33,55-58].2018年,Casini等[33]筛选并表征了能够提高CRISPR/Cas9系统编辑效率的Cas9 REC3结构域突变库并组合了四种有利突变后,获得了一种保真性比野生型SpCas9提高了79倍的突变体蛋白evoCas9,同时编辑效率接近野生型SpCas9.除了对Cas9蛋白进行优化,对gRNA不断进行工程化改造也能使CRISPR/Cas9系统的性能得到显著提升.2014年,Fu等[44]证明了间隔区截短后的gRNA的脱靶率明显低于全长的gRNA(20 nt),最大降低幅度达5000倍.2019年,Kocak等[21]在工程化设计的sgRNA上的间隔区加入一“发夹”形RNA二级结构,该结构能够在发生脱靶编辑时在脱靶位点处提高gRNA或crRNA链入侵的热力学屏障而并不影响中靶效率,使CRISPR系统的特异性增加了两个数量级.2020年,Rose等[59]基于过短的gRNA(间隔区小于16 nt)与Cas9形成的RNP只能结合却不能切割靶位点特性设计的失活RNA(dead-RNAs,dRNAs)可保护多个脱靶位点,从而使得SpCas9的靶向特异性提高了40倍.相较于工程化改造的高特异性Cas9突变体来说,通过缩短gRNA的间隔区来提高靶向特异性的方法在设计和应用方面均更易用和灵活,预计将成为未来的研究热点方向. ...
Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression
5
2013
... CRISPR系统利用单链向导RNA(single guide RNA,sgRNA或gRNA)靶向目的位点,随后Cas9等核酸酶切断目的区域的双链核酸,诱导细胞内源DNA损伤修复机制工作,最终借助该机制达到编辑遗传信息的目的[10].CRISPR/Cas9系统在发挥基因敲除、基因插入、单碱基编辑等主要编辑手段时主要依靠的是细胞内源的非同源末端连接修复机制(non-homologous end joining,NHEJ)、微同源介导末端连接机制(microhomology-mediated end joining,MMEJ)或同源重组修复机制(homology directed repair,HDR)[11].NHEJ系统进行双链断裂(double-strand breaks, DSBs)修复时,首先Ku异源二聚体识别并结合至两断裂末端,随后由DNA聚合酶和核酸外切酶对该处进行加工处理,最后进行末端连接形成双链.在对末端进行修饰时,由于外切酶和聚合酶结合顺序的不确定性则造成修复位点碱基缺失和插入突变等不同情况,致使目的基因失去活性[12].也有报道将Cas9蛋白靶向目的基因或染色体上两处位点,则会造成目的基因内或染色体上整段序列的缺失[4,13-15].MMEJ在修复双链断裂时的断裂末端比对过程中使用的微同源序列易导致原始断裂侧翼缺失,进而造成染色体异常如缺失、易位、倒位和其他复杂的重排.Cas9蛋白造成DNA双链断裂后,在损伤序列的同源供体DNA如姐妹染色单体存在的情况下,HDR则以同源DNA序列为模板进行无错修复.若人为地提供一段含有外源基因或替换了特定位置核苷酸的同源供体DNA(donor),则可利用HDR实现精准的基因插入或单碱基编辑(图1)[16].随着CRISPR/Cas9技术的普及和发展,其基本的基因编辑能力和性能已不能满足在基因组工程和各个领域中的应用.例如,除了受限于CRISPR/Cas9系统本身的PAM(protospacer adjacent motif)依赖性和脱靶效应,该系统在应用于代谢工程中时,还被其编辑靶点的数量和效率所制约[17],而应用在人类疾病基因疗法时还需进一步提高该系统的保真性、安全性等因素[18].因此,自张锋团队首次应用CRISPR/Cas9系统在哺乳动物细胞中实现多重编辑后,国内外的科研人员围绕该系统的多元化应用展开了研究,如通过优化Cas9蛋白拓宽该系统的识别范围并提高其靶向特异性[19-21]、通过融合效应结构域的方式使该系统获得动态可控性或进行转录调控[22-24]、为了进行表观基因组编辑或在非HDR介导的细胞中进行单碱基编辑将功能缺失的Cas9蛋白与甲基转移酶或脱氨酶融合等[25-26].2019年,哈佛大学David R. Liu团队开发的先导编辑(prime editing,PE)作为基于gRNA工程的多功能精准基因编辑工具,可以在不引入靶基因双链断裂及同源片段的条件下介导靶向的插入、缺失及所有碱基编辑,大大拓展了CRISPR/Cas9系统多功能基因组编辑的范围和能力[27].多重CRISPR技术是通过同时引入多个gRNA或多种直系同源Cas9蛋白,从而实现基因组上多靶点的同时靶向或多功能正交编辑.该技术不仅极大提高了基因编辑和转录调控的范围和效率,更促进了强大的生物工程应用.因此,如今越来越多的CRISPR/Cas9工具不仅可以实现定点改造,还可以实现多靶点的同时改造.发展趋势证明了基于RNA靶向性的CRISPR/Cas9技术凭借简单可编程性和广泛适用性具有强大优势,同时凸显出CRISPR/Cas9系统与TALENs、ZFNs等相比的优势[28]. ...
... Functional optimization through fusion expression of effector proteins with CRISPR/Cas9 system
Table 2| 功能 | 效应蛋白或gRNA工程 | 系统 | 特征 | 参考文献 |
|---|
| 表达调控 | 无 | dCas9 | 原核生物基因抑制表达,真核生物基因抑制效率低 | [22] |
| KRAB,MXI1,TUP1抑制结构域 | dCas9 | 可在真核生物中抑制基因表达 | [62-63] |
| KRAB-MeCP1融合抑制结构域 | dCas9 | 在哺乳动物细胞中高效抑制目的基因表达 | [64] |
| p65,VP64激活结构域 | dCas9 | 激活基因表达,效率较低 | [65-66] |
| VP64-p65-Rta(VPR)融合激活结构域 | dCas9 | 与VP64相比提高了22~320倍;多重基因激活表达;刺激iPSCs神经元分化 | [67] |
| gRNA工程;gRNA上携带携带两个MS2发夹二级结构 | dCas9 | 每个发夹结构招募一个MS2-p65-HSF1融合激活蛋白;与VP64相比,使10个靶基因上调2倍以上 | [68] |
| SPY系统(包括SpyTag和SpyCatcher)和Med2激活结构域 | dCas9 | 招募多拷贝SpyCatcher和Med2融合蛋白使酿酒酵母目的基因表达提高34.9倍 | [69] |
| SunTag系统(包括GCN4肽和抗GCN4抗体的单链可变片段scFv)和MIG1抑制结构域 | dCpf1 | 招募多拷贝ScFv与MIG1融合蛋白使酿酒酵母目的基因受到95%的抑制 | [69] |
| 表观基因组编辑 | KRAB-DNA甲基化酶催化结构域融合蛋白 | dCas9 | 融合抑制因子和甲基化酶;造成目的基因78%的稳定沉默 | [70] |
| 两侧分别融合KRAB和Dnmt3A-Dnmt3L融合DNA甲基化酶结构域 | dCas9 | 在干细胞分裂分化为神经细胞后仍维持DNA甲基化和基因抑制 | [71] |
| TET1甲基胞嘧啶双加氧酶1催化结构域 | dCas9 | dCas9- TET1靶向至BRCA1基因启动子后成功地上调其表达水平 | [72] |
| 人类组蛋白乙酰基转移酶p300催化核心 | dCas9 | 催化组蛋白H3 第27位赖氨酸残基的乙酰基化;基因表达上调 | [73] |
| 单碱基编辑 | 胞嘧啶脱氨酶,腺嘌呤脱氨酶 | nCas9 | 在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换 | [30,74-75] |
| CBE-UGI融合蛋白 | nCas9 | UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率 | [30] |
| 单链DNA结合结构域ssDBD融合到胞嘧啶脱氨酶与nCas9之间 | nCas9 | 通过延长ssDNA的暴露时间极大地提高CBEs的编辑效率;使CBEs的编辑窗口范围得到了提高 | [76] |
| 组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶 | nCas9 | 首次实现大肠杆菌中C到A 87.2%和哺乳动物细胞C到G 5.3%至53.0%的编辑效率 | [77] |
| 先导编辑 | 通过gRNA工程改造的pegRNA;与逆转录酶融合 | nCas9 | 改变pegRNA可实现精确的碱基替换、基因敲除和插入 | [27] |
| 通过gRNA工程改造的pegRNA;与逆转录酶和Rad51融合 | nCas9 | 融合单链DNA结合蛋白Rad51使PE2的编辑效率最高提升2.6倍 | [78] |
在CRISPR/Cas9的基因转录调控系统中,对Cas9蛋白工程化的dCas9分别在RuvC和HNH结构域发生突变使得RNP复合物仅能发挥靶序列识别功能,而无法切断核苷酸双链[29].nCas9(Cas9 nickase)是仅能使靶DNA发生单链断裂(single-strand break,SSB)的Cas9突变体,其中nCas9(H840A)和nCas9(D10A)分别是Cas9在HNH和RuvC结构域发生突变后的突变体蛋白,从而能够使该结构域失去内切酶活性,使DNA单链断裂,产生“缺口”[22,39].将dCas9或nCas9与各种效应蛋白如转录激活因子或抑制因子、表观遗传修饰因子、碱基编辑器甚至具有特定功能的酶等融合后,研究人员便能够借助CRISPR/Cas9系统对靶序列实现特定生物学功能,如表征转录基因乃至基因组上非编码区的功能、通过将碱基编辑器与nCas9融合能够在HDR活性低的细胞中进行精确高效的单碱基编辑等.因此本节将重点回顾与总结多种多样的效应蛋白为CRISPR/Cas9系统带来的多元化优化和发展,以及赋予该系统的多种新功能. ...
... 在CRISPR/Cas9的基因转录调控系统中,对Cas9蛋白工程化的dCas9分别在RuvC和HNH结构域发生突变使得RNP复合物仅能发挥靶序列识别功能,而无法切断核苷酸双链[29].nCas9(Cas9 nickase)是仅能使靶DNA发生单链断裂(single-strand break,SSB)的Cas9突变体,其中nCas9(H840A)和nCas9(D10A)分别是Cas9在HNH和RuvC结构域发生突变后的突变体蛋白,从而能够使该结构域失去内切酶活性,使DNA单链断裂,产生“缺口”[22,39].将dCas9或nCas9与各种效应蛋白如转录激活因子或抑制因子、表观遗传修饰因子、碱基编辑器甚至具有特定功能的酶等融合后,研究人员便能够借助CRISPR/Cas9系统对靶序列实现特定生物学功能,如表征转录基因乃至基因组上非编码区的功能、通过将碱基编辑器与nCas9融合能够在HDR活性低的细胞中进行精确高效的单碱基编辑等.因此本节将重点回顾与总结多种多样的效应蛋白为CRISPR/Cas9系统带来的多元化优化和发展,以及赋予该系统的多种新功能. ...
... 仅将dCas9蛋白靶向至目的基因上游时,dCas9蛋白对RNA聚合酶的空间位阻效应便可在一定程度上抑制基因表达[22],而该效应抑制表达的方式仅在原核细胞中有较高的效率,在真核细胞中由于其内部复杂的调控机制使得抑制效率较低[22,62,89].将dCas9蛋白与转录抑制结构域如KRAB[62]、MXI1[62]和TUP1[63]等蛋白融合表达则可提高CRISPRi系统在真核细胞中的抑制效率.其中KRAB锌指蛋白(KRAB-ZFP)属于哺乳动物细胞中最大的转录调节因子家族[90],该蛋白能够通过诱导靶序列的染色质结构变化从而抑制基因转录[91].KRAB与dCas9融合后被靶向至启动子区、5′非翻译区以及近端或远端增强子元件区后可以有效地沉默单个基因或非编码RNA[62,92-94].2018年,Yeo等[64]表征了20多种已知的能够诱导基因沉默的转录因子的抑制效率后,将获得的最高效的6种抑制因子——MeCP2、SIN3A、HDT1、MBD2B、NIPP1和HP1A分别与通用抑制因子KRAB融合形成双重抑制因子,再与dCas9蛋白C端融合表达并表征CRISPRi系统的抑制效率,最终获得了比单独dCas9抑制效率提高了60倍的双重抑制因子融合蛋白dCas9-KRAB-MeCP2.而之后研究人员继续表征的三重抑制因子融合蛋白可能由于功能冗余或蛋白体量较大从而影响抑制因子正常工作导致抑制效率不如双重抑制因子,证明了通过无限累加融合的方式提高融合蛋白的调控效率是不可行的[64]. ...
... [22,62,89].将dCas9蛋白与转录抑制结构域如KRAB[62]、MXI1[62]和TUP1[63]等蛋白融合表达则可提高CRISPRi系统在真核细胞中的抑制效率.其中KRAB锌指蛋白(KRAB-ZFP)属于哺乳动物细胞中最大的转录调节因子家族[90],该蛋白能够通过诱导靶序列的染色质结构变化从而抑制基因转录[91].KRAB与dCas9融合后被靶向至启动子区、5′非翻译区以及近端或远端增强子元件区后可以有效地沉默单个基因或非编码RNA[62,92-94].2018年,Yeo等[64]表征了20多种已知的能够诱导基因沉默的转录因子的抑制效率后,将获得的最高效的6种抑制因子——MeCP2、SIN3A、HDT1、MBD2B、NIPP1和HP1A分别与通用抑制因子KRAB融合形成双重抑制因子,再与dCas9蛋白C端融合表达并表征CRISPRi系统的抑制效率,最终获得了比单独dCas9抑制效率提高了60倍的双重抑制因子融合蛋白dCas9-KRAB-MeCP2.而之后研究人员继续表征的三重抑制因子融合蛋白可能由于功能冗余或蛋白体量较大从而影响抑制因子正常工作导致抑制效率不如双重抑制因子,证明了通过无限累加融合的方式提高融合蛋白的调控效率是不可行的[64]. ...
RNA-guided gene activation by CRISPR-Cas9-based transcription factors
1
2013
... 将dCas9与各种转录激活因子融合后,则可在gRNA的靶向作用下精确激活目的基因,该系统称为CRISPR激活系统或CRISPRa.最初的CRISPRa系统中dCas9仅融合了单个反式激活因子p65(也被称为RelA,是构成NF-κB转录因子家族的五种成员之一)或VP64 (由单纯疱疹病毒蛋白VP16的四个重复结构域构成),这些早期工作共同构成了最初的可编程dCas9基因激活工具箱,但效果差强人意[23-28,39-44,62,65-66].为了不断提高CRISPRa系统的激活效率,研究人员尝试了用各种方式将更复杂或多拷贝的激活结构域应用于CRISPRa.2015年,Chavez等[67]通过蛋白质理性设计,构建了三重融合激活因子VP64-p65-Rta(VPR)并应用于CRISPRa系统,其中Rta是来源于EB病毒(epstein-barr virus)的反式激活因子.与dCas9-VP64相比,dCas9-VPR使人胚胎肾细胞293(HEK 293T cells)的内源靶基因表达上调了22~320倍,同时证明了该系统也能够在多重基因的激活表达与刺激人诱导多功能干细胞(induced pluripotent stem cells,iPSCs)的神经元分化上发挥作用.同年,张锋研究团队[68]开发的基于CRISPRa的协同激活介导系统(synergistic activation mediator system, SAM)通过修饰sgRNA使其携带了两个MS2发夹二级结构,每个发夹结构都能招募一个MS2-p65-HSF1融合蛋白,其中包括反式激活因子p65、MS2结合蛋白以及热休克蛋白1(heat shock protein 1),最终证明该修饰后的gRNA与dCas9-VP64的协同作用能够使10个靶基因的表达均上调2倍以上.2022年,山东大学候进课题组[69]构建的CRISPR介导的蛋白质标签信号放大系统首次将SPY系统(包括SpyTag和SpyCatcher)和SunTag系统分别引入dCas9介导的CRISPRa和dCpf1(一种丧失核酸酶活性的Cas9同源蛋白)介导的CRISPRi系统并进行骨架长度和转录因子优化后,成功使酿酒酵母内目的基因转录水平分别提高了34.9倍以及受到几乎完全的抑制.其中,SunTag系统是由若干个拷贝的GCN4肽以及作为标签的抗GCN4抗体的单链可变片段scFv组成[95],与dCas9融合的多拷贝GCN4肽通过招募多拷贝的转录因子与GCN4抗体标签的融合蛋白从而进行转录调控,该系统除了与CRISPR系统结合后用于碱基编辑以及基因调控,还在哺乳动物和植物细胞的DNA的甲基化和去甲基化诱导中发挥作用[96-100];而SPY系统作为一种广泛使用的蛋白质组装系统首次在酿酒酵母的转录调控中得到了应用[69].可见在dCas9与转录因子融合的基础上,CRISPRa与CRISPRi系统能够利用融合多重结构域或融合蛋白质骨架系统的方式提高调控效率.而通过利用RNA与蛋白质间的相互作用,将多个适配子融合在gRNA中使其同时含有目的基因靶向序列和转录因子招募序列的方式能够产生与dCas9融合多个转录激活或抑制结构域的策略类似的效果[101].此外,针对不同细胞乃至不同dCas9同源蛋白,CRISPRa与CRISPRi系统均需对不同转录激活或抑制结构域乃至其组合进行优化才能发挥该系统的最大效能. ...
CRISPR RNA-guided activation of endogenous human genes
1
2013
... CRISPR系统利用单链向导RNA(single guide RNA,sgRNA或gRNA)靶向目的位点,随后Cas9等核酸酶切断目的区域的双链核酸,诱导细胞内源DNA损伤修复机制工作,最终借助该机制达到编辑遗传信息的目的[10].CRISPR/Cas9系统在发挥基因敲除、基因插入、单碱基编辑等主要编辑手段时主要依靠的是细胞内源的非同源末端连接修复机制(non-homologous end joining,NHEJ)、微同源介导末端连接机制(microhomology-mediated end joining,MMEJ)或同源重组修复机制(homology directed repair,HDR)[11].NHEJ系统进行双链断裂(double-strand breaks, DSBs)修复时,首先Ku异源二聚体识别并结合至两断裂末端,随后由DNA聚合酶和核酸外切酶对该处进行加工处理,最后进行末端连接形成双链.在对末端进行修饰时,由于外切酶和聚合酶结合顺序的不确定性则造成修复位点碱基缺失和插入突变等不同情况,致使目的基因失去活性[12].也有报道将Cas9蛋白靶向目的基因或染色体上两处位点,则会造成目的基因内或染色体上整段序列的缺失[4,13-15].MMEJ在修复双链断裂时的断裂末端比对过程中使用的微同源序列易导致原始断裂侧翼缺失,进而造成染色体异常如缺失、易位、倒位和其他复杂的重排.Cas9蛋白造成DNA双链断裂后,在损伤序列的同源供体DNA如姐妹染色单体存在的情况下,HDR则以同源DNA序列为模板进行无错修复.若人为地提供一段含有外源基因或替换了特定位置核苷酸的同源供体DNA(donor),则可利用HDR实现精准的基因插入或单碱基编辑(图1)[16].随着CRISPR/Cas9技术的普及和发展,其基本的基因编辑能力和性能已不能满足在基因组工程和各个领域中的应用.例如,除了受限于CRISPR/Cas9系统本身的PAM(protospacer adjacent motif)依赖性和脱靶效应,该系统在应用于代谢工程中时,还被其编辑靶点的数量和效率所制约[17],而应用在人类疾病基因疗法时还需进一步提高该系统的保真性、安全性等因素[18].因此,自张锋团队首次应用CRISPR/Cas9系统在哺乳动物细胞中实现多重编辑后,国内外的科研人员围绕该系统的多元化应用展开了研究,如通过优化Cas9蛋白拓宽该系统的识别范围并提高其靶向特异性[19-21]、通过融合效应结构域的方式使该系统获得动态可控性或进行转录调控[22-24]、为了进行表观基因组编辑或在非HDR介导的细胞中进行单碱基编辑将功能缺失的Cas9蛋白与甲基转移酶或脱氨酶融合等[25-26].2019年,哈佛大学David R. Liu团队开发的先导编辑(prime editing,PE)作为基于gRNA工程的多功能精准基因编辑工具,可以在不引入靶基因双链断裂及同源片段的条件下介导靶向的插入、缺失及所有碱基编辑,大大拓展了CRISPR/Cas9系统多功能基因组编辑的范围和能力[27].多重CRISPR技术是通过同时引入多个gRNA或多种直系同源Cas9蛋白,从而实现基因组上多靶点的同时靶向或多功能正交编辑.该技术不仅极大提高了基因编辑和转录调控的范围和效率,更促进了强大的生物工程应用.因此,如今越来越多的CRISPR/Cas9工具不仅可以实现定点改造,还可以实现多靶点的同时改造.发展趋势证明了基于RNA靶向性的CRISPR/Cas9技术凭借简单可编程性和广泛适用性具有强大优势,同时凸显出CRISPR/Cas9系统与TALENs、ZFNs等相比的优势[28]. ...
CRISPR technologies for precise epigenome editing
1
2021
... CRISPR系统利用单链向导RNA(single guide RNA,sgRNA或gRNA)靶向目的位点,随后Cas9等核酸酶切断目的区域的双链核酸,诱导细胞内源DNA损伤修复机制工作,最终借助该机制达到编辑遗传信息的目的[10].CRISPR/Cas9系统在发挥基因敲除、基因插入、单碱基编辑等主要编辑手段时主要依靠的是细胞内源的非同源末端连接修复机制(non-homologous end joining,NHEJ)、微同源介导末端连接机制(microhomology-mediated end joining,MMEJ)或同源重组修复机制(homology directed repair,HDR)[11].NHEJ系统进行双链断裂(double-strand breaks, DSBs)修复时,首先Ku异源二聚体识别并结合至两断裂末端,随后由DNA聚合酶和核酸外切酶对该处进行加工处理,最后进行末端连接形成双链.在对末端进行修饰时,由于外切酶和聚合酶结合顺序的不确定性则造成修复位点碱基缺失和插入突变等不同情况,致使目的基因失去活性[12].也有报道将Cas9蛋白靶向目的基因或染色体上两处位点,则会造成目的基因内或染色体上整段序列的缺失[4,13-15].MMEJ在修复双链断裂时的断裂末端比对过程中使用的微同源序列易导致原始断裂侧翼缺失,进而造成染色体异常如缺失、易位、倒位和其他复杂的重排.Cas9蛋白造成DNA双链断裂后,在损伤序列的同源供体DNA如姐妹染色单体存在的情况下,HDR则以同源DNA序列为模板进行无错修复.若人为地提供一段含有外源基因或替换了特定位置核苷酸的同源供体DNA(donor),则可利用HDR实现精准的基因插入或单碱基编辑(图1)[16].随着CRISPR/Cas9技术的普及和发展,其基本的基因编辑能力和性能已不能满足在基因组工程和各个领域中的应用.例如,除了受限于CRISPR/Cas9系统本身的PAM(protospacer adjacent motif)依赖性和脱靶效应,该系统在应用于代谢工程中时,还被其编辑靶点的数量和效率所制约[17],而应用在人类疾病基因疗法时还需进一步提高该系统的保真性、安全性等因素[18].因此,自张锋团队首次应用CRISPR/Cas9系统在哺乳动物细胞中实现多重编辑后,国内外的科研人员围绕该系统的多元化应用展开了研究,如通过优化Cas9蛋白拓宽该系统的识别范围并提高其靶向特异性[19-21]、通过融合效应结构域的方式使该系统获得动态可控性或进行转录调控[22-24]、为了进行表观基因组编辑或在非HDR介导的细胞中进行单碱基编辑将功能缺失的Cas9蛋白与甲基转移酶或脱氨酶融合等[25-26].2019年,哈佛大学David R. Liu团队开发的先导编辑(prime editing,PE)作为基于gRNA工程的多功能精准基因编辑工具,可以在不引入靶基因双链断裂及同源片段的条件下介导靶向的插入、缺失及所有碱基编辑,大大拓展了CRISPR/Cas9系统多功能基因组编辑的范围和能力[27].多重CRISPR技术是通过同时引入多个gRNA或多种直系同源Cas9蛋白,从而实现基因组上多靶点的同时靶向或多功能正交编辑.该技术不仅极大提高了基因编辑和转录调控的范围和效率,更促进了强大的生物工程应用.因此,如今越来越多的CRISPR/Cas9工具不仅可以实现定点改造,还可以实现多靶点的同时改造.发展趋势证明了基于RNA靶向性的CRISPR/Cas9技术凭借简单可编程性和广泛适用性具有强大优势,同时凸显出CRISPR/Cas9系统与TALENs、ZFNs等相比的优势[28]. ...
Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors
1
2020
... CRISPR系统利用单链向导RNA(single guide RNA,sgRNA或gRNA)靶向目的位点,随后Cas9等核酸酶切断目的区域的双链核酸,诱导细胞内源DNA损伤修复机制工作,最终借助该机制达到编辑遗传信息的目的[10].CRISPR/Cas9系统在发挥基因敲除、基因插入、单碱基编辑等主要编辑手段时主要依靠的是细胞内源的非同源末端连接修复机制(non-homologous end joining,NHEJ)、微同源介导末端连接机制(microhomology-mediated end joining,MMEJ)或同源重组修复机制(homology directed repair,HDR)[11].NHEJ系统进行双链断裂(double-strand breaks, DSBs)修复时,首先Ku异源二聚体识别并结合至两断裂末端,随后由DNA聚合酶和核酸外切酶对该处进行加工处理,最后进行末端连接形成双链.在对末端进行修饰时,由于外切酶和聚合酶结合顺序的不确定性则造成修复位点碱基缺失和插入突变等不同情况,致使目的基因失去活性[12].也有报道将Cas9蛋白靶向目的基因或染色体上两处位点,则会造成目的基因内或染色体上整段序列的缺失[4,13-15].MMEJ在修复双链断裂时的断裂末端比对过程中使用的微同源序列易导致原始断裂侧翼缺失,进而造成染色体异常如缺失、易位、倒位和其他复杂的重排.Cas9蛋白造成DNA双链断裂后,在损伤序列的同源供体DNA如姐妹染色单体存在的情况下,HDR则以同源DNA序列为模板进行无错修复.若人为地提供一段含有外源基因或替换了特定位置核苷酸的同源供体DNA(donor),则可利用HDR实现精准的基因插入或单碱基编辑(图1)[16].随着CRISPR/Cas9技术的普及和发展,其基本的基因编辑能力和性能已不能满足在基因组工程和各个领域中的应用.例如,除了受限于CRISPR/Cas9系统本身的PAM(protospacer adjacent motif)依赖性和脱靶效应,该系统在应用于代谢工程中时,还被其编辑靶点的数量和效率所制约[17],而应用在人类疾病基因疗法时还需进一步提高该系统的保真性、安全性等因素[18].因此,自张锋团队首次应用CRISPR/Cas9系统在哺乳动物细胞中实现多重编辑后,国内外的科研人员围绕该系统的多元化应用展开了研究,如通过优化Cas9蛋白拓宽该系统的识别范围并提高其靶向特异性[19-21]、通过融合效应结构域的方式使该系统获得动态可控性或进行转录调控[22-24]、为了进行表观基因组编辑或在非HDR介导的细胞中进行单碱基编辑将功能缺失的Cas9蛋白与甲基转移酶或脱氨酶融合等[25-26].2019年,哈佛大学David R. Liu团队开发的先导编辑(prime editing,PE)作为基于gRNA工程的多功能精准基因编辑工具,可以在不引入靶基因双链断裂及同源片段的条件下介导靶向的插入、缺失及所有碱基编辑,大大拓展了CRISPR/Cas9系统多功能基因组编辑的范围和能力[27].多重CRISPR技术是通过同时引入多个gRNA或多种直系同源Cas9蛋白,从而实现基因组上多靶点的同时靶向或多功能正交编辑.该技术不仅极大提高了基因编辑和转录调控的范围和效率,更促进了强大的生物工程应用.因此,如今越来越多的CRISPR/Cas9工具不仅可以实现定点改造,还可以实现多靶点的同时改造.发展趋势证明了基于RNA靶向性的CRISPR/Cas9技术凭借简单可编程性和广泛适用性具有强大优势,同时凸显出CRISPR/Cas9系统与TALENs、ZFNs等相比的优势[28]. ...
Search-and-replace genome editing without double-strand breaks or donor DNA
2
2019
... CRISPR系统利用单链向导RNA(single guide RNA,sgRNA或gRNA)靶向目的位点,随后Cas9等核酸酶切断目的区域的双链核酸,诱导细胞内源DNA损伤修复机制工作,最终借助该机制达到编辑遗传信息的目的[10].CRISPR/Cas9系统在发挥基因敲除、基因插入、单碱基编辑等主要编辑手段时主要依靠的是细胞内源的非同源末端连接修复机制(non-homologous end joining,NHEJ)、微同源介导末端连接机制(microhomology-mediated end joining,MMEJ)或同源重组修复机制(homology directed repair,HDR)[11].NHEJ系统进行双链断裂(double-strand breaks, DSBs)修复时,首先Ku异源二聚体识别并结合至两断裂末端,随后由DNA聚合酶和核酸外切酶对该处进行加工处理,最后进行末端连接形成双链.在对末端进行修饰时,由于外切酶和聚合酶结合顺序的不确定性则造成修复位点碱基缺失和插入突变等不同情况,致使目的基因失去活性[12].也有报道将Cas9蛋白靶向目的基因或染色体上两处位点,则会造成目的基因内或染色体上整段序列的缺失[4,13-15].MMEJ在修复双链断裂时的断裂末端比对过程中使用的微同源序列易导致原始断裂侧翼缺失,进而造成染色体异常如缺失、易位、倒位和其他复杂的重排.Cas9蛋白造成DNA双链断裂后,在损伤序列的同源供体DNA如姐妹染色单体存在的情况下,HDR则以同源DNA序列为模板进行无错修复.若人为地提供一段含有外源基因或替换了特定位置核苷酸的同源供体DNA(donor),则可利用HDR实现精准的基因插入或单碱基编辑(图1)[16].随着CRISPR/Cas9技术的普及和发展,其基本的基因编辑能力和性能已不能满足在基因组工程和各个领域中的应用.例如,除了受限于CRISPR/Cas9系统本身的PAM(protospacer adjacent motif)依赖性和脱靶效应,该系统在应用于代谢工程中时,还被其编辑靶点的数量和效率所制约[17],而应用在人类疾病基因疗法时还需进一步提高该系统的保真性、安全性等因素[18].因此,自张锋团队首次应用CRISPR/Cas9系统在哺乳动物细胞中实现多重编辑后,国内外的科研人员围绕该系统的多元化应用展开了研究,如通过优化Cas9蛋白拓宽该系统的识别范围并提高其靶向特异性[19-21]、通过融合效应结构域的方式使该系统获得动态可控性或进行转录调控[22-24]、为了进行表观基因组编辑或在非HDR介导的细胞中进行单碱基编辑将功能缺失的Cas9蛋白与甲基转移酶或脱氨酶融合等[25-26].2019年,哈佛大学David R. Liu团队开发的先导编辑(prime editing,PE)作为基于gRNA工程的多功能精准基因编辑工具,可以在不引入靶基因双链断裂及同源片段的条件下介导靶向的插入、缺失及所有碱基编辑,大大拓展了CRISPR/Cas9系统多功能基因组编辑的范围和能力[27].多重CRISPR技术是通过同时引入多个gRNA或多种直系同源Cas9蛋白,从而实现基因组上多靶点的同时靶向或多功能正交编辑.该技术不仅极大提高了基因编辑和转录调控的范围和效率,更促进了强大的生物工程应用.因此,如今越来越多的CRISPR/Cas9工具不仅可以实现定点改造,还可以实现多靶点的同时改造.发展趋势证明了基于RNA靶向性的CRISPR/Cas9技术凭借简单可编程性和广泛适用性具有强大优势,同时凸显出CRISPR/Cas9系统与TALENs、ZFNs等相比的优势[28]. ...
... Functional optimization through fusion expression of effector proteins with CRISPR/Cas9 system
Table 2| 功能 | 效应蛋白或gRNA工程 | 系统 | 特征 | 参考文献 |
|---|
| 表达调控 | 无 | dCas9 | 原核生物基因抑制表达,真核生物基因抑制效率低 | [22] |
| KRAB,MXI1,TUP1抑制结构域 | dCas9 | 可在真核生物中抑制基因表达 | [62-63] |
| KRAB-MeCP1融合抑制结构域 | dCas9 | 在哺乳动物细胞中高效抑制目的基因表达 | [64] |
| p65,VP64激活结构域 | dCas9 | 激活基因表达,效率较低 | [65-66] |
| VP64-p65-Rta(VPR)融合激活结构域 | dCas9 | 与VP64相比提高了22~320倍;多重基因激活表达;刺激iPSCs神经元分化 | [67] |
| gRNA工程;gRNA上携带携带两个MS2发夹二级结构 | dCas9 | 每个发夹结构招募一个MS2-p65-HSF1融合激活蛋白;与VP64相比,使10个靶基因上调2倍以上 | [68] |
| SPY系统(包括SpyTag和SpyCatcher)和Med2激活结构域 | dCas9 | 招募多拷贝SpyCatcher和Med2融合蛋白使酿酒酵母目的基因表达提高34.9倍 | [69] |
| SunTag系统(包括GCN4肽和抗GCN4抗体的单链可变片段scFv)和MIG1抑制结构域 | dCpf1 | 招募多拷贝ScFv与MIG1融合蛋白使酿酒酵母目的基因受到95%的抑制 | [69] |
| 表观基因组编辑 | KRAB-DNA甲基化酶催化结构域融合蛋白 | dCas9 | 融合抑制因子和甲基化酶;造成目的基因78%的稳定沉默 | [70] |
| 两侧分别融合KRAB和Dnmt3A-Dnmt3L融合DNA甲基化酶结构域 | dCas9 | 在干细胞分裂分化为神经细胞后仍维持DNA甲基化和基因抑制 | [71] |
| TET1甲基胞嘧啶双加氧酶1催化结构域 | dCas9 | dCas9- TET1靶向至BRCA1基因启动子后成功地上调其表达水平 | [72] |
| 人类组蛋白乙酰基转移酶p300催化核心 | dCas9 | 催化组蛋白H3 第27位赖氨酸残基的乙酰基化;基因表达上调 | [73] |
| 单碱基编辑 | 胞嘧啶脱氨酶,腺嘌呤脱氨酶 | nCas9 | 在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换 | [30,74-75] |
| CBE-UGI融合蛋白 | nCas9 | UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率 | [30] |
| 单链DNA结合结构域ssDBD融合到胞嘧啶脱氨酶与nCas9之间 | nCas9 | 通过延长ssDNA的暴露时间极大地提高CBEs的编辑效率;使CBEs的编辑窗口范围得到了提高 | [76] |
| 组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶 | nCas9 | 首次实现大肠杆菌中C到A 87.2%和哺乳动物细胞C到G 5.3%至53.0%的编辑效率 | [77] |
| 先导编辑 | 通过gRNA工程改造的pegRNA;与逆转录酶融合 | nCas9 | 改变pegRNA可实现精确的碱基替换、基因敲除和插入 | [27] |
| 通过gRNA工程改造的pegRNA;与逆转录酶和Rad51融合 | nCas9 | 融合单链DNA结合蛋白Rad51使PE2的编辑效率最高提升2.6倍 | [78] |
在CRISPR/Cas9的基因转录调控系统中,对Cas9蛋白工程化的dCas9分别在RuvC和HNH结构域发生突变使得RNP复合物仅能发挥靶序列识别功能,而无法切断核苷酸双链[29].nCas9(Cas9 nickase)是仅能使靶DNA发生单链断裂(single-strand break,SSB)的Cas9突变体,其中nCas9(H840A)和nCas9(D10A)分别是Cas9在HNH和RuvC结构域发生突变后的突变体蛋白,从而能够使该结构域失去内切酶活性,使DNA单链断裂,产生“缺口”[22,39].将dCas9或nCas9与各种效应蛋白如转录激活因子或抑制因子、表观遗传修饰因子、碱基编辑器甚至具有特定功能的酶等融合后,研究人员便能够借助CRISPR/Cas9系统对靶序列实现特定生物学功能,如表征转录基因乃至基因组上非编码区的功能、通过将碱基编辑器与nCas9融合能够在HDR活性低的细胞中进行精确高效的单碱基编辑等.因此本节将重点回顾与总结多种多样的效应蛋白为CRISPR/Cas9系统带来的多元化优化和发展,以及赋予该系统的多种新功能. ...
The CRISPR-Cas toolbox and gene editing technologies
3
2022
... CRISPR系统利用单链向导RNA(single guide RNA,sgRNA或gRNA)靶向目的位点,随后Cas9等核酸酶切断目的区域的双链核酸,诱导细胞内源DNA损伤修复机制工作,最终借助该机制达到编辑遗传信息的目的[10].CRISPR/Cas9系统在发挥基因敲除、基因插入、单碱基编辑等主要编辑手段时主要依靠的是细胞内源的非同源末端连接修复机制(non-homologous end joining,NHEJ)、微同源介导末端连接机制(microhomology-mediated end joining,MMEJ)或同源重组修复机制(homology directed repair,HDR)[11].NHEJ系统进行双链断裂(double-strand breaks, DSBs)修复时,首先Ku异源二聚体识别并结合至两断裂末端,随后由DNA聚合酶和核酸外切酶对该处进行加工处理,最后进行末端连接形成双链.在对末端进行修饰时,由于外切酶和聚合酶结合顺序的不确定性则造成修复位点碱基缺失和插入突变等不同情况,致使目的基因失去活性[12].也有报道将Cas9蛋白靶向目的基因或染色体上两处位点,则会造成目的基因内或染色体上整段序列的缺失[4,13-15].MMEJ在修复双链断裂时的断裂末端比对过程中使用的微同源序列易导致原始断裂侧翼缺失,进而造成染色体异常如缺失、易位、倒位和其他复杂的重排.Cas9蛋白造成DNA双链断裂后,在损伤序列的同源供体DNA如姐妹染色单体存在的情况下,HDR则以同源DNA序列为模板进行无错修复.若人为地提供一段含有外源基因或替换了特定位置核苷酸的同源供体DNA(donor),则可利用HDR实现精准的基因插入或单碱基编辑(图1)[16].随着CRISPR/Cas9技术的普及和发展,其基本的基因编辑能力和性能已不能满足在基因组工程和各个领域中的应用.例如,除了受限于CRISPR/Cas9系统本身的PAM(protospacer adjacent motif)依赖性和脱靶效应,该系统在应用于代谢工程中时,还被其编辑靶点的数量和效率所制约[17],而应用在人类疾病基因疗法时还需进一步提高该系统的保真性、安全性等因素[18].因此,自张锋团队首次应用CRISPR/Cas9系统在哺乳动物细胞中实现多重编辑后,国内外的科研人员围绕该系统的多元化应用展开了研究,如通过优化Cas9蛋白拓宽该系统的识别范围并提高其靶向特异性[19-21]、通过融合效应结构域的方式使该系统获得动态可控性或进行转录调控[22-24]、为了进行表观基因组编辑或在非HDR介导的细胞中进行单碱基编辑将功能缺失的Cas9蛋白与甲基转移酶或脱氨酶融合等[25-26].2019年,哈佛大学David R. Liu团队开发的先导编辑(prime editing,PE)作为基于gRNA工程的多功能精准基因编辑工具,可以在不引入靶基因双链断裂及同源片段的条件下介导靶向的插入、缺失及所有碱基编辑,大大拓展了CRISPR/Cas9系统多功能基因组编辑的范围和能力[27].多重CRISPR技术是通过同时引入多个gRNA或多种直系同源Cas9蛋白,从而实现基因组上多靶点的同时靶向或多功能正交编辑.该技术不仅极大提高了基因编辑和转录调控的范围和效率,更促进了强大的生物工程应用.因此,如今越来越多的CRISPR/Cas9工具不仅可以实现定点改造,还可以实现多靶点的同时改造.发展趋势证明了基于RNA靶向性的CRISPR/Cas9技术凭借简单可编程性和广泛适用性具有强大优势,同时凸显出CRISPR/Cas9系统与TALENs、ZFNs等相比的优势[28]. ...
... 将dCas9与各种转录激活因子融合后,则可在gRNA的靶向作用下精确激活目的基因,该系统称为CRISPR激活系统或CRISPRa.最初的CRISPRa系统中dCas9仅融合了单个反式激活因子p65(也被称为RelA,是构成NF-κB转录因子家族的五种成员之一)或VP64 (由单纯疱疹病毒蛋白VP16的四个重复结构域构成),这些早期工作共同构成了最初的可编程dCas9基因激活工具箱,但效果差强人意[23-28,39-44,62,65-66].为了不断提高CRISPRa系统的激活效率,研究人员尝试了用各种方式将更复杂或多拷贝的激活结构域应用于CRISPRa.2015年,Chavez等[67]通过蛋白质理性设计,构建了三重融合激活因子VP64-p65-Rta(VPR)并应用于CRISPRa系统,其中Rta是来源于EB病毒(epstein-barr virus)的反式激活因子.与dCas9-VP64相比,dCas9-VPR使人胚胎肾细胞293(HEK 293T cells)的内源靶基因表达上调了22~320倍,同时证明了该系统也能够在多重基因的激活表达与刺激人诱导多功能干细胞(induced pluripotent stem cells,iPSCs)的神经元分化上发挥作用.同年,张锋研究团队[68]开发的基于CRISPRa的协同激活介导系统(synergistic activation mediator system, SAM)通过修饰sgRNA使其携带了两个MS2发夹二级结构,每个发夹结构都能招募一个MS2-p65-HSF1融合蛋白,其中包括反式激活因子p65、MS2结合蛋白以及热休克蛋白1(heat shock protein 1),最终证明该修饰后的gRNA与dCas9-VP64的协同作用能够使10个靶基因的表达均上调2倍以上.2022年,山东大学候进课题组[69]构建的CRISPR介导的蛋白质标签信号放大系统首次将SPY系统(包括SpyTag和SpyCatcher)和SunTag系统分别引入dCas9介导的CRISPRa和dCpf1(一种丧失核酸酶活性的Cas9同源蛋白)介导的CRISPRi系统并进行骨架长度和转录因子优化后,成功使酿酒酵母内目的基因转录水平分别提高了34.9倍以及受到几乎完全的抑制.其中,SunTag系统是由若干个拷贝的GCN4肽以及作为标签的抗GCN4抗体的单链可变片段scFv组成[95],与dCas9融合的多拷贝GCN4肽通过招募多拷贝的转录因子与GCN4抗体标签的融合蛋白从而进行转录调控,该系统除了与CRISPR系统结合后用于碱基编辑以及基因调控,还在哺乳动物和植物细胞的DNA的甲基化和去甲基化诱导中发挥作用[96-100];而SPY系统作为一种广泛使用的蛋白质组装系统首次在酿酒酵母的转录调控中得到了应用[69].可见在dCas9与转录因子融合的基础上,CRISPRa与CRISPRi系统能够利用融合多重结构域或融合蛋白质骨架系统的方式提高调控效率.而通过利用RNA与蛋白质间的相互作用,将多个适配子融合在gRNA中使其同时含有目的基因靶向序列和转录因子招募序列的方式能够产生与dCas9融合多个转录激活或抑制结构域的策略类似的效果[101].此外,针对不同细胞乃至不同dCas9同源蛋白,CRISPRa与CRISPRi系统均需对不同转录激活或抑制结构域乃至其组合进行优化才能发挥该系统的最大效能. ...
... 为了改善CRIPSR/Cas9系统的碱基编辑工具箱,研究人员通过将nCas9与胞嘧啶脱氨酶(cytosine deaminase)或腺嘌呤脱氨酶(adenine deaminase)融合的方式在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换(图2),其中相应的融合蛋白分别被称为胞嘧啶碱基编辑器(cytosine base editors,CBEs)[30,74]和腺嘌呤碱基编辑器(adenine base editors,ABEs)[75].CBEs中的胞嘧啶脱氨酶能使被nCas9释放的单链DNA(single strand DNA,ssDNA)上的胞嘧啶C转变为尿嘧啶U,随后该位置产生的U:G错配碱基对被修复为T:A碱基对;相应的,ABEs中的腺嘌呤脱氨酶使腺嘌呤A水解脱氨为次黄嘌呤I,随后DNA聚合酶能将次黄嘌呤I识别为鸟嘌呤G进行复制,互补链上原来与腺嘌呤A配对的胸腺嘧啶T则会相应变成胞嘧啶C,至此完成碱基置换全过程[107].由于不存在天然的ssDNA腺嘌呤脱氨酶[75],Kim等[108]通过定向进化改造得到一种大肠杆菌(Escherichia coli)tRNA来源的腺嘌呤脱氨酶ecTadA(E.coli TadA)突变体TadA*.由于在天然环境(大肠杆菌)中TadA是以同型二聚体的方式发挥作用,所以最初的TadA*-nCas9融合蛋白在哺乳动物细胞中的编辑效率并不高,直到研究人员将野生型TadA单体、进化的突变体TadA*及nCas9整合到一起(TadA- TadA*- nCas9),才使ABE在哺乳动物中的编辑效率大大提高.2018年,David R. Liu团队[109]工程化改造并优化了ABE7.10后获得的ABEmax最高可获得前者7.9倍的编辑效率,是目前为止在哺乳动物中编辑效率最高的ABEs.为了方便体内编辑,该团队[110]在2022年7月发表的紧凑型ABEs递送系统通过将小型腺嘌呤编辑器SaABE8e及其PAM变体SaKKH-ABE8e的基因长度最小化以及AAV组件基因长度最小化后使得ABEs能够进行单AAV递送,且在肝脏(66%)、心脏(33%)和肌肉细胞(22%)中的编辑效率均高于双AAV递送系统.同年,吉林大学李占军团队和赖学良团队[111]合作开发的紧凑型的Cas9(Cje2Cas9和Cje3Cas9)能够与sgRNA一起包装到单个AAV中,并在成年小鼠肝脏中以12%的效率成功编辑Pcsk9基因,最终有效降低了小鼠体内的胆固醇水平.高效的单AAV递送系统的开发不仅确立了其相对于双AAV递送系统的优势,还促进了CRISPR/Cas9系统作为体内碱基编辑工具时的研究和治疗应用,具有巨大的临床治疗价值.为了改善编辑效率,Zhang等[76]通过将单链DNA结合结构域(single-stranded DNA-binding domain,ssDBD)融合到胞嘧啶脱氨酶与nCas9之间从而延长ssDNA的暴露时间,可在不增加插入缺失突变(indels)、细胞毒性或脱靶效应等负面效应概率的条件下极大地提高CBEs的编辑效率.ssDBD的融合还使CBEs的编辑窗口范围得到了提高,从而拓宽了其靶向作用范围及用于疾病建模和基因治疗的适用范围.David R. Liu团队[30]在CBE的基础上融合了尿嘧啶糖基化酶抑制剂(uracil glycosylase inhibitor,UGI),作为一种体量较小的蛋白(9.5kDa),UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率.该团队还发现通过招募多拷贝的脱氨酶或更改其空间位置以优化编辑窗口能有效提高编辑效率和特异性.此外,与nCas9融合的脱氨酶若工程化改造后具有序列偏好性,则可通过降低旁观者编辑效应从而提高编辑精度.尽管ABEs和CBEs碱基编辑器的性能不断提升,A:T到G:C和C:G到T:A碱基对的单向固定替换仍限制着BEs作为单碱基改变工具使用时的灵活性.2020年,Zhao等[77]通过组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶(uracil-DNA glycosylase,Ung)实现了大肠杆菌中C到A 87.2%的编辑效率和哺乳动物细胞C到G 5.3%~53.0%的编辑效率.先导编辑作为一种多功能编辑手段,虽然同时具备可编程性和高精确度,但对于单碱基编辑的效率仍与BEs有较大差距[28].2021年,Song等[78]构建的hyPE2在PE2的基础上融合加入了一个ssDBD结构域Rad51,使得PE2的编辑效率最高提升了2.6倍,同时该团队还通过深度学习开发了能够预测不同的hyPE2与PE2相比编辑效率提升倍数的计算模型PEselector,该工具的网址为http://deepcrispr.info/PEselector.2022年,Velimirovic等[112]通过高通量肽链自编辑测序(peptide self-editing sequencing assay,PepSEq)测出12000个85个氨基酸的肽段对PE效率的影响,从而通过肽链融合提高PE的效率.综上所述,基于CRIPSR/Cas9系统的碱基编辑器编辑效率、编辑精度的不断提高以及编辑窗口乃至碱基固定替换模式的扩展相对于大量已知且不断增加的遗传病相关点突变数量仍捉襟见肘,各种脱氨酶赋予CRIPSR/Cas9系统的碱基编辑能力在其他结构域如UGI和ssDBD加持下不仅能够发挥CRIPSR系统高效率、高灵活、易编程的优势,也能够充分发挥脱氨酶的效能,但仍需进一步优化它们以有效地转换碱基和在无旁观者编辑效应的条件下进行目的碱基替换. ...
Editing the epigenome: technologies for programmable transcription and epigenetic modulation
3
2016
... Major Cas9 variants and orthologues as well as their features
Table 1| 变体及直系同源 | 改造位点或方式 | PAM | 特性 | 参考文献 |
|---|
| SpCas9 | 原生酿脓链球菌Cas9 | NGG | 1368个氨基酸 | [29] |
| dCas9 | D10A,H840A | NGG | 核酸酶活性失活 | [29] |
| nCas9(D10A) | D10A | NGG | 切口酶,非靶向链切割活性失活 | [30] |
| nCas9(H840A) | H840A | NGG | 切口酶,靶向链切割活性失活 | [30] |
| SpCas9NG | R1335V,L1111R,D1135V,G1218R,E1219F,A1322R,T1337R | NG | PAM改变;通过Cas9理性设计获得 | [31] |
| VRERSpCas9 | D1135A,G1218R,R1335E,T1337R | NGCG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| VQRSpCas9 | D1135V,R1335Q,T1337R | NGAN,NGNG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| EQRSpCas9 | D1335E,R1335Q,T1337R | NGAG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| xCas9 | A262T,R324L,S409I,E480K,E543D,M694I,E1219V | NG,GAA,GAT | PAM改变;通过噬菌体辅助持续进化获得 | [20] |
| SpG | D1135L,S1136W,G1218K,E1219Q,R1335Q,T1337R | NGN | PAM改变;通过SpCas9理性设计获得 | [32] |
| SpRY | SpG,A61R,L1111R,A1322R,N1317R,R1333P | NRN,NYN | PAM改变;通过对SpG进一步理性设计获得 | [32] |
| evoCas9 | M495V, Y515N, K526E, R661Q | NGG | 保真性提高,编辑效率接近天然SpCas9 | [33] |
| fCas9 | SpdCas9与FokI融合 | NGG | 保真性提高;通过将FokI核酸酶与dCas9融合获得 | [34] |
| StCas9 | 原生嗜热链球菌Cas9 | NNAGAAW | 1121个氨基酸 | [35] |
| NmCas9 | 原生脑膜炎奈瑟菌Cas9 | NNNNGATT | 1082个氨基酸 | [36] |
| SaCas9 | 原生金黄色葡萄球菌Cas9 | NNGRRT | 1053个氨基酸;基因组编辑效率与SpCas9相当 | [37] |
| CjCas9 | 原生曲状杆菌Cas9 | NNNVRYM | 984个氨基酸 | [38] |
注:V代表A、C或G;R代表A或G;Y代表C或T;M代表A或C. ...
... [
29]
| nCas9(D10A) | D10A | NGG | 切口酶,非靶向链切割活性失活 | [30] |
| nCas9(H840A) | H840A | NGG | 切口酶,靶向链切割活性失活 | [30] |
| SpCas9NG | R1335V,L1111R,D1135V,G1218R,E1219F,A1322R,T1337R | NG | PAM改变;通过Cas9理性设计获得 | [31] |
| VRERSpCas9 | D1135A,G1218R,R1335E,T1337R | NGCG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| VQRSpCas9 | D1135V,R1335Q,T1337R | NGAN,NGNG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| EQRSpCas9 | D1335E,R1335Q,T1337R | NGAG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| xCas9 | A262T,R324L,S409I,E480K,E543D,M694I,E1219V | NG,GAA,GAT | PAM改变;通过噬菌体辅助持续进化获得 | [20] |
| SpG | D1135L,S1136W,G1218K,E1219Q,R1335Q,T1337R | NGN | PAM改变;通过SpCas9理性设计获得 | [32] |
| SpRY | SpG,A61R,L1111R,A1322R,N1317R,R1333P | NRN,NYN | PAM改变;通过对SpG进一步理性设计获得 | [32] |
| evoCas9 | M495V, Y515N, K526E, R661Q | NGG | 保真性提高,编辑效率接近天然SpCas9 | [33] |
| fCas9 | SpdCas9与FokI融合 | NGG | 保真性提高;通过将FokI核酸酶与dCas9融合获得 | [34] |
| StCas9 | 原生嗜热链球菌Cas9 | NNAGAAW | 1121个氨基酸 | [35] |
| NmCas9 | 原生脑膜炎奈瑟菌Cas9 | NNNNGATT | 1082个氨基酸 | [36] |
| SaCas9 | 原生金黄色葡萄球菌Cas9 | NNGRRT | 1053个氨基酸;基因组编辑效率与SpCas9相当 | [37] |
| CjCas9 | 原生曲状杆菌Cas9 | NNNVRYM | 984个氨基酸 | [38] |
注:V代表A、C或G;R代表A或G;Y代表C或T;M代表A或C. ...
... 在CRISPR/Cas9的基因转录调控系统中,对Cas9蛋白工程化的dCas9分别在RuvC和HNH结构域发生突变使得RNP复合物仅能发挥靶序列识别功能,而无法切断核苷酸双链[29].nCas9(Cas9 nickase)是仅能使靶DNA发生单链断裂(single-strand break,SSB)的Cas9突变体,其中nCas9(H840A)和nCas9(D10A)分别是Cas9在HNH和RuvC结构域发生突变后的突变体蛋白,从而能够使该结构域失去内切酶活性,使DNA单链断裂,产生“缺口”[22,39].将dCas9或nCas9与各种效应蛋白如转录激活因子或抑制因子、表观遗传修饰因子、碱基编辑器甚至具有特定功能的酶等融合后,研究人员便能够借助CRISPR/Cas9系统对靶序列实现特定生物学功能,如表征转录基因乃至基因组上非编码区的功能、通过将碱基编辑器与nCas9融合能够在HDR活性低的细胞中进行精确高效的单碱基编辑等.因此本节将重点回顾与总结多种多样的效应蛋白为CRISPR/Cas9系统带来的多元化优化和发展,以及赋予该系统的多种新功能. ...
Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage
7
2016
... Major Cas9 variants and orthologues as well as their features
Table 1| 变体及直系同源 | 改造位点或方式 | PAM | 特性 | 参考文献 |
|---|
| SpCas9 | 原生酿脓链球菌Cas9 | NGG | 1368个氨基酸 | [29] |
| dCas9 | D10A,H840A | NGG | 核酸酶活性失活 | [29] |
| nCas9(D10A) | D10A | NGG | 切口酶,非靶向链切割活性失活 | [30] |
| nCas9(H840A) | H840A | NGG | 切口酶,靶向链切割活性失活 | [30] |
| SpCas9NG | R1335V,L1111R,D1135V,G1218R,E1219F,A1322R,T1337R | NG | PAM改变;通过Cas9理性设计获得 | [31] |
| VRERSpCas9 | D1135A,G1218R,R1335E,T1337R | NGCG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| VQRSpCas9 | D1135V,R1335Q,T1337R | NGAN,NGNG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| EQRSpCas9 | D1335E,R1335Q,T1337R | NGAG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| xCas9 | A262T,R324L,S409I,E480K,E543D,M694I,E1219V | NG,GAA,GAT | PAM改变;通过噬菌体辅助持续进化获得 | [20] |
| SpG | D1135L,S1136W,G1218K,E1219Q,R1335Q,T1337R | NGN | PAM改变;通过SpCas9理性设计获得 | [32] |
| SpRY | SpG,A61R,L1111R,A1322R,N1317R,R1333P | NRN,NYN | PAM改变;通过对SpG进一步理性设计获得 | [32] |
| evoCas9 | M495V, Y515N, K526E, R661Q | NGG | 保真性提高,编辑效率接近天然SpCas9 | [33] |
| fCas9 | SpdCas9与FokI融合 | NGG | 保真性提高;通过将FokI核酸酶与dCas9融合获得 | [34] |
| StCas9 | 原生嗜热链球菌Cas9 | NNAGAAW | 1121个氨基酸 | [35] |
| NmCas9 | 原生脑膜炎奈瑟菌Cas9 | NNNNGATT | 1082个氨基酸 | [36] |
| SaCas9 | 原生金黄色葡萄球菌Cas9 | NNGRRT | 1053个氨基酸;基因组编辑效率与SpCas9相当 | [37] |
| CjCas9 | 原生曲状杆菌Cas9 | NNNVRYM | 984个氨基酸 | [38] |
注:V代表A、C或G;R代表A或G;Y代表C或T;M代表A或C. ...
... [
30]
| SpCas9NG | R1335V,L1111R,D1135V,G1218R,E1219F,A1322R,T1337R | NG | PAM改变;通过Cas9理性设计获得 | [31] |
| VRERSpCas9 | D1135A,G1218R,R1335E,T1337R | NGCG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| VQRSpCas9 | D1135V,R1335Q,T1337R | NGAN,NGNG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| EQRSpCas9 | D1335E,R1335Q,T1337R | NGAG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| xCas9 | A262T,R324L,S409I,E480K,E543D,M694I,E1219V | NG,GAA,GAT | PAM改变;通过噬菌体辅助持续进化获得 | [20] |
| SpG | D1135L,S1136W,G1218K,E1219Q,R1335Q,T1337R | NGN | PAM改变;通过SpCas9理性设计获得 | [32] |
| SpRY | SpG,A61R,L1111R,A1322R,N1317R,R1333P | NRN,NYN | PAM改变;通过对SpG进一步理性设计获得 | [32] |
| evoCas9 | M495V, Y515N, K526E, R661Q | NGG | 保真性提高,编辑效率接近天然SpCas9 | [33] |
| fCas9 | SpdCas9与FokI融合 | NGG | 保真性提高;通过将FokI核酸酶与dCas9融合获得 | [34] |
| StCas9 | 原生嗜热链球菌Cas9 | NNAGAAW | 1121个氨基酸 | [35] |
| NmCas9 | 原生脑膜炎奈瑟菌Cas9 | NNNNGATT | 1082个氨基酸 | [36] |
| SaCas9 | 原生金黄色葡萄球菌Cas9 | NNGRRT | 1053个氨基酸;基因组编辑效率与SpCas9相当 | [37] |
| CjCas9 | 原生曲状杆菌Cas9 | NNNVRYM | 984个氨基酸 | [38] |
注:V代表A、C或G;R代表A或G;Y代表C或T;M代表A或C. ...
... Functional optimization through fusion expression of effector proteins with CRISPR/Cas9 system
Table 2| 功能 | 效应蛋白或gRNA工程 | 系统 | 特征 | 参考文献 |
|---|
| 表达调控 | 无 | dCas9 | 原核生物基因抑制表达,真核生物基因抑制效率低 | [22] |
| KRAB,MXI1,TUP1抑制结构域 | dCas9 | 可在真核生物中抑制基因表达 | [62-63] |
| KRAB-MeCP1融合抑制结构域 | dCas9 | 在哺乳动物细胞中高效抑制目的基因表达 | [64] |
| p65,VP64激活结构域 | dCas9 | 激活基因表达,效率较低 | [65-66] |
| VP64-p65-Rta(VPR)融合激活结构域 | dCas9 | 与VP64相比提高了22~320倍;多重基因激活表达;刺激iPSCs神经元分化 | [67] |
| gRNA工程;gRNA上携带携带两个MS2发夹二级结构 | dCas9 | 每个发夹结构招募一个MS2-p65-HSF1融合激活蛋白;与VP64相比,使10个靶基因上调2倍以上 | [68] |
| SPY系统(包括SpyTag和SpyCatcher)和Med2激活结构域 | dCas9 | 招募多拷贝SpyCatcher和Med2融合蛋白使酿酒酵母目的基因表达提高34.9倍 | [69] |
| SunTag系统(包括GCN4肽和抗GCN4抗体的单链可变片段scFv)和MIG1抑制结构域 | dCpf1 | 招募多拷贝ScFv与MIG1融合蛋白使酿酒酵母目的基因受到95%的抑制 | [69] |
| 表观基因组编辑 | KRAB-DNA甲基化酶催化结构域融合蛋白 | dCas9 | 融合抑制因子和甲基化酶;造成目的基因78%的稳定沉默 | [70] |
| 两侧分别融合KRAB和Dnmt3A-Dnmt3L融合DNA甲基化酶结构域 | dCas9 | 在干细胞分裂分化为神经细胞后仍维持DNA甲基化和基因抑制 | [71] |
| TET1甲基胞嘧啶双加氧酶1催化结构域 | dCas9 | dCas9- TET1靶向至BRCA1基因启动子后成功地上调其表达水平 | [72] |
| 人类组蛋白乙酰基转移酶p300催化核心 | dCas9 | 催化组蛋白H3 第27位赖氨酸残基的乙酰基化;基因表达上调 | [73] |
| 单碱基编辑 | 胞嘧啶脱氨酶,腺嘌呤脱氨酶 | nCas9 | 在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换 | [30,74-75] |
| CBE-UGI融合蛋白 | nCas9 | UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率 | [30] |
| 单链DNA结合结构域ssDBD融合到胞嘧啶脱氨酶与nCas9之间 | nCas9 | 通过延长ssDNA的暴露时间极大地提高CBEs的编辑效率;使CBEs的编辑窗口范围得到了提高 | [76] |
| 组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶 | nCas9 | 首次实现大肠杆菌中C到A 87.2%和哺乳动物细胞C到G 5.3%至53.0%的编辑效率 | [77] |
| 先导编辑 | 通过gRNA工程改造的pegRNA;与逆转录酶融合 | nCas9 | 改变pegRNA可实现精确的碱基替换、基因敲除和插入 | [27] |
| 通过gRNA工程改造的pegRNA;与逆转录酶和Rad51融合 | nCas9 | 融合单链DNA结合蛋白Rad51使PE2的编辑效率最高提升2.6倍 | [78] |
在CRISPR/Cas9的基因转录调控系统中,对Cas9蛋白工程化的dCas9分别在RuvC和HNH结构域发生突变使得RNP复合物仅能发挥靶序列识别功能,而无法切断核苷酸双链[29].nCas9(Cas9 nickase)是仅能使靶DNA发生单链断裂(single-strand break,SSB)的Cas9突变体,其中nCas9(H840A)和nCas9(D10A)分别是Cas9在HNH和RuvC结构域发生突变后的突变体蛋白,从而能够使该结构域失去内切酶活性,使DNA单链断裂,产生“缺口”[22,39].将dCas9或nCas9与各种效应蛋白如转录激活因子或抑制因子、表观遗传修饰因子、碱基编辑器甚至具有特定功能的酶等融合后,研究人员便能够借助CRISPR/Cas9系统对靶序列实现特定生物学功能,如表征转录基因乃至基因组上非编码区的功能、通过将碱基编辑器与nCas9融合能够在HDR活性低的细胞中进行精确高效的单碱基编辑等.因此本节将重点回顾与总结多种多样的效应蛋白为CRISPR/Cas9系统带来的多元化优化和发展,以及赋予该系统的多种新功能. ...
... [
30]
| 单链DNA结合结构域ssDBD融合到胞嘧啶脱氨酶与nCas9之间 | nCas9 | 通过延长ssDNA的暴露时间极大地提高CBEs的编辑效率;使CBEs的编辑窗口范围得到了提高 | [76] |
| 组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶 | nCas9 | 首次实现大肠杆菌中C到A 87.2%和哺乳动物细胞C到G 5.3%至53.0%的编辑效率 | [77] |
| 先导编辑 | 通过gRNA工程改造的pegRNA;与逆转录酶融合 | nCas9 | 改变pegRNA可实现精确的碱基替换、基因敲除和插入 | [27] |
| 通过gRNA工程改造的pegRNA;与逆转录酶和Rad51融合 | nCas9 | 融合单链DNA结合蛋白Rad51使PE2的编辑效率最高提升2.6倍 | [78] |
在CRISPR/Cas9的基因转录调控系统中,对Cas9蛋白工程化的dCas9分别在RuvC和HNH结构域发生突变使得RNP复合物仅能发挥靶序列识别功能,而无法切断核苷酸双链[29].nCas9(Cas9 nickase)是仅能使靶DNA发生单链断裂(single-strand break,SSB)的Cas9突变体,其中nCas9(H840A)和nCas9(D10A)分别是Cas9在HNH和RuvC结构域发生突变后的突变体蛋白,从而能够使该结构域失去内切酶活性,使DNA单链断裂,产生“缺口”[22,39].将dCas9或nCas9与各种效应蛋白如转录激活因子或抑制因子、表观遗传修饰因子、碱基编辑器甚至具有特定功能的酶等融合后,研究人员便能够借助CRISPR/Cas9系统对靶序列实现特定生物学功能,如表征转录基因乃至基因组上非编码区的功能、通过将碱基编辑器与nCas9融合能够在HDR活性低的细胞中进行精确高效的单碱基编辑等.因此本节将重点回顾与总结多种多样的效应蛋白为CRISPR/Cas9系统带来的多元化优化和发展,以及赋予该系统的多种新功能. ...
... 通过使用基于CRISPR/dCas9系统的一系列融合蛋白工具可以实现诸多靶向的表观遗传修饰,如DNA甲基化和去甲基化、组蛋白乙酰基化/去乙酰化和甲基化/去甲基化等,从而调控基因的表达并在许多细胞过程、癌症治疗及细胞工厂中发挥关键作用(图2)[30].其中DNA甲基化如5′胞嘧啶甲基化(5mC)是一种稳定的抑制性化学修饰调控.2016年,研究人员将dCas9、KRAB转录抑制因子以及DNA甲基化酶(DNA methyltransferase)的催化结构域融合蛋白基因与7个gRNA在K562细胞中共表达后获得了β2-微球蛋白启动子-增强子78%的稳定沉默[70].2021年,Nunez等[71]开发的CRISPRoff甲基化修饰工具能够在细胞分裂及分化过程中稳定维持靶基因的甲基化并抑制其表达水平.DNA去甲基化是将甲基胞嘧啶双加氧酶1(methylcytosine dioxygenase 1,TET1)的肽链C端催化结构域与dCas9融合后,再靶向至目的基因启动子或增强子区,从而实现目的基因的表达上调.2016年,Choudhury等[72]将dCas9- TET1靶向至乳腺癌基因1(breast cancer 1,BRCA1)启动子后成功地上调其表达水平.BRCA1作为抑制恶性肿瘤发生的关键基因之一,其成功的靶向上调为研究癌变新疗法带来新的思路,证明了通过去甲基化靶向激活人类肿瘤抑制基因的疗法是可行的.同样的,将dCas9与人类组蛋白乙酰基转移酶p300的催化核心融合后靶向至启动子和增强子区从而催化组蛋白H3 第27位赖氨酸残基的乙酰基化也会造成较强的基因上调[73].2019年,Chen等[102]通过提高麦角生物碱合成基因簇(ergot alkaloid synthesis gene cluster)的组蛋白乙酰化水平,使麦角菌Cp-1菌株(Claviceps pururea Cp-1 strain)中的麦角生物碱含量提高了一倍.通过融合dCas9与适当的表观遗传效应蛋白,CRISPR表观遗传工程工具则可利用CRISPR系统稳健地靶向性实现各种所需的表观遗传修饰,而不同工具间的适当组合,理论上讲可以为表观遗传学工具箱带来更多新型和功能复杂的应用策略,如利用与CRISPRa和CRISPRi优化策略类似的方式优化CRISPR表观遗传工程工具箱的特异性、稳定性和性能[103]. ...
... 为了改善CRIPSR/Cas9系统的碱基编辑工具箱,研究人员通过将nCas9与胞嘧啶脱氨酶(cytosine deaminase)或腺嘌呤脱氨酶(adenine deaminase)融合的方式在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换(图2),其中相应的融合蛋白分别被称为胞嘧啶碱基编辑器(cytosine base editors,CBEs)[30,74]和腺嘌呤碱基编辑器(adenine base editors,ABEs)[75].CBEs中的胞嘧啶脱氨酶能使被nCas9释放的单链DNA(single strand DNA,ssDNA)上的胞嘧啶C转变为尿嘧啶U,随后该位置产生的U:G错配碱基对被修复为T:A碱基对;相应的,ABEs中的腺嘌呤脱氨酶使腺嘌呤A水解脱氨为次黄嘌呤I,随后DNA聚合酶能将次黄嘌呤I识别为鸟嘌呤G进行复制,互补链上原来与腺嘌呤A配对的胸腺嘧啶T则会相应变成胞嘧啶C,至此完成碱基置换全过程[107].由于不存在天然的ssDNA腺嘌呤脱氨酶[75],Kim等[108]通过定向进化改造得到一种大肠杆菌(Escherichia coli)tRNA来源的腺嘌呤脱氨酶ecTadA(E.coli TadA)突变体TadA*.由于在天然环境(大肠杆菌)中TadA是以同型二聚体的方式发挥作用,所以最初的TadA*-nCas9融合蛋白在哺乳动物细胞中的编辑效率并不高,直到研究人员将野生型TadA单体、进化的突变体TadA*及nCas9整合到一起(TadA- TadA*- nCas9),才使ABE在哺乳动物中的编辑效率大大提高.2018年,David R. Liu团队[109]工程化改造并优化了ABE7.10后获得的ABEmax最高可获得前者7.9倍的编辑效率,是目前为止在哺乳动物中编辑效率最高的ABEs.为了方便体内编辑,该团队[110]在2022年7月发表的紧凑型ABEs递送系统通过将小型腺嘌呤编辑器SaABE8e及其PAM变体SaKKH-ABE8e的基因长度最小化以及AAV组件基因长度最小化后使得ABEs能够进行单AAV递送,且在肝脏(66%)、心脏(33%)和肌肉细胞(22%)中的编辑效率均高于双AAV递送系统.同年,吉林大学李占军团队和赖学良团队[111]合作开发的紧凑型的Cas9(Cje2Cas9和Cje3Cas9)能够与sgRNA一起包装到单个AAV中,并在成年小鼠肝脏中以12%的效率成功编辑Pcsk9基因,最终有效降低了小鼠体内的胆固醇水平.高效的单AAV递送系统的开发不仅确立了其相对于双AAV递送系统的优势,还促进了CRISPR/Cas9系统作为体内碱基编辑工具时的研究和治疗应用,具有巨大的临床治疗价值.为了改善编辑效率,Zhang等[76]通过将单链DNA结合结构域(single-stranded DNA-binding domain,ssDBD)融合到胞嘧啶脱氨酶与nCas9之间从而延长ssDNA的暴露时间,可在不增加插入缺失突变(indels)、细胞毒性或脱靶效应等负面效应概率的条件下极大地提高CBEs的编辑效率.ssDBD的融合还使CBEs的编辑窗口范围得到了提高,从而拓宽了其靶向作用范围及用于疾病建模和基因治疗的适用范围.David R. Liu团队[30]在CBE的基础上融合了尿嘧啶糖基化酶抑制剂(uracil glycosylase inhibitor,UGI),作为一种体量较小的蛋白(9.5kDa),UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率.该团队还发现通过招募多拷贝的脱氨酶或更改其空间位置以优化编辑窗口能有效提高编辑效率和特异性.此外,与nCas9融合的脱氨酶若工程化改造后具有序列偏好性,则可通过降低旁观者编辑效应从而提高编辑精度.尽管ABEs和CBEs碱基编辑器的性能不断提升,A:T到G:C和C:G到T:A碱基对的单向固定替换仍限制着BEs作为单碱基改变工具使用时的灵活性.2020年,Zhao等[77]通过组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶(uracil-DNA glycosylase,Ung)实现了大肠杆菌中C到A 87.2%的编辑效率和哺乳动物细胞C到G 5.3%~53.0%的编辑效率.先导编辑作为一种多功能编辑手段,虽然同时具备可编程性和高精确度,但对于单碱基编辑的效率仍与BEs有较大差距[28].2021年,Song等[78]构建的hyPE2在PE2的基础上融合加入了一个ssDBD结构域Rad51,使得PE2的编辑效率最高提升了2.6倍,同时该团队还通过深度学习开发了能够预测不同的hyPE2与PE2相比编辑效率提升倍数的计算模型PEselector,该工具的网址为http://deepcrispr.info/PEselector.2022年,Velimirovic等[112]通过高通量肽链自编辑测序(peptide self-editing sequencing assay,PepSEq)测出12000个85个氨基酸的肽段对PE效率的影响,从而通过肽链融合提高PE的效率.综上所述,基于CRIPSR/Cas9系统的碱基编辑器编辑效率、编辑精度的不断提高以及编辑窗口乃至碱基固定替换模式的扩展相对于大量已知且不断增加的遗传病相关点突变数量仍捉襟见肘,各种脱氨酶赋予CRIPSR/Cas9系统的碱基编辑能力在其他结构域如UGI和ssDBD加持下不仅能够发挥CRIPSR系统高效率、高灵活、易编程的优势,也能够充分发挥脱氨酶的效能,但仍需进一步优化它们以有效地转换碱基和在无旁观者编辑效应的条件下进行目的碱基替换. ...
... [30]在CBE的基础上融合了尿嘧啶糖基化酶抑制剂(uracil glycosylase inhibitor,UGI),作为一种体量较小的蛋白(9.5kDa),UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率.该团队还发现通过招募多拷贝的脱氨酶或更改其空间位置以优化编辑窗口能有效提高编辑效率和特异性.此外,与nCas9融合的脱氨酶若工程化改造后具有序列偏好性,则可通过降低旁观者编辑效应从而提高编辑精度.尽管ABEs和CBEs碱基编辑器的性能不断提升,A:T到G:C和C:G到T:A碱基对的单向固定替换仍限制着BEs作为单碱基改变工具使用时的灵活性.2020年,Zhao等[77]通过组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶(uracil-DNA glycosylase,Ung)实现了大肠杆菌中C到A 87.2%的编辑效率和哺乳动物细胞C到G 5.3%~53.0%的编辑效率.先导编辑作为一种多功能编辑手段,虽然同时具备可编程性和高精确度,但对于单碱基编辑的效率仍与BEs有较大差距[28].2021年,Song等[78]构建的hyPE2在PE2的基础上融合加入了一个ssDBD结构域Rad51,使得PE2的编辑效率最高提升了2.6倍,同时该团队还通过深度学习开发了能够预测不同的hyPE2与PE2相比编辑效率提升倍数的计算模型PEselector,该工具的网址为http://deepcrispr.info/PEselector.2022年,Velimirovic等[112]通过高通量肽链自编辑测序(peptide self-editing sequencing assay,PepSEq)测出12000个85个氨基酸的肽段对PE效率的影响,从而通过肽链融合提高PE的效率.综上所述,基于CRIPSR/Cas9系统的碱基编辑器编辑效率、编辑精度的不断提高以及编辑窗口乃至碱基固定替换模式的扩展相对于大量已知且不断增加的遗传病相关点突变数量仍捉襟见肘,各种脱氨酶赋予CRIPSR/Cas9系统的碱基编辑能力在其他结构域如UGI和ssDBD加持下不仅能够发挥CRIPSR系统高效率、高灵活、易编程的优势,也能够充分发挥脱氨酶的效能,但仍需进一步优化它们以有效地转换碱基和在无旁观者编辑效应的条件下进行目的碱基替换. ...
Engineered CRISPR-Cas9 nuclease with expanded targeting space
2
2018
... Major Cas9 variants and orthologues as well as their features
Table 1| 变体及直系同源 | 改造位点或方式 | PAM | 特性 | 参考文献 |
|---|
| SpCas9 | 原生酿脓链球菌Cas9 | NGG | 1368个氨基酸 | [29] |
| dCas9 | D10A,H840A | NGG | 核酸酶活性失活 | [29] |
| nCas9(D10A) | D10A | NGG | 切口酶,非靶向链切割活性失活 | [30] |
| nCas9(H840A) | H840A | NGG | 切口酶,靶向链切割活性失活 | [30] |
| SpCas9NG | R1335V,L1111R,D1135V,G1218R,E1219F,A1322R,T1337R | NG | PAM改变;通过Cas9理性设计获得 | [31] |
| VRERSpCas9 | D1135A,G1218R,R1335E,T1337R | NGCG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| VQRSpCas9 | D1135V,R1335Q,T1337R | NGAN,NGNG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| EQRSpCas9 | D1335E,R1335Q,T1337R | NGAG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| xCas9 | A262T,R324L,S409I,E480K,E543D,M694I,E1219V | NG,GAA,GAT | PAM改变;通过噬菌体辅助持续进化获得 | [20] |
| SpG | D1135L,S1136W,G1218K,E1219Q,R1335Q,T1337R | NGN | PAM改变;通过SpCas9理性设计获得 | [32] |
| SpRY | SpG,A61R,L1111R,A1322R,N1317R,R1333P | NRN,NYN | PAM改变;通过对SpG进一步理性设计获得 | [32] |
| evoCas9 | M495V, Y515N, K526E, R661Q | NGG | 保真性提高,编辑效率接近天然SpCas9 | [33] |
| fCas9 | SpdCas9与FokI融合 | NGG | 保真性提高;通过将FokI核酸酶与dCas9融合获得 | [34] |
| StCas9 | 原生嗜热链球菌Cas9 | NNAGAAW | 1121个氨基酸 | [35] |
| NmCas9 | 原生脑膜炎奈瑟菌Cas9 | NNNNGATT | 1082个氨基酸 | [36] |
| SaCas9 | 原生金黄色葡萄球菌Cas9 | NNGRRT | 1053个氨基酸;基因组编辑效率与SpCas9相当 | [37] |
| CjCas9 | 原生曲状杆菌Cas9 | NNNVRYM | 984个氨基酸 | [38] |
注:V代表A、C或G;R代表A或G;Y代表C或T;M代表A或C. ...
... 尽管CRISPR/Cas9系统已在多种生物和细胞的基因组上得到了应用[46-51],Cas9对于PAM序列NGG的严格识别、复杂基因组中gRNA潜在的脱靶效应以及该系统作为医疗手段使用时的保真性仍大大限制其在更广范围下的应用与开发.如在人类基因组DNA上平均每8个碱基对存在一个NGG位点[52],Fu等[53]在人类细胞中表征了CRISPR/Cas9系统的脱靶效应,结果显示该系统的脱靶率高达66%.因此,研究人员通过工程化Cas9蛋白或gRNA等方式,使CRISPR/Cas9系统的靶向范围、靶向特异性和保真性均得到了拓展和提高[19-20].2018年,张锋团队和Osamu Nureki团队[31]通过对SpCas9进行理性设计获得了能够识别NG PAM序列的突变体蛋白SpCas9-NG,并证明了其在人类细胞中的基因组编辑能力以及与胞苷脱氨酶融合后仍具有单碱基编辑能力.2020年,David R. Liu团队[54]利用噬菌体辅助持续进化(phage-assisted continuous evolution,PACE)方法获得了三种识别非G PAM序列的SpCas9突变体,从而进一步提高了CRISPR/Cas9系统的靶向范围.Walton等[32]通过改造与PAM序列直接作用的氨基酸位点获得了能稳定识别NRN PAM序列的突变体蛋白SpRY,同时其能以较低效率识别NYN PAM序列,使其在靶点识别上几乎不受PAM限制.Hu等[20]应用PACE方法获得一种能够识别NG、GAA及GAT等PAM序列的SpCas9突变体xCas9,同时其能以优于SpCas9的特异性靶向NGG,且对各种PAM的靶向能力均在人类细胞中的基因编辑、转录调控以及胞嘧啶与腺嘌呤核苷酸的碱基修饰中得到了应用.2014年,Guilinger等[34]将FokⅠ核酸酶与核酸酶活性失活的Cas9蛋白(catalytically inactive Cas9,dCas9)融合后得到了一种融合蛋白fCas9,只有两个fCas9单体同时结合在靶基因附近时才能切割目的基因,使其在人类细胞中进行基因编辑的特异性比野生型Cas9提高了140倍.多种研究已报道了将SpCas9或sgRNA工程化后,通过质粒载体携带并表达CRISPR/Cas9系统或通过RNP递送的方式均能够在基因组范围上降低CRISPR/Cas9系统的脱靶效应,并显著提高该系统的保真性[33,55-58].2018年,Casini等[33]筛选并表征了能够提高CRISPR/Cas9系统编辑效率的Cas9 REC3结构域突变库并组合了四种有利突变后,获得了一种保真性比野生型SpCas9提高了79倍的突变体蛋白evoCas9,同时编辑效率接近野生型SpCas9.除了对Cas9蛋白进行优化,对gRNA不断进行工程化改造也能使CRISPR/Cas9系统的性能得到显著提升.2014年,Fu等[44]证明了间隔区截短后的gRNA的脱靶率明显低于全长的gRNA(20 nt),最大降低幅度达5000倍.2019年,Kocak等[21]在工程化设计的sgRNA上的间隔区加入一“发夹”形RNA二级结构,该结构能够在发生脱靶编辑时在脱靶位点处提高gRNA或crRNA链入侵的热力学屏障而并不影响中靶效率,使CRISPR系统的特异性增加了两个数量级.2020年,Rose等[59]基于过短的gRNA(间隔区小于16 nt)与Cas9形成的RNP只能结合却不能切割靶位点特性设计的失活RNA(dead-RNAs,dRNAs)可保护多个脱靶位点,从而使得SpCas9的靶向特异性提高了40倍.相较于工程化改造的高特异性Cas9突变体来说,通过缩短gRNA的间隔区来提高靶向特异性的方法在设计和应用方面均更易用和灵活,预计将成为未来的研究热点方向. ...
Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants
3
2020
... Major Cas9 variants and orthologues as well as their features
Table 1| 变体及直系同源 | 改造位点或方式 | PAM | 特性 | 参考文献 |
|---|
| SpCas9 | 原生酿脓链球菌Cas9 | NGG | 1368个氨基酸 | [29] |
| dCas9 | D10A,H840A | NGG | 核酸酶活性失活 | [29] |
| nCas9(D10A) | D10A | NGG | 切口酶,非靶向链切割活性失活 | [30] |
| nCas9(H840A) | H840A | NGG | 切口酶,靶向链切割活性失活 | [30] |
| SpCas9NG | R1335V,L1111R,D1135V,G1218R,E1219F,A1322R,T1337R | NG | PAM改变;通过Cas9理性设计获得 | [31] |
| VRERSpCas9 | D1135A,G1218R,R1335E,T1337R | NGCG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| VQRSpCas9 | D1135V,R1335Q,T1337R | NGAN,NGNG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| EQRSpCas9 | D1335E,R1335Q,T1337R | NGAG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| xCas9 | A262T,R324L,S409I,E480K,E543D,M694I,E1219V | NG,GAA,GAT | PAM改变;通过噬菌体辅助持续进化获得 | [20] |
| SpG | D1135L,S1136W,G1218K,E1219Q,R1335Q,T1337R | NGN | PAM改变;通过SpCas9理性设计获得 | [32] |
| SpRY | SpG,A61R,L1111R,A1322R,N1317R,R1333P | NRN,NYN | PAM改变;通过对SpG进一步理性设计获得 | [32] |
| evoCas9 | M495V, Y515N, K526E, R661Q | NGG | 保真性提高,编辑效率接近天然SpCas9 | [33] |
| fCas9 | SpdCas9与FokI融合 | NGG | 保真性提高;通过将FokI核酸酶与dCas9融合获得 | [34] |
| StCas9 | 原生嗜热链球菌Cas9 | NNAGAAW | 1121个氨基酸 | [35] |
| NmCas9 | 原生脑膜炎奈瑟菌Cas9 | NNNNGATT | 1082个氨基酸 | [36] |
| SaCas9 | 原生金黄色葡萄球菌Cas9 | NNGRRT | 1053个氨基酸;基因组编辑效率与SpCas9相当 | [37] |
| CjCas9 | 原生曲状杆菌Cas9 | NNNVRYM | 984个氨基酸 | [38] |
注:V代表A、C或G;R代表A或G;Y代表C或T;M代表A或C. ...
... [
32]
| evoCas9 | M495V, Y515N, K526E, R661Q | NGG | 保真性提高,编辑效率接近天然SpCas9 | [33] |
| fCas9 | SpdCas9与FokI融合 | NGG | 保真性提高;通过将FokI核酸酶与dCas9融合获得 | [34] |
| StCas9 | 原生嗜热链球菌Cas9 | NNAGAAW | 1121个氨基酸 | [35] |
| NmCas9 | 原生脑膜炎奈瑟菌Cas9 | NNNNGATT | 1082个氨基酸 | [36] |
| SaCas9 | 原生金黄色葡萄球菌Cas9 | NNGRRT | 1053个氨基酸;基因组编辑效率与SpCas9相当 | [37] |
| CjCas9 | 原生曲状杆菌Cas9 | NNNVRYM | 984个氨基酸 | [38] |
注:V代表A、C或G;R代表A或G;Y代表C或T;M代表A或C. ...
... 尽管CRISPR/Cas9系统已在多种生物和细胞的基因组上得到了应用[46-51],Cas9对于PAM序列NGG的严格识别、复杂基因组中gRNA潜在的脱靶效应以及该系统作为医疗手段使用时的保真性仍大大限制其在更广范围下的应用与开发.如在人类基因组DNA上平均每8个碱基对存在一个NGG位点[52],Fu等[53]在人类细胞中表征了CRISPR/Cas9系统的脱靶效应,结果显示该系统的脱靶率高达66%.因此,研究人员通过工程化Cas9蛋白或gRNA等方式,使CRISPR/Cas9系统的靶向范围、靶向特异性和保真性均得到了拓展和提高[19-20].2018年,张锋团队和Osamu Nureki团队[31]通过对SpCas9进行理性设计获得了能够识别NG PAM序列的突变体蛋白SpCas9-NG,并证明了其在人类细胞中的基因组编辑能力以及与胞苷脱氨酶融合后仍具有单碱基编辑能力.2020年,David R. Liu团队[54]利用噬菌体辅助持续进化(phage-assisted continuous evolution,PACE)方法获得了三种识别非G PAM序列的SpCas9突变体,从而进一步提高了CRISPR/Cas9系统的靶向范围.Walton等[32]通过改造与PAM序列直接作用的氨基酸位点获得了能稳定识别NRN PAM序列的突变体蛋白SpRY,同时其能以较低效率识别NYN PAM序列,使其在靶点识别上几乎不受PAM限制.Hu等[20]应用PACE方法获得一种能够识别NG、GAA及GAT等PAM序列的SpCas9突变体xCas9,同时其能以优于SpCas9的特异性靶向NGG,且对各种PAM的靶向能力均在人类细胞中的基因编辑、转录调控以及胞嘧啶与腺嘌呤核苷酸的碱基修饰中得到了应用.2014年,Guilinger等[34]将FokⅠ核酸酶与核酸酶活性失活的Cas9蛋白(catalytically inactive Cas9,dCas9)融合后得到了一种融合蛋白fCas9,只有两个fCas9单体同时结合在靶基因附近时才能切割目的基因,使其在人类细胞中进行基因编辑的特异性比野生型Cas9提高了140倍.多种研究已报道了将SpCas9或sgRNA工程化后,通过质粒载体携带并表达CRISPR/Cas9系统或通过RNP递送的方式均能够在基因组范围上降低CRISPR/Cas9系统的脱靶效应,并显著提高该系统的保真性[33,55-58].2018年,Casini等[33]筛选并表征了能够提高CRISPR/Cas9系统编辑效率的Cas9 REC3结构域突变库并组合了四种有利突变后,获得了一种保真性比野生型SpCas9提高了79倍的突变体蛋白evoCas9,同时编辑效率接近野生型SpCas9.除了对Cas9蛋白进行优化,对gRNA不断进行工程化改造也能使CRISPR/Cas9系统的性能得到显著提升.2014年,Fu等[44]证明了间隔区截短后的gRNA的脱靶率明显低于全长的gRNA(20 nt),最大降低幅度达5000倍.2019年,Kocak等[21]在工程化设计的sgRNA上的间隔区加入一“发夹”形RNA二级结构,该结构能够在发生脱靶编辑时在脱靶位点处提高gRNA或crRNA链入侵的热力学屏障而并不影响中靶效率,使CRISPR系统的特异性增加了两个数量级.2020年,Rose等[59]基于过短的gRNA(间隔区小于16 nt)与Cas9形成的RNP只能结合却不能切割靶位点特性设计的失活RNA(dead-RNAs,dRNAs)可保护多个脱靶位点,从而使得SpCas9的靶向特异性提高了40倍.相较于工程化改造的高特异性Cas9突变体来说,通过缩短gRNA的间隔区来提高靶向特异性的方法在设计和应用方面均更易用和灵活,预计将成为未来的研究热点方向. ...
A highly specific SpCas9 variant is identified by in vivo screening in yeast
3
2018
... Major Cas9 variants and orthologues as well as their features
Table 1| 变体及直系同源 | 改造位点或方式 | PAM | 特性 | 参考文献 |
|---|
| SpCas9 | 原生酿脓链球菌Cas9 | NGG | 1368个氨基酸 | [29] |
| dCas9 | D10A,H840A | NGG | 核酸酶活性失活 | [29] |
| nCas9(D10A) | D10A | NGG | 切口酶,非靶向链切割活性失活 | [30] |
| nCas9(H840A) | H840A | NGG | 切口酶,靶向链切割活性失活 | [30] |
| SpCas9NG | R1335V,L1111R,D1135V,G1218R,E1219F,A1322R,T1337R | NG | PAM改变;通过Cas9理性设计获得 | [31] |
| VRERSpCas9 | D1135A,G1218R,R1335E,T1337R | NGCG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| VQRSpCas9 | D1135V,R1335Q,T1337R | NGAN,NGNG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| EQRSpCas9 | D1335E,R1335Q,T1337R | NGAG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| xCas9 | A262T,R324L,S409I,E480K,E543D,M694I,E1219V | NG,GAA,GAT | PAM改变;通过噬菌体辅助持续进化获得 | [20] |
| SpG | D1135L,S1136W,G1218K,E1219Q,R1335Q,T1337R | NGN | PAM改变;通过SpCas9理性设计获得 | [32] |
| SpRY | SpG,A61R,L1111R,A1322R,N1317R,R1333P | NRN,NYN | PAM改变;通过对SpG进一步理性设计获得 | [32] |
| evoCas9 | M495V, Y515N, K526E, R661Q | NGG | 保真性提高,编辑效率接近天然SpCas9 | [33] |
| fCas9 | SpdCas9与FokI融合 | NGG | 保真性提高;通过将FokI核酸酶与dCas9融合获得 | [34] |
| StCas9 | 原生嗜热链球菌Cas9 | NNAGAAW | 1121个氨基酸 | [35] |
| NmCas9 | 原生脑膜炎奈瑟菌Cas9 | NNNNGATT | 1082个氨基酸 | [36] |
| SaCas9 | 原生金黄色葡萄球菌Cas9 | NNGRRT | 1053个氨基酸;基因组编辑效率与SpCas9相当 | [37] |
| CjCas9 | 原生曲状杆菌Cas9 | NNNVRYM | 984个氨基酸 | [38] |
注:V代表A、C或G;R代表A或G;Y代表C或T;M代表A或C. ...
... 尽管CRISPR/Cas9系统已在多种生物和细胞的基因组上得到了应用[46-51],Cas9对于PAM序列NGG的严格识别、复杂基因组中gRNA潜在的脱靶效应以及该系统作为医疗手段使用时的保真性仍大大限制其在更广范围下的应用与开发.如在人类基因组DNA上平均每8个碱基对存在一个NGG位点[52],Fu等[53]在人类细胞中表征了CRISPR/Cas9系统的脱靶效应,结果显示该系统的脱靶率高达66%.因此,研究人员通过工程化Cas9蛋白或gRNA等方式,使CRISPR/Cas9系统的靶向范围、靶向特异性和保真性均得到了拓展和提高[19-20].2018年,张锋团队和Osamu Nureki团队[31]通过对SpCas9进行理性设计获得了能够识别NG PAM序列的突变体蛋白SpCas9-NG,并证明了其在人类细胞中的基因组编辑能力以及与胞苷脱氨酶融合后仍具有单碱基编辑能力.2020年,David R. Liu团队[54]利用噬菌体辅助持续进化(phage-assisted continuous evolution,PACE)方法获得了三种识别非G PAM序列的SpCas9突变体,从而进一步提高了CRISPR/Cas9系统的靶向范围.Walton等[32]通过改造与PAM序列直接作用的氨基酸位点获得了能稳定识别NRN PAM序列的突变体蛋白SpRY,同时其能以较低效率识别NYN PAM序列,使其在靶点识别上几乎不受PAM限制.Hu等[20]应用PACE方法获得一种能够识别NG、GAA及GAT等PAM序列的SpCas9突变体xCas9,同时其能以优于SpCas9的特异性靶向NGG,且对各种PAM的靶向能力均在人类细胞中的基因编辑、转录调控以及胞嘧啶与腺嘌呤核苷酸的碱基修饰中得到了应用.2014年,Guilinger等[34]将FokⅠ核酸酶与核酸酶活性失活的Cas9蛋白(catalytically inactive Cas9,dCas9)融合后得到了一种融合蛋白fCas9,只有两个fCas9单体同时结合在靶基因附近时才能切割目的基因,使其在人类细胞中进行基因编辑的特异性比野生型Cas9提高了140倍.多种研究已报道了将SpCas9或sgRNA工程化后,通过质粒载体携带并表达CRISPR/Cas9系统或通过RNP递送的方式均能够在基因组范围上降低CRISPR/Cas9系统的脱靶效应,并显著提高该系统的保真性[33,55-58].2018年,Casini等[33]筛选并表征了能够提高CRISPR/Cas9系统编辑效率的Cas9 REC3结构域突变库并组合了四种有利突变后,获得了一种保真性比野生型SpCas9提高了79倍的突变体蛋白evoCas9,同时编辑效率接近野生型SpCas9.除了对Cas9蛋白进行优化,对gRNA不断进行工程化改造也能使CRISPR/Cas9系统的性能得到显著提升.2014年,Fu等[44]证明了间隔区截短后的gRNA的脱靶率明显低于全长的gRNA(20 nt),最大降低幅度达5000倍.2019年,Kocak等[21]在工程化设计的sgRNA上的间隔区加入一“发夹”形RNA二级结构,该结构能够在发生脱靶编辑时在脱靶位点处提高gRNA或crRNA链入侵的热力学屏障而并不影响中靶效率,使CRISPR系统的特异性增加了两个数量级.2020年,Rose等[59]基于过短的gRNA(间隔区小于16 nt)与Cas9形成的RNP只能结合却不能切割靶位点特性设计的失活RNA(dead-RNAs,dRNAs)可保护多个脱靶位点,从而使得SpCas9的靶向特异性提高了40倍.相较于工程化改造的高特异性Cas9突变体来说,通过缩短gRNA的间隔区来提高靶向特异性的方法在设计和应用方面均更易用和灵活,预计将成为未来的研究热点方向. ...
... [33]筛选并表征了能够提高CRISPR/Cas9系统编辑效率的Cas9 REC3结构域突变库并组合了四种有利突变后,获得了一种保真性比野生型SpCas9提高了79倍的突变体蛋白evoCas9,同时编辑效率接近野生型SpCas9.除了对Cas9蛋白进行优化,对gRNA不断进行工程化改造也能使CRISPR/Cas9系统的性能得到显著提升.2014年,Fu等[44]证明了间隔区截短后的gRNA的脱靶率明显低于全长的gRNA(20 nt),最大降低幅度达5000倍.2019年,Kocak等[21]在工程化设计的sgRNA上的间隔区加入一“发夹”形RNA二级结构,该结构能够在发生脱靶编辑时在脱靶位点处提高gRNA或crRNA链入侵的热力学屏障而并不影响中靶效率,使CRISPR系统的特异性增加了两个数量级.2020年,Rose等[59]基于过短的gRNA(间隔区小于16 nt)与Cas9形成的RNP只能结合却不能切割靶位点特性设计的失活RNA(dead-RNAs,dRNAs)可保护多个脱靶位点,从而使得SpCas9的靶向特异性提高了40倍.相较于工程化改造的高特异性Cas9突变体来说,通过缩短gRNA的间隔区来提高靶向特异性的方法在设计和应用方面均更易用和灵活,预计将成为未来的研究热点方向. ...
Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification
2
2014
... Major Cas9 variants and orthologues as well as their features
Table 1| 变体及直系同源 | 改造位点或方式 | PAM | 特性 | 参考文献 |
|---|
| SpCas9 | 原生酿脓链球菌Cas9 | NGG | 1368个氨基酸 | [29] |
| dCas9 | D10A,H840A | NGG | 核酸酶活性失活 | [29] |
| nCas9(D10A) | D10A | NGG | 切口酶,非靶向链切割活性失活 | [30] |
| nCas9(H840A) | H840A | NGG | 切口酶,靶向链切割活性失活 | [30] |
| SpCas9NG | R1335V,L1111R,D1135V,G1218R,E1219F,A1322R,T1337R | NG | PAM改变;通过Cas9理性设计获得 | [31] |
| VRERSpCas9 | D1135A,G1218R,R1335E,T1337R | NGCG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| VQRSpCas9 | D1135V,R1335Q,T1337R | NGAN,NGNG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| EQRSpCas9 | D1335E,R1335Q,T1337R | NGAG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| xCas9 | A262T,R324L,S409I,E480K,E543D,M694I,E1219V | NG,GAA,GAT | PAM改变;通过噬菌体辅助持续进化获得 | [20] |
| SpG | D1135L,S1136W,G1218K,E1219Q,R1335Q,T1337R | NGN | PAM改变;通过SpCas9理性设计获得 | [32] |
| SpRY | SpG,A61R,L1111R,A1322R,N1317R,R1333P | NRN,NYN | PAM改变;通过对SpG进一步理性设计获得 | [32] |
| evoCas9 | M495V, Y515N, K526E, R661Q | NGG | 保真性提高,编辑效率接近天然SpCas9 | [33] |
| fCas9 | SpdCas9与FokI融合 | NGG | 保真性提高;通过将FokI核酸酶与dCas9融合获得 | [34] |
| StCas9 | 原生嗜热链球菌Cas9 | NNAGAAW | 1121个氨基酸 | [35] |
| NmCas9 | 原生脑膜炎奈瑟菌Cas9 | NNNNGATT | 1082个氨基酸 | [36] |
| SaCas9 | 原生金黄色葡萄球菌Cas9 | NNGRRT | 1053个氨基酸;基因组编辑效率与SpCas9相当 | [37] |
| CjCas9 | 原生曲状杆菌Cas9 | NNNVRYM | 984个氨基酸 | [38] |
注:V代表A、C或G;R代表A或G;Y代表C或T;M代表A或C. ...
... 尽管CRISPR/Cas9系统已在多种生物和细胞的基因组上得到了应用[46-51],Cas9对于PAM序列NGG的严格识别、复杂基因组中gRNA潜在的脱靶效应以及该系统作为医疗手段使用时的保真性仍大大限制其在更广范围下的应用与开发.如在人类基因组DNA上平均每8个碱基对存在一个NGG位点[52],Fu等[53]在人类细胞中表征了CRISPR/Cas9系统的脱靶效应,结果显示该系统的脱靶率高达66%.因此,研究人员通过工程化Cas9蛋白或gRNA等方式,使CRISPR/Cas9系统的靶向范围、靶向特异性和保真性均得到了拓展和提高[19-20].2018年,张锋团队和Osamu Nureki团队[31]通过对SpCas9进行理性设计获得了能够识别NG PAM序列的突变体蛋白SpCas9-NG,并证明了其在人类细胞中的基因组编辑能力以及与胞苷脱氨酶融合后仍具有单碱基编辑能力.2020年,David R. Liu团队[54]利用噬菌体辅助持续进化(phage-assisted continuous evolution,PACE)方法获得了三种识别非G PAM序列的SpCas9突变体,从而进一步提高了CRISPR/Cas9系统的靶向范围.Walton等[32]通过改造与PAM序列直接作用的氨基酸位点获得了能稳定识别NRN PAM序列的突变体蛋白SpRY,同时其能以较低效率识别NYN PAM序列,使其在靶点识别上几乎不受PAM限制.Hu等[20]应用PACE方法获得一种能够识别NG、GAA及GAT等PAM序列的SpCas9突变体xCas9,同时其能以优于SpCas9的特异性靶向NGG,且对各种PAM的靶向能力均在人类细胞中的基因编辑、转录调控以及胞嘧啶与腺嘌呤核苷酸的碱基修饰中得到了应用.2014年,Guilinger等[34]将FokⅠ核酸酶与核酸酶活性失活的Cas9蛋白(catalytically inactive Cas9,dCas9)融合后得到了一种融合蛋白fCas9,只有两个fCas9单体同时结合在靶基因附近时才能切割目的基因,使其在人类细胞中进行基因编辑的特异性比野生型Cas9提高了140倍.多种研究已报道了将SpCas9或sgRNA工程化后,通过质粒载体携带并表达CRISPR/Cas9系统或通过RNP递送的方式均能够在基因组范围上降低CRISPR/Cas9系统的脱靶效应,并显著提高该系统的保真性[33,55-58].2018年,Casini等[33]筛选并表征了能够提高CRISPR/Cas9系统编辑效率的Cas9 REC3结构域突变库并组合了四种有利突变后,获得了一种保真性比野生型SpCas9提高了79倍的突变体蛋白evoCas9,同时编辑效率接近野生型SpCas9.除了对Cas9蛋白进行优化,对gRNA不断进行工程化改造也能使CRISPR/Cas9系统的性能得到显著提升.2014年,Fu等[44]证明了间隔区截短后的gRNA的脱靶率明显低于全长的gRNA(20 nt),最大降低幅度达5000倍.2019年,Kocak等[21]在工程化设计的sgRNA上的间隔区加入一“发夹”形RNA二级结构,该结构能够在发生脱靶编辑时在脱靶位点处提高gRNA或crRNA链入侵的热力学屏障而并不影响中靶效率,使CRISPR系统的特异性增加了两个数量级.2020年,Rose等[59]基于过短的gRNA(间隔区小于16 nt)与Cas9形成的RNP只能结合却不能切割靶位点特性设计的失活RNA(dead-RNAs,dRNAs)可保护多个脱靶位点,从而使得SpCas9的靶向特异性提高了40倍.相较于工程化改造的高特异性Cas9突变体来说,通过缩短gRNA的间隔区来提高靶向特异性的方法在设计和应用方面均更易用和灵活,预计将成为未来的研究热点方向. ...
Phage response to CRISPR-encoded resistance in Streptococcus thermophilus
2
2008
... Major Cas9 variants and orthologues as well as their features
Table 1| 变体及直系同源 | 改造位点或方式 | PAM | 特性 | 参考文献 |
|---|
| SpCas9 | 原生酿脓链球菌Cas9 | NGG | 1368个氨基酸 | [29] |
| dCas9 | D10A,H840A | NGG | 核酸酶活性失活 | [29] |
| nCas9(D10A) | D10A | NGG | 切口酶,非靶向链切割活性失活 | [30] |
| nCas9(H840A) | H840A | NGG | 切口酶,靶向链切割活性失活 | [30] |
| SpCas9NG | R1335V,L1111R,D1135V,G1218R,E1219F,A1322R,T1337R | NG | PAM改变;通过Cas9理性设计获得 | [31] |
| VRERSpCas9 | D1135A,G1218R,R1335E,T1337R | NGCG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| VQRSpCas9 | D1135V,R1335Q,T1337R | NGAN,NGNG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| EQRSpCas9 | D1335E,R1335Q,T1337R | NGAG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| xCas9 | A262T,R324L,S409I,E480K,E543D,M694I,E1219V | NG,GAA,GAT | PAM改变;通过噬菌体辅助持续进化获得 | [20] |
| SpG | D1135L,S1136W,G1218K,E1219Q,R1335Q,T1337R | NGN | PAM改变;通过SpCas9理性设计获得 | [32] |
| SpRY | SpG,A61R,L1111R,A1322R,N1317R,R1333P | NRN,NYN | PAM改变;通过对SpG进一步理性设计获得 | [32] |
| evoCas9 | M495V, Y515N, K526E, R661Q | NGG | 保真性提高,编辑效率接近天然SpCas9 | [33] |
| fCas9 | SpdCas9与FokI融合 | NGG | 保真性提高;通过将FokI核酸酶与dCas9融合获得 | [34] |
| StCas9 | 原生嗜热链球菌Cas9 | NNAGAAW | 1121个氨基酸 | [35] |
| NmCas9 | 原生脑膜炎奈瑟菌Cas9 | NNNNGATT | 1082个氨基酸 | [36] |
| SaCas9 | 原生金黄色葡萄球菌Cas9 | NNGRRT | 1053个氨基酸;基因组编辑效率与SpCas9相当 | [37] |
| CjCas9 | 原生曲状杆菌Cas9 | NNNVRYM | 984个氨基酸 | [38] |
注:V代表A、C或G;R代表A或G;Y代表C或T;M代表A或C. ...
... 除了开发Cas9的突变体以弥补和拓展CRISPR/Cas9系统的不足与应用范围外,对Cas9 直系同源蛋白进行挖掘和开发也能够使该系统识别范围得到极大扩展.如来自嗜热链球菌(Streptococcus thermophilus)的StCas9能识别NNGAAW(其中W代表A或T)的PAM序列[4,35],来自脑膜炎奈瑟菌(Neisseria meningitidis)的NmCas9识别NNNNGATT序列[36,60-61],来自金黄色葡萄球菌(Staphylococcus aureus)的SaCas9识别NNGRRT序列(其中R代表A或G)[37],这些SpCas9同源蛋白或其组合应用均已在细菌和哺乳动物细胞中得到了开发.此外,SaCas9在肽链长度较小的条件下(1053个氨基酸,SpCas9含有1368个氨基酸)拥有与SpCas9相当的编辑效率,这使其在大小受限制的传递载体如腺相关病毒(adeno-associated virus,AAV)中得到广泛应用.来自曲状杆菌(Campylobacter jejuni)的CjCas9多肽链上仅含984个氨基酸,能识别PAM序列NNNVRYM,已被报道可以通过AAV递送至小鼠肌纤维和视网膜色素上皮细胞(retinal pigment epithelium,RPE)中并成功进行基因编辑[38].工程化的新型Cas9突变体、gRNA与Cas9直系同源的不断发掘为CRISPR/Cas9系统在各种背景下的应用提供了源源不断的新方法和工具,也证明了该系统作为第三代“基因剪刀”目前仍然处于高速发展阶段.该系统中存在的PAM依赖性、脱靶率较高等问题都需要利用进化工程或计算机辅助理性设计等方式对Cas9蛋白进行进一步探索和改造才能得到解决,使其能够在更多的场景下最大程度地发挥自身高灵活性、高精准性、易编程性的优势. ...
Orthogonal Cas9 proteins for RNA-guided gene regulation and editing
3
2013
... Major Cas9 variants and orthologues as well as their features
Table 1| 变体及直系同源 | 改造位点或方式 | PAM | 特性 | 参考文献 |
|---|
| SpCas9 | 原生酿脓链球菌Cas9 | NGG | 1368个氨基酸 | [29] |
| dCas9 | D10A,H840A | NGG | 核酸酶活性失活 | [29] |
| nCas9(D10A) | D10A | NGG | 切口酶,非靶向链切割活性失活 | [30] |
| nCas9(H840A) | H840A | NGG | 切口酶,靶向链切割活性失活 | [30] |
| SpCas9NG | R1335V,L1111R,D1135V,G1218R,E1219F,A1322R,T1337R | NG | PAM改变;通过Cas9理性设计获得 | [31] |
| VRERSpCas9 | D1135A,G1218R,R1335E,T1337R | NGCG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| VQRSpCas9 | D1135V,R1335Q,T1337R | NGAN,NGNG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| EQRSpCas9 | D1335E,R1335Q,T1337R | NGAG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| xCas9 | A262T,R324L,S409I,E480K,E543D,M694I,E1219V | NG,GAA,GAT | PAM改变;通过噬菌体辅助持续进化获得 | [20] |
| SpG | D1135L,S1136W,G1218K,E1219Q,R1335Q,T1337R | NGN | PAM改变;通过SpCas9理性设计获得 | [32] |
| SpRY | SpG,A61R,L1111R,A1322R,N1317R,R1333P | NRN,NYN | PAM改变;通过对SpG进一步理性设计获得 | [32] |
| evoCas9 | M495V, Y515N, K526E, R661Q | NGG | 保真性提高,编辑效率接近天然SpCas9 | [33] |
| fCas9 | SpdCas9与FokI融合 | NGG | 保真性提高;通过将FokI核酸酶与dCas9融合获得 | [34] |
| StCas9 | 原生嗜热链球菌Cas9 | NNAGAAW | 1121个氨基酸 | [35] |
| NmCas9 | 原生脑膜炎奈瑟菌Cas9 | NNNNGATT | 1082个氨基酸 | [36] |
| SaCas9 | 原生金黄色葡萄球菌Cas9 | NNGRRT | 1053个氨基酸;基因组编辑效率与SpCas9相当 | [37] |
| CjCas9 | 原生曲状杆菌Cas9 | NNNVRYM | 984个氨基酸 | [38] |
注:V代表A、C或G;R代表A或G;Y代表C或T;M代表A或C. ...
... 除了开发Cas9的突变体以弥补和拓展CRISPR/Cas9系统的不足与应用范围外,对Cas9 直系同源蛋白进行挖掘和开发也能够使该系统识别范围得到极大扩展.如来自嗜热链球菌(Streptococcus thermophilus)的StCas9能识别NNGAAW(其中W代表A或T)的PAM序列[4,35],来自脑膜炎奈瑟菌(Neisseria meningitidis)的NmCas9识别NNNNGATT序列[36,60-61],来自金黄色葡萄球菌(Staphylococcus aureus)的SaCas9识别NNGRRT序列(其中R代表A或G)[37],这些SpCas9同源蛋白或其组合应用均已在细菌和哺乳动物细胞中得到了开发.此外,SaCas9在肽链长度较小的条件下(1053个氨基酸,SpCas9含有1368个氨基酸)拥有与SpCas9相当的编辑效率,这使其在大小受限制的传递载体如腺相关病毒(adeno-associated virus,AAV)中得到广泛应用.来自曲状杆菌(Campylobacter jejuni)的CjCas9多肽链上仅含984个氨基酸,能识别PAM序列NNNVRYM,已被报道可以通过AAV递送至小鼠肌纤维和视网膜色素上皮细胞(retinal pigment epithelium,RPE)中并成功进行基因编辑[38].工程化的新型Cas9突变体、gRNA与Cas9直系同源的不断发掘为CRISPR/Cas9系统在各种背景下的应用提供了源源不断的新方法和工具,也证明了该系统作为第三代“基因剪刀”目前仍然处于高速发展阶段.该系统中存在的PAM依赖性、脱靶率较高等问题都需要利用进化工程或计算机辅助理性设计等方式对Cas9蛋白进行进一步探索和改造才能得到解决,使其能够在更多的场景下最大程度地发挥自身高灵活性、高精准性、易编程性的优势. ...
... 基于gRNA的多表达、多个同源Cas9或dCas9蛋白同时表达则可实现CRISPR系统的多路复用正交编辑或多功能组合代谢工程[36,116].自2013年Jiang等[117]证明了肺炎链球菌原生CRISPR/Cas9系统具备多重应用潜力后,研究人员基于gRNA多表达策略在大肠杆菌、酿酒酵母等模式生物体中开展了多项CRISPR系统的多路复用应用研究,从而在合成生物学及生物制造领域实现对底盘细胞的改造,最终提升目标产品产量或改变细胞特性(表3). ...
In vivo genome editing using Staphylococcus aureus Cas9
2
2015
... Major Cas9 variants and orthologues as well as their features
Table 1| 变体及直系同源 | 改造位点或方式 | PAM | 特性 | 参考文献 |
|---|
| SpCas9 | 原生酿脓链球菌Cas9 | NGG | 1368个氨基酸 | [29] |
| dCas9 | D10A,H840A | NGG | 核酸酶活性失活 | [29] |
| nCas9(D10A) | D10A | NGG | 切口酶,非靶向链切割活性失活 | [30] |
| nCas9(H840A) | H840A | NGG | 切口酶,靶向链切割活性失活 | [30] |
| SpCas9NG | R1335V,L1111R,D1135V,G1218R,E1219F,A1322R,T1337R | NG | PAM改变;通过Cas9理性设计获得 | [31] |
| VRERSpCas9 | D1135A,G1218R,R1335E,T1337R | NGCG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| VQRSpCas9 | D1135V,R1335Q,T1337R | NGAN,NGNG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| EQRSpCas9 | D1335E,R1335Q,T1337R | NGAG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| xCas9 | A262T,R324L,S409I,E480K,E543D,M694I,E1219V | NG,GAA,GAT | PAM改变;通过噬菌体辅助持续进化获得 | [20] |
| SpG | D1135L,S1136W,G1218K,E1219Q,R1335Q,T1337R | NGN | PAM改变;通过SpCas9理性设计获得 | [32] |
| SpRY | SpG,A61R,L1111R,A1322R,N1317R,R1333P | NRN,NYN | PAM改变;通过对SpG进一步理性设计获得 | [32] |
| evoCas9 | M495V, Y515N, K526E, R661Q | NGG | 保真性提高,编辑效率接近天然SpCas9 | [33] |
| fCas9 | SpdCas9与FokI融合 | NGG | 保真性提高;通过将FokI核酸酶与dCas9融合获得 | [34] |
| StCas9 | 原生嗜热链球菌Cas9 | NNAGAAW | 1121个氨基酸 | [35] |
| NmCas9 | 原生脑膜炎奈瑟菌Cas9 | NNNNGATT | 1082个氨基酸 | [36] |
| SaCas9 | 原生金黄色葡萄球菌Cas9 | NNGRRT | 1053个氨基酸;基因组编辑效率与SpCas9相当 | [37] |
| CjCas9 | 原生曲状杆菌Cas9 | NNNVRYM | 984个氨基酸 | [38] |
注:V代表A、C或G;R代表A或G;Y代表C或T;M代表A或C. ...
... 除了开发Cas9的突变体以弥补和拓展CRISPR/Cas9系统的不足与应用范围外,对Cas9 直系同源蛋白进行挖掘和开发也能够使该系统识别范围得到极大扩展.如来自嗜热链球菌(Streptococcus thermophilus)的StCas9能识别NNGAAW(其中W代表A或T)的PAM序列[4,35],来自脑膜炎奈瑟菌(Neisseria meningitidis)的NmCas9识别NNNNGATT序列[36,60-61],来自金黄色葡萄球菌(Staphylococcus aureus)的SaCas9识别NNGRRT序列(其中R代表A或G)[37],这些SpCas9同源蛋白或其组合应用均已在细菌和哺乳动物细胞中得到了开发.此外,SaCas9在肽链长度较小的条件下(1053个氨基酸,SpCas9含有1368个氨基酸)拥有与SpCas9相当的编辑效率,这使其在大小受限制的传递载体如腺相关病毒(adeno-associated virus,AAV)中得到广泛应用.来自曲状杆菌(Campylobacter jejuni)的CjCas9多肽链上仅含984个氨基酸,能识别PAM序列NNNVRYM,已被报道可以通过AAV递送至小鼠肌纤维和视网膜色素上皮细胞(retinal pigment epithelium,RPE)中并成功进行基因编辑[38].工程化的新型Cas9突变体、gRNA与Cas9直系同源的不断发掘为CRISPR/Cas9系统在各种背景下的应用提供了源源不断的新方法和工具,也证明了该系统作为第三代“基因剪刀”目前仍然处于高速发展阶段.该系统中存在的PAM依赖性、脱靶率较高等问题都需要利用进化工程或计算机辅助理性设计等方式对Cas9蛋白进行进一步探索和改造才能得到解决,使其能够在更多的场景下最大程度地发挥自身高灵活性、高精准性、易编程性的优势. ...
In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni
2
2017
... Major Cas9 variants and orthologues as well as their features
Table 1| 变体及直系同源 | 改造位点或方式 | PAM | 特性 | 参考文献 |
|---|
| SpCas9 | 原生酿脓链球菌Cas9 | NGG | 1368个氨基酸 | [29] |
| dCas9 | D10A,H840A | NGG | 核酸酶活性失活 | [29] |
| nCas9(D10A) | D10A | NGG | 切口酶,非靶向链切割活性失活 | [30] |
| nCas9(H840A) | H840A | NGG | 切口酶,靶向链切割活性失活 | [30] |
| SpCas9NG | R1335V,L1111R,D1135V,G1218R,E1219F,A1322R,T1337R | NG | PAM改变;通过Cas9理性设计获得 | [31] |
| VRERSpCas9 | D1135A,G1218R,R1335E,T1337R | NGCG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| VQRSpCas9 | D1135V,R1335Q,T1337R | NGAN,NGNG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| EQRSpCas9 | D1335E,R1335Q,T1337R | NGAG | PAM改变;通过细菌选择性定向进化获得 | [19] |
| xCas9 | A262T,R324L,S409I,E480K,E543D,M694I,E1219V | NG,GAA,GAT | PAM改变;通过噬菌体辅助持续进化获得 | [20] |
| SpG | D1135L,S1136W,G1218K,E1219Q,R1335Q,T1337R | NGN | PAM改变;通过SpCas9理性设计获得 | [32] |
| SpRY | SpG,A61R,L1111R,A1322R,N1317R,R1333P | NRN,NYN | PAM改变;通过对SpG进一步理性设计获得 | [32] |
| evoCas9 | M495V, Y515N, K526E, R661Q | NGG | 保真性提高,编辑效率接近天然SpCas9 | [33] |
| fCas9 | SpdCas9与FokI融合 | NGG | 保真性提高;通过将FokI核酸酶与dCas9融合获得 | [34] |
| StCas9 | 原生嗜热链球菌Cas9 | NNAGAAW | 1121个氨基酸 | [35] |
| NmCas9 | 原生脑膜炎奈瑟菌Cas9 | NNNNGATT | 1082个氨基酸 | [36] |
| SaCas9 | 原生金黄色葡萄球菌Cas9 | NNGRRT | 1053个氨基酸;基因组编辑效率与SpCas9相当 | [37] |
| CjCas9 | 原生曲状杆菌Cas9 | NNNVRYM | 984个氨基酸 | [38] |
注:V代表A、C或G;R代表A或G;Y代表C或T;M代表A或C. ...
... 除了开发Cas9的突变体以弥补和拓展CRISPR/Cas9系统的不足与应用范围外,对Cas9 直系同源蛋白进行挖掘和开发也能够使该系统识别范围得到极大扩展.如来自嗜热链球菌(Streptococcus thermophilus)的StCas9能识别NNGAAW(其中W代表A或T)的PAM序列[4,35],来自脑膜炎奈瑟菌(Neisseria meningitidis)的NmCas9识别NNNNGATT序列[36,60-61],来自金黄色葡萄球菌(Staphylococcus aureus)的SaCas9识别NNGRRT序列(其中R代表A或G)[37],这些SpCas9同源蛋白或其组合应用均已在细菌和哺乳动物细胞中得到了开发.此外,SaCas9在肽链长度较小的条件下(1053个氨基酸,SpCas9含有1368个氨基酸)拥有与SpCas9相当的编辑效率,这使其在大小受限制的传递载体如腺相关病毒(adeno-associated virus,AAV)中得到广泛应用.来自曲状杆菌(Campylobacter jejuni)的CjCas9多肽链上仅含984个氨基酸,能识别PAM序列NNNVRYM,已被报道可以通过AAV递送至小鼠肌纤维和视网膜色素上皮细胞(retinal pigment epithelium,RPE)中并成功进行基因编辑[38].工程化的新型Cas9突变体、gRNA与Cas9直系同源的不断发掘为CRISPR/Cas9系统在各种背景下的应用提供了源源不断的新方法和工具,也证明了该系统作为第三代“基因剪刀”目前仍然处于高速发展阶段.该系统中存在的PAM依赖性、脱靶率较高等问题都需要利用进化工程或计算机辅助理性设计等方式对Cas9蛋白进行进一步探索和改造才能得到解决,使其能够在更多的场景下最大程度地发挥自身高灵活性、高精准性、易编程性的优势. ...
A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity
5
2012
... 酿脓链球菌(Streptococcus pyogenes)Cas9(SpCas9)是第一种被应用于哺乳动物细胞基因组编辑的Cas9蛋白[4,39-40],目前仍是CRISPR系统在各应用领域中最常用的Cas9蛋白.在发挥靶序列切割的功能时,SpCas9、CRISPR RNA(crRNA)、反式激活CRISPR RNA(trans-activating crRNA, tracrRNA)结合形成核糖核蛋白复合物(ribonucleoprotein,RNP)共同参与这一过程.在天然CRISPR/Cas9系统中,crRNA作为识别模块,其上的间隔区段能够识别与之互补的特异性靶序列(常见20个核苷酸长度),而靶序列下游必须有三个碱基的PAM序列NGG(N代表A、G、C和T中的任一碱基);tracrRNA和crRNA上的核苷酸链有能互补配对的部分,配对后促使RNP形成以及激活Cas9蛋白[41-42].在目前通用的CRISPR/Cas9系统中,crRNA与tracrRNA被融合成一条sgRNA发挥相同的作用[39].也有研究发现截短gRNA的间隔区段(如14 nt的间隔区段)能够使RNP保持识别和结合靶序列的能力,但无法引入DSB[43];而当gRNA的间隔区段长度在16~20 nt时均可有效进行基因编辑,且17~20 nt的gRNA编辑效率无显著性差异[44-45].最后,位于Cas9蛋白上的HNH和RuvC结构域发挥核酸内切酶活性,分别切断靶向链和非靶向链,最终使靶DNA形成平末端的双链断裂,实现“打靶”[39,42]. ...
... [39].也有研究发现截短gRNA的间隔区段(如14 nt的间隔区段)能够使RNP保持识别和结合靶序列的能力,但无法引入DSB[43];而当gRNA的间隔区段长度在16~20 nt时均可有效进行基因编辑,且17~20 nt的gRNA编辑效率无显著性差异[44-45].最后,位于Cas9蛋白上的HNH和RuvC结构域发挥核酸内切酶活性,分别切断靶向链和非靶向链,最终使靶DNA形成平末端的双链断裂,实现“打靶”[39,42]. ...
... [39,42]. ...
... 在CRISPR/Cas9的基因转录调控系统中,对Cas9蛋白工程化的dCas9分别在RuvC和HNH结构域发生突变使得RNP复合物仅能发挥靶序列识别功能,而无法切断核苷酸双链[29].nCas9(Cas9 nickase)是仅能使靶DNA发生单链断裂(single-strand break,SSB)的Cas9突变体,其中nCas9(H840A)和nCas9(D10A)分别是Cas9在HNH和RuvC结构域发生突变后的突变体蛋白,从而能够使该结构域失去内切酶活性,使DNA单链断裂,产生“缺口”[22,39].将dCas9或nCas9与各种效应蛋白如转录激活因子或抑制因子、表观遗传修饰因子、碱基编辑器甚至具有特定功能的酶等融合后,研究人员便能够借助CRISPR/Cas9系统对靶序列实现特定生物学功能,如表征转录基因乃至基因组上非编码区的功能、通过将碱基编辑器与nCas9融合能够在HDR活性低的细胞中进行精确高效的单碱基编辑等.因此本节将重点回顾与总结多种多样的效应蛋白为CRISPR/Cas9系统带来的多元化优化和发展,以及赋予该系统的多种新功能. ...
... 将dCas9与各种转录激活因子融合后,则可在gRNA的靶向作用下精确激活目的基因,该系统称为CRISPR激活系统或CRISPRa.最初的CRISPRa系统中dCas9仅融合了单个反式激活因子p65(也被称为RelA,是构成NF-κB转录因子家族的五种成员之一)或VP64 (由单纯疱疹病毒蛋白VP16的四个重复结构域构成),这些早期工作共同构成了最初的可编程dCas9基因激活工具箱,但效果差强人意[23-28,39-44,62,65-66].为了不断提高CRISPRa系统的激活效率,研究人员尝试了用各种方式将更复杂或多拷贝的激活结构域应用于CRISPRa.2015年,Chavez等[67]通过蛋白质理性设计,构建了三重融合激活因子VP64-p65-Rta(VPR)并应用于CRISPRa系统,其中Rta是来源于EB病毒(epstein-barr virus)的反式激活因子.与dCas9-VP64相比,dCas9-VPR使人胚胎肾细胞293(HEK 293T cells)的内源靶基因表达上调了22~320倍,同时证明了该系统也能够在多重基因的激活表达与刺激人诱导多功能干细胞(induced pluripotent stem cells,iPSCs)的神经元分化上发挥作用.同年,张锋研究团队[68]开发的基于CRISPRa的协同激活介导系统(synergistic activation mediator system, SAM)通过修饰sgRNA使其携带了两个MS2发夹二级结构,每个发夹结构都能招募一个MS2-p65-HSF1融合蛋白,其中包括反式激活因子p65、MS2结合蛋白以及热休克蛋白1(heat shock protein 1),最终证明该修饰后的gRNA与dCas9-VP64的协同作用能够使10个靶基因的表达均上调2倍以上.2022年,山东大学候进课题组[69]构建的CRISPR介导的蛋白质标签信号放大系统首次将SPY系统(包括SpyTag和SpyCatcher)和SunTag系统分别引入dCas9介导的CRISPRa和dCpf1(一种丧失核酸酶活性的Cas9同源蛋白)介导的CRISPRi系统并进行骨架长度和转录因子优化后,成功使酿酒酵母内目的基因转录水平分别提高了34.9倍以及受到几乎完全的抑制.其中,SunTag系统是由若干个拷贝的GCN4肽以及作为标签的抗GCN4抗体的单链可变片段scFv组成[95],与dCas9融合的多拷贝GCN4肽通过招募多拷贝的转录因子与GCN4抗体标签的融合蛋白从而进行转录调控,该系统除了与CRISPR系统结合后用于碱基编辑以及基因调控,还在哺乳动物和植物细胞的DNA的甲基化和去甲基化诱导中发挥作用[96-100];而SPY系统作为一种广泛使用的蛋白质组装系统首次在酿酒酵母的转录调控中得到了应用[69].可见在dCas9与转录因子融合的基础上,CRISPRa与CRISPRi系统能够利用融合多重结构域或融合蛋白质骨架系统的方式提高调控效率.而通过利用RNA与蛋白质间的相互作用,将多个适配子融合在gRNA中使其同时含有目的基因靶向序列和转录因子招募序列的方式能够产生与dCas9融合多个转录激活或抑制结构域的策略类似的效果[101].此外,针对不同细胞乃至不同dCas9同源蛋白,CRISPRa与CRISPRi系统均需对不同转录激活或抑制结构域乃至其组合进行优化才能发挥该系统的最大效能. ...
RNA-guided human genome engineering via Cas9
1
2013
... 酿脓链球菌(Streptococcus pyogenes)Cas9(SpCas9)是第一种被应用于哺乳动物细胞基因组编辑的Cas9蛋白[4,39-40],目前仍是CRISPR系统在各应用领域中最常用的Cas9蛋白.在发挥靶序列切割的功能时,SpCas9、CRISPR RNA(crRNA)、反式激活CRISPR RNA(trans-activating crRNA, tracrRNA)结合形成核糖核蛋白复合物(ribonucleoprotein,RNP)共同参与这一过程.在天然CRISPR/Cas9系统中,crRNA作为识别模块,其上的间隔区段能够识别与之互补的特异性靶序列(常见20个核苷酸长度),而靶序列下游必须有三个碱基的PAM序列NGG(N代表A、G、C和T中的任一碱基);tracrRNA和crRNA上的核苷酸链有能互补配对的部分,配对后促使RNP形成以及激活Cas9蛋白[41-42].在目前通用的CRISPR/Cas9系统中,crRNA与tracrRNA被融合成一条sgRNA发挥相同的作用[39].也有研究发现截短gRNA的间隔区段(如14 nt的间隔区段)能够使RNP保持识别和结合靶序列的能力,但无法引入DSB[43];而当gRNA的间隔区段长度在16~20 nt时均可有效进行基因编辑,且17~20 nt的gRNA编辑效率无显著性差异[44-45].最后,位于Cas9蛋白上的HNH和RuvC结构域发挥核酸内切酶活性,分别切断靶向链和非靶向链,最终使靶DNA形成平末端的双链断裂,实现“打靶”[39,42]. ...
CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III
1
2011
... 酿脓链球菌(Streptococcus pyogenes)Cas9(SpCas9)是第一种被应用于哺乳动物细胞基因组编辑的Cas9蛋白[4,39-40],目前仍是CRISPR系统在各应用领域中最常用的Cas9蛋白.在发挥靶序列切割的功能时,SpCas9、CRISPR RNA(crRNA)、反式激活CRISPR RNA(trans-activating crRNA, tracrRNA)结合形成核糖核蛋白复合物(ribonucleoprotein,RNP)共同参与这一过程.在天然CRISPR/Cas9系统中,crRNA作为识别模块,其上的间隔区段能够识别与之互补的特异性靶序列(常见20个核苷酸长度),而靶序列下游必须有三个碱基的PAM序列NGG(N代表A、G、C和T中的任一碱基);tracrRNA和crRNA上的核苷酸链有能互补配对的部分,配对后促使RNP形成以及激活Cas9蛋白[41-42].在目前通用的CRISPR/Cas9系统中,crRNA与tracrRNA被融合成一条sgRNA发挥相同的作用[39].也有研究发现截短gRNA的间隔区段(如14 nt的间隔区段)能够使RNP保持识别和结合靶序列的能力,但无法引入DSB[43];而当gRNA的间隔区段长度在16~20 nt时均可有效进行基因编辑,且17~20 nt的gRNA编辑效率无显著性差异[44-45].最后,位于Cas9蛋白上的HNH和RuvC结构域发挥核酸内切酶活性,分别切断靶向链和非靶向链,最终使靶DNA形成平末端的双链断裂,实现“打靶”[39,42]. ...
Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria
2
2012
... 酿脓链球菌(Streptococcus pyogenes)Cas9(SpCas9)是第一种被应用于哺乳动物细胞基因组编辑的Cas9蛋白[4,39-40],目前仍是CRISPR系统在各应用领域中最常用的Cas9蛋白.在发挥靶序列切割的功能时,SpCas9、CRISPR RNA(crRNA)、反式激活CRISPR RNA(trans-activating crRNA, tracrRNA)结合形成核糖核蛋白复合物(ribonucleoprotein,RNP)共同参与这一过程.在天然CRISPR/Cas9系统中,crRNA作为识别模块,其上的间隔区段能够识别与之互补的特异性靶序列(常见20个核苷酸长度),而靶序列下游必须有三个碱基的PAM序列NGG(N代表A、G、C和T中的任一碱基);tracrRNA和crRNA上的核苷酸链有能互补配对的部分,配对后促使RNP形成以及激活Cas9蛋白[41-42].在目前通用的CRISPR/Cas9系统中,crRNA与tracrRNA被融合成一条sgRNA发挥相同的作用[39].也有研究发现截短gRNA的间隔区段(如14 nt的间隔区段)能够使RNP保持识别和结合靶序列的能力,但无法引入DSB[43];而当gRNA的间隔区段长度在16~20 nt时均可有效进行基因编辑,且17~20 nt的gRNA编辑效率无显著性差异[44-45].最后,位于Cas9蛋白上的HNH和RuvC结构域发挥核酸内切酶活性,分别切断靶向链和非靶向链,最终使靶DNA形成平末端的双链断裂,实现“打靶”[39,42]. ...
... ,42]. ...
A single Cas9-VPR nuclease for simultaneous gene activation, repression, and editing in Saccharomyces cerevisiae
3
2020
... 酿脓链球菌(Streptococcus pyogenes)Cas9(SpCas9)是第一种被应用于哺乳动物细胞基因组编辑的Cas9蛋白[4,39-40],目前仍是CRISPR系统在各应用领域中最常用的Cas9蛋白.在发挥靶序列切割的功能时,SpCas9、CRISPR RNA(crRNA)、反式激活CRISPR RNA(trans-activating crRNA, tracrRNA)结合形成核糖核蛋白复合物(ribonucleoprotein,RNP)共同参与这一过程.在天然CRISPR/Cas9系统中,crRNA作为识别模块,其上的间隔区段能够识别与之互补的特异性靶序列(常见20个核苷酸长度),而靶序列下游必须有三个碱基的PAM序列NGG(N代表A、G、C和T中的任一碱基);tracrRNA和crRNA上的核苷酸链有能互补配对的部分,配对后促使RNP形成以及激活Cas9蛋白[41-42].在目前通用的CRISPR/Cas9系统中,crRNA与tracrRNA被融合成一条sgRNA发挥相同的作用[39].也有研究发现截短gRNA的间隔区段(如14 nt的间隔区段)能够使RNP保持识别和结合靶序列的能力,但无法引入DSB[43];而当gRNA的间隔区段长度在16~20 nt时均可有效进行基因编辑,且17~20 nt的gRNA编辑效率无显著性差异[44-45].最后,位于Cas9蛋白上的HNH和RuvC结构域发挥核酸内切酶活性,分别切断靶向链和非靶向链,最终使靶DNA形成平末端的双链断裂,实现“打靶”[39,42]. ...
... Research on the applications of the multiplexed CRISPR/Cas9 system
Table 3| 宿主细胞 | 多重CRISPR策略 | 靶向目标数量 | 应用/概念验证 | 参考文献 |
|---|
| 哺乳动物细胞 | 2个间隔区通过天然CRISPR序列自我加工 | 2个靶标(1个基因) | 对1个靶基因敲除,效率为1.6% | [4] |
| 酿酒酵母 | Casy4核酸酶加工 | 3个基因 | 同时对3个不同启动子进行激活,使得3个报告基因荧光强度提升了2倍 | [113] |
| 酿酒酵母 | 基于Csy4核酸酶加工的多gRNA快速组装 | 12个靶标(3个基因) | 通过12个gRNA靶向使得3个报告基因荧光强度分别被抑制了92%、81%和95% | [114] |
| 酿酒酵母 | 基于tRNA转录本切割机制的多gRNA表达系统 | 8个基因 | 通过两轮8个基因的敲除获得了30倍的游离脂肪酸产量 | [115] |
| 酿酒酵母 | 多个gRNA表达盒通过pol Ⅲ启动子启动 | 3个基因 | 将适配子与gRNA融合进行多基因调控,获得了不同的紫罗兰素生物合成产物 | [118] |
| 酿酒酵母 | 基于Csy4核酸酶加工的多gRNA快速组装策略及多Cas9正交组合调控 | 3个基因 | 通过组合调控使两个报告基因表达分别被抑制和激活5倍,同时以95%的效率敲除第三个基因 | [119] |
| 酿酒酵母 | 通过质粒表达多个gRNA表达盒以及截短gRNA用于调控 | 3个基因 | 通过对三个基因分别进行编辑、激活和抑制获得了α-檀香烯2.66倍的产量 | [43] |
| 马克思克鲁维酵母(Kluyveromyces marxianus) | 基于tRNA转录本切割机制的多gRNA表达系统 | 6个靶标(4个基因) | 通过对4个基因进行调控,使乙酸乙酯的产量提升了3.8倍 | [120] |
| 大肠杆菌 | 多个gRNA表达盒通过质粒表达 | 3个基因 | 通过对3个基因进行组合调控得到了2.3倍苹果酸产量菌株 | [121] |
| 大肠杆菌 | 多个gRNA表达盒通过质粒表达 | 3个基因 | 对苹果酸合成途径基因进行组合调控后获得了2.3倍苹果酸产量菌株 | [121] |
| 枯草芽孢杆菌(Bacillus subtilis) | 多个gRNA表达盒整合至基因组 | 3个基因 | 通过多基因动态组合调控提高了N-乙酰葡糖胺产量 | [122] |
| 天蓝色链霉菌(Streptomyces coelicolor) | 多个gRNA表达盒与dCas9在质粒上表达 | 4个基因 | 通过对4个靶基因同时进行抑制,使其mRNA表达量降为对照的2%~32% | [123] |
2015年,Zalatan等[118]通过多sgRNA表达盒策略及sgRNA工程将适配体与识别区结合,将配体与转录因子融合表达后同时对3个目标基因进行激活或抑制调控,最终在酿酒酵母中获得了不同的紫罗兰素生物合成途径产物.Lobs等[120]在对一株马克思克鲁维酵母(Kluyveromyces marxianus)中乙酸酯的主要代谢途径和前体物质进行鉴定后,利用CRISPRi系统对ACO2b、SDH2、RIP1和MSS51四个基因进行多重调控,最终获得了乙酸乙酯3.8倍的提升.2017年,Lian等[119]在表征了多种同源Cas9蛋白的基因敲除、转录激活和转录抑制效率后,优化并组合三种同源蛋白最终构建了一套完全正交的三功能CRISPR代谢调控系统.由于正交的Cas9同源蛋白所识别的PAM序列不同,发挥不同功能的同源Cas9之间互不干扰,使得两个被调控报告基因的表达强度均被激活和抑制了5倍,并使第三个报告基因的敲除效率达到了95%.该系统通过一步组合调控使得改造的酿酒酵母中胡萝卜素产量提升了3倍,同时使表面葡聚糖酶展示量提高了2.5倍.原核生物具有相对较低的代谢网络复杂性,同时其无核膜包被的拟核能够实现转录与翻译同时进行,这使得原核生物(如大肠杆菌)成为多基因操作及表达调控的模式生物[122-123].2018年,Gao等[121]通过多重CRISPRi系统对苹果酸生物合成途径中的基因进行组合调控后得到了2.3倍的苹果酸产量菌株. ...
... 此外,通过修饰gRNA如使其融合适配子或改变gRNA识别区长度,就可能实现CRISPR系统的多重编辑或调控.2020年,Fan等[124]通过研究证实了不同长度的gRNA-DNA互补区会对HNH和RuvC结构域切割靶向链和非靶向链的能力产生影响,并基于此开发了一种使两个靶基因分别产生DSB和C-T碱基转换的双功能编辑系统.同年,浙江大学徐志南组[43]通过gRNA工程构建了一种能同时进行基因激活、抑制和编辑的三功能CRISPR系统(CRISPR-ARE).该系统在仅使用Cas9-VPR一种CRISPR融合蛋白的条件下,利用截短的14bp gRNA实现靶基因的激活和抑制,并利用正常的20bp gRNA实现目标基因敲除.尽管目前的gRNA多表达策略已逐渐成熟,对于gRNA在细胞内的多路复用机制及其对Cas9竞争作用的了解仍有待加强.多gRNA策略(包括Csy4、核酶和tRNA等)的处理效率将在确定理论上的gRNA数量上限方面发挥关键作用,在未来的研究中,创建和处理长阵列的策略将会得到优化和加强. ...
Improving CRISPR-Cas nuclease specificity using truncated guide RNAs
3
2014
... 酿脓链球菌(Streptococcus pyogenes)Cas9(SpCas9)是第一种被应用于哺乳动物细胞基因组编辑的Cas9蛋白[4,39-40],目前仍是CRISPR系统在各应用领域中最常用的Cas9蛋白.在发挥靶序列切割的功能时,SpCas9、CRISPR RNA(crRNA)、反式激活CRISPR RNA(trans-activating crRNA, tracrRNA)结合形成核糖核蛋白复合物(ribonucleoprotein,RNP)共同参与这一过程.在天然CRISPR/Cas9系统中,crRNA作为识别模块,其上的间隔区段能够识别与之互补的特异性靶序列(常见20个核苷酸长度),而靶序列下游必须有三个碱基的PAM序列NGG(N代表A、G、C和T中的任一碱基);tracrRNA和crRNA上的核苷酸链有能互补配对的部分,配对后促使RNP形成以及激活Cas9蛋白[41-42].在目前通用的CRISPR/Cas9系统中,crRNA与tracrRNA被融合成一条sgRNA发挥相同的作用[39].也有研究发现截短gRNA的间隔区段(如14 nt的间隔区段)能够使RNP保持识别和结合靶序列的能力,但无法引入DSB[43];而当gRNA的间隔区段长度在16~20 nt时均可有效进行基因编辑,且17~20 nt的gRNA编辑效率无显著性差异[44-45].最后,位于Cas9蛋白上的HNH和RuvC结构域发挥核酸内切酶活性,分别切断靶向链和非靶向链,最终使靶DNA形成平末端的双链断裂,实现“打靶”[39,42]. ...
... 尽管CRISPR/Cas9系统已在多种生物和细胞的基因组上得到了应用[46-51],Cas9对于PAM序列NGG的严格识别、复杂基因组中gRNA潜在的脱靶效应以及该系统作为医疗手段使用时的保真性仍大大限制其在更广范围下的应用与开发.如在人类基因组DNA上平均每8个碱基对存在一个NGG位点[52],Fu等[53]在人类细胞中表征了CRISPR/Cas9系统的脱靶效应,结果显示该系统的脱靶率高达66%.因此,研究人员通过工程化Cas9蛋白或gRNA等方式,使CRISPR/Cas9系统的靶向范围、靶向特异性和保真性均得到了拓展和提高[19-20].2018年,张锋团队和Osamu Nureki团队[31]通过对SpCas9进行理性设计获得了能够识别NG PAM序列的突变体蛋白SpCas9-NG,并证明了其在人类细胞中的基因组编辑能力以及与胞苷脱氨酶融合后仍具有单碱基编辑能力.2020年,David R. Liu团队[54]利用噬菌体辅助持续进化(phage-assisted continuous evolution,PACE)方法获得了三种识别非G PAM序列的SpCas9突变体,从而进一步提高了CRISPR/Cas9系统的靶向范围.Walton等[32]通过改造与PAM序列直接作用的氨基酸位点获得了能稳定识别NRN PAM序列的突变体蛋白SpRY,同时其能以较低效率识别NYN PAM序列,使其在靶点识别上几乎不受PAM限制.Hu等[20]应用PACE方法获得一种能够识别NG、GAA及GAT等PAM序列的SpCas9突变体xCas9,同时其能以优于SpCas9的特异性靶向NGG,且对各种PAM的靶向能力均在人类细胞中的基因编辑、转录调控以及胞嘧啶与腺嘌呤核苷酸的碱基修饰中得到了应用.2014年,Guilinger等[34]将FokⅠ核酸酶与核酸酶活性失活的Cas9蛋白(catalytically inactive Cas9,dCas9)融合后得到了一种融合蛋白fCas9,只有两个fCas9单体同时结合在靶基因附近时才能切割目的基因,使其在人类细胞中进行基因编辑的特异性比野生型Cas9提高了140倍.多种研究已报道了将SpCas9或sgRNA工程化后,通过质粒载体携带并表达CRISPR/Cas9系统或通过RNP递送的方式均能够在基因组范围上降低CRISPR/Cas9系统的脱靶效应,并显著提高该系统的保真性[33,55-58].2018年,Casini等[33]筛选并表征了能够提高CRISPR/Cas9系统编辑效率的Cas9 REC3结构域突变库并组合了四种有利突变后,获得了一种保真性比野生型SpCas9提高了79倍的突变体蛋白evoCas9,同时编辑效率接近野生型SpCas9.除了对Cas9蛋白进行优化,对gRNA不断进行工程化改造也能使CRISPR/Cas9系统的性能得到显著提升.2014年,Fu等[44]证明了间隔区截短后的gRNA的脱靶率明显低于全长的gRNA(20 nt),最大降低幅度达5000倍.2019年,Kocak等[21]在工程化设计的sgRNA上的间隔区加入一“发夹”形RNA二级结构,该结构能够在发生脱靶编辑时在脱靶位点处提高gRNA或crRNA链入侵的热力学屏障而并不影响中靶效率,使CRISPR系统的特异性增加了两个数量级.2020年,Rose等[59]基于过短的gRNA(间隔区小于16 nt)与Cas9形成的RNP只能结合却不能切割靶位点特性设计的失活RNA(dead-RNAs,dRNAs)可保护多个脱靶位点,从而使得SpCas9的靶向特异性提高了40倍.相较于工程化改造的高特异性Cas9突变体来说,通过缩短gRNA的间隔区来提高靶向特异性的方法在设计和应用方面均更易用和灵活,预计将成为未来的研究热点方向. ...
... 将dCas9与各种转录激活因子融合后,则可在gRNA的靶向作用下精确激活目的基因,该系统称为CRISPR激活系统或CRISPRa.最初的CRISPRa系统中dCas9仅融合了单个反式激活因子p65(也被称为RelA,是构成NF-κB转录因子家族的五种成员之一)或VP64 (由单纯疱疹病毒蛋白VP16的四个重复结构域构成),这些早期工作共同构成了最初的可编程dCas9基因激活工具箱,但效果差强人意[23-28,39-44,62,65-66].为了不断提高CRISPRa系统的激活效率,研究人员尝试了用各种方式将更复杂或多拷贝的激活结构域应用于CRISPRa.2015年,Chavez等[67]通过蛋白质理性设计,构建了三重融合激活因子VP64-p65-Rta(VPR)并应用于CRISPRa系统,其中Rta是来源于EB病毒(epstein-barr virus)的反式激活因子.与dCas9-VP64相比,dCas9-VPR使人胚胎肾细胞293(HEK 293T cells)的内源靶基因表达上调了22~320倍,同时证明了该系统也能够在多重基因的激活表达与刺激人诱导多功能干细胞(induced pluripotent stem cells,iPSCs)的神经元分化上发挥作用.同年,张锋研究团队[68]开发的基于CRISPRa的协同激活介导系统(synergistic activation mediator system, SAM)通过修饰sgRNA使其携带了两个MS2发夹二级结构,每个发夹结构都能招募一个MS2-p65-HSF1融合蛋白,其中包括反式激活因子p65、MS2结合蛋白以及热休克蛋白1(heat shock protein 1),最终证明该修饰后的gRNA与dCas9-VP64的协同作用能够使10个靶基因的表达均上调2倍以上.2022年,山东大学候进课题组[69]构建的CRISPR介导的蛋白质标签信号放大系统首次将SPY系统(包括SpyTag和SpyCatcher)和SunTag系统分别引入dCas9介导的CRISPRa和dCpf1(一种丧失核酸酶活性的Cas9同源蛋白)介导的CRISPRi系统并进行骨架长度和转录因子优化后,成功使酿酒酵母内目的基因转录水平分别提高了34.9倍以及受到几乎完全的抑制.其中,SunTag系统是由若干个拷贝的GCN4肽以及作为标签的抗GCN4抗体的单链可变片段scFv组成[95],与dCas9融合的多拷贝GCN4肽通过招募多拷贝的转录因子与GCN4抗体标签的融合蛋白从而进行转录调控,该系统除了与CRISPR系统结合后用于碱基编辑以及基因调控,还在哺乳动物和植物细胞的DNA的甲基化和去甲基化诱导中发挥作用[96-100];而SPY系统作为一种广泛使用的蛋白质组装系统首次在酿酒酵母的转录调控中得到了应用[69].可见在dCas9与转录因子融合的基础上,CRISPRa与CRISPRi系统能够利用融合多重结构域或融合蛋白质骨架系统的方式提高调控效率.而通过利用RNA与蛋白质间的相互作用,将多个适配子融合在gRNA中使其同时含有目的基因靶向序列和转录因子招募序列的方式能够产生与dCas9融合多个转录激活或抑制结构域的策略类似的效果[101].此外,针对不同细胞乃至不同dCas9同源蛋白,CRISPRa与CRISPRi系统均需对不同转录激活或抑制结构域乃至其组合进行优化才能发挥该系统的最大效能. ...
Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases
1
2014
... 酿脓链球菌(Streptococcus pyogenes)Cas9(SpCas9)是第一种被应用于哺乳动物细胞基因组编辑的Cas9蛋白[4,39-40],目前仍是CRISPR系统在各应用领域中最常用的Cas9蛋白.在发挥靶序列切割的功能时,SpCas9、CRISPR RNA(crRNA)、反式激活CRISPR RNA(trans-activating crRNA, tracrRNA)结合形成核糖核蛋白复合物(ribonucleoprotein,RNP)共同参与这一过程.在天然CRISPR/Cas9系统中,crRNA作为识别模块,其上的间隔区段能够识别与之互补的特异性靶序列(常见20个核苷酸长度),而靶序列下游必须有三个碱基的PAM序列NGG(N代表A、G、C和T中的任一碱基);tracrRNA和crRNA上的核苷酸链有能互补配对的部分,配对后促使RNP形成以及激活Cas9蛋白[41-42].在目前通用的CRISPR/Cas9系统中,crRNA与tracrRNA被融合成一条sgRNA发挥相同的作用[39].也有研究发现截短gRNA的间隔区段(如14 nt的间隔区段)能够使RNP保持识别和结合靶序列的能力,但无法引入DSB[43];而当gRNA的间隔区段长度在16~20 nt时均可有效进行基因编辑,且17~20 nt的gRNA编辑效率无显著性差异[44-45].最后,位于Cas9蛋白上的HNH和RuvC结构域发挥核酸内切酶活性,分别切断靶向链和非靶向链,最终使靶DNA形成平末端的双链断裂,实现“打靶”[39,42]. ...
A CRISPR-Cas9 system for genetic engineering of filamentous fungi
1
2015
... 尽管CRISPR/Cas9系统已在多种生物和细胞的基因组上得到了应用[46-51],Cas9对于PAM序列NGG的严格识别、复杂基因组中gRNA潜在的脱靶效应以及该系统作为医疗手段使用时的保真性仍大大限制其在更广范围下的应用与开发.如在人类基因组DNA上平均每8个碱基对存在一个NGG位点[52],Fu等[53]在人类细胞中表征了CRISPR/Cas9系统的脱靶效应,结果显示该系统的脱靶率高达66%.因此,研究人员通过工程化Cas9蛋白或gRNA等方式,使CRISPR/Cas9系统的靶向范围、靶向特异性和保真性均得到了拓展和提高[19-20].2018年,张锋团队和Osamu Nureki团队[31]通过对SpCas9进行理性设计获得了能够识别NG PAM序列的突变体蛋白SpCas9-NG,并证明了其在人类细胞中的基因组编辑能力以及与胞苷脱氨酶融合后仍具有单碱基编辑能力.2020年,David R. Liu团队[54]利用噬菌体辅助持续进化(phage-assisted continuous evolution,PACE)方法获得了三种识别非G PAM序列的SpCas9突变体,从而进一步提高了CRISPR/Cas9系统的靶向范围.Walton等[32]通过改造与PAM序列直接作用的氨基酸位点获得了能稳定识别NRN PAM序列的突变体蛋白SpRY,同时其能以较低效率识别NYN PAM序列,使其在靶点识别上几乎不受PAM限制.Hu等[20]应用PACE方法获得一种能够识别NG、GAA及GAT等PAM序列的SpCas9突变体xCas9,同时其能以优于SpCas9的特异性靶向NGG,且对各种PAM的靶向能力均在人类细胞中的基因编辑、转录调控以及胞嘧啶与腺嘌呤核苷酸的碱基修饰中得到了应用.2014年,Guilinger等[34]将FokⅠ核酸酶与核酸酶活性失活的Cas9蛋白(catalytically inactive Cas9,dCas9)融合后得到了一种融合蛋白fCas9,只有两个fCas9单体同时结合在靶基因附近时才能切割目的基因,使其在人类细胞中进行基因编辑的特异性比野生型Cas9提高了140倍.多种研究已报道了将SpCas9或sgRNA工程化后,通过质粒载体携带并表达CRISPR/Cas9系统或通过RNP递送的方式均能够在基因组范围上降低CRISPR/Cas9系统的脱靶效应,并显著提高该系统的保真性[33,55-58].2018年,Casini等[33]筛选并表征了能够提高CRISPR/Cas9系统编辑效率的Cas9 REC3结构域突变库并组合了四种有利突变后,获得了一种保真性比野生型SpCas9提高了79倍的突变体蛋白evoCas9,同时编辑效率接近野生型SpCas9.除了对Cas9蛋白进行优化,对gRNA不断进行工程化改造也能使CRISPR/Cas9系统的性能得到显著提升.2014年,Fu等[44]证明了间隔区截短后的gRNA的脱靶率明显低于全长的gRNA(20 nt),最大降低幅度达5000倍.2019年,Kocak等[21]在工程化设计的sgRNA上的间隔区加入一“发夹”形RNA二级结构,该结构能够在发生脱靶编辑时在脱靶位点处提高gRNA或crRNA链入侵的热力学屏障而并不影响中靶效率,使CRISPR系统的特异性增加了两个数量级.2020年,Rose等[59]基于过短的gRNA(间隔区小于16 nt)与Cas9形成的RNP只能结合却不能切割靶位点特性设计的失活RNA(dead-RNAs,dRNAs)可保护多个脱靶位点,从而使得SpCas9的靶向特异性提高了40倍.相较于工程化改造的高特异性Cas9突变体来说,通过缩短gRNA的间隔区来提高靶向特异性的方法在设计和应用方面均更易用和灵活,预计将成为未来的研究热点方向. ...
CRISPR technologies for bacterial systems: current achievements and future directions
0
2016
Heritable genome editing in C. elegans via a CRISPR-Cas9 system
0
2013
Generation of gene-modified mice via Cas9/RNA-mediated gene targeting
0
2013
Efficient genome editing in zebrafish using a CRISPR-Cas system
0
2013
Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice
1
2013
... 尽管CRISPR/Cas9系统已在多种生物和细胞的基因组上得到了应用[46-51],Cas9对于PAM序列NGG的严格识别、复杂基因组中gRNA潜在的脱靶效应以及该系统作为医疗手段使用时的保真性仍大大限制其在更广范围下的应用与开发.如在人类基因组DNA上平均每8个碱基对存在一个NGG位点[52],Fu等[53]在人类细胞中表征了CRISPR/Cas9系统的脱靶效应,结果显示该系统的脱靶率高达66%.因此,研究人员通过工程化Cas9蛋白或gRNA等方式,使CRISPR/Cas9系统的靶向范围、靶向特异性和保真性均得到了拓展和提高[19-20].2018年,张锋团队和Osamu Nureki团队[31]通过对SpCas9进行理性设计获得了能够识别NG PAM序列的突变体蛋白SpCas9-NG,并证明了其在人类细胞中的基因组编辑能力以及与胞苷脱氨酶融合后仍具有单碱基编辑能力.2020年,David R. Liu团队[54]利用噬菌体辅助持续进化(phage-assisted continuous evolution,PACE)方法获得了三种识别非G PAM序列的SpCas9突变体,从而进一步提高了CRISPR/Cas9系统的靶向范围.Walton等[32]通过改造与PAM序列直接作用的氨基酸位点获得了能稳定识别NRN PAM序列的突变体蛋白SpRY,同时其能以较低效率识别NYN PAM序列,使其在靶点识别上几乎不受PAM限制.Hu等[20]应用PACE方法获得一种能够识别NG、GAA及GAT等PAM序列的SpCas9突变体xCas9,同时其能以优于SpCas9的特异性靶向NGG,且对各种PAM的靶向能力均在人类细胞中的基因编辑、转录调控以及胞嘧啶与腺嘌呤核苷酸的碱基修饰中得到了应用.2014年,Guilinger等[34]将FokⅠ核酸酶与核酸酶活性失活的Cas9蛋白(catalytically inactive Cas9,dCas9)融合后得到了一种融合蛋白fCas9,只有两个fCas9单体同时结合在靶基因附近时才能切割目的基因,使其在人类细胞中进行基因编辑的特异性比野生型Cas9提高了140倍.多种研究已报道了将SpCas9或sgRNA工程化后,通过质粒载体携带并表达CRISPR/Cas9系统或通过RNP递送的方式均能够在基因组范围上降低CRISPR/Cas9系统的脱靶效应,并显著提高该系统的保真性[33,55-58].2018年,Casini等[33]筛选并表征了能够提高CRISPR/Cas9系统编辑效率的Cas9 REC3结构域突变库并组合了四种有利突变后,获得了一种保真性比野生型SpCas9提高了79倍的突变体蛋白evoCas9,同时编辑效率接近野生型SpCas9.除了对Cas9蛋白进行优化,对gRNA不断进行工程化改造也能使CRISPR/Cas9系统的性能得到显著提升.2014年,Fu等[44]证明了间隔区截短后的gRNA的脱靶率明显低于全长的gRNA(20 nt),最大降低幅度达5000倍.2019年,Kocak等[21]在工程化设计的sgRNA上的间隔区加入一“发夹”形RNA二级结构,该结构能够在发生脱靶编辑时在脱靶位点处提高gRNA或crRNA链入侵的热力学屏障而并不影响中靶效率,使CRISPR系统的特异性增加了两个数量级.2020年,Rose等[59]基于过短的gRNA(间隔区小于16 nt)与Cas9形成的RNP只能结合却不能切割靶位点特性设计的失活RNA(dead-RNAs,dRNAs)可保护多个脱靶位点,从而使得SpCas9的靶向特异性提高了40倍.相较于工程化改造的高特异性Cas9突变体来说,通过缩短gRNA的间隔区来提高靶向特异性的方法在设计和应用方面均更易用和灵活,预计将成为未来的研究热点方向. ...
Development and applications of CRISPR-Cas9 for genome engineering
1
2014
... 尽管CRISPR/Cas9系统已在多种生物和细胞的基因组上得到了应用[46-51],Cas9对于PAM序列NGG的严格识别、复杂基因组中gRNA潜在的脱靶效应以及该系统作为医疗手段使用时的保真性仍大大限制其在更广范围下的应用与开发.如在人类基因组DNA上平均每8个碱基对存在一个NGG位点[52],Fu等[53]在人类细胞中表征了CRISPR/Cas9系统的脱靶效应,结果显示该系统的脱靶率高达66%.因此,研究人员通过工程化Cas9蛋白或gRNA等方式,使CRISPR/Cas9系统的靶向范围、靶向特异性和保真性均得到了拓展和提高[19-20].2018年,张锋团队和Osamu Nureki团队[31]通过对SpCas9进行理性设计获得了能够识别NG PAM序列的突变体蛋白SpCas9-NG,并证明了其在人类细胞中的基因组编辑能力以及与胞苷脱氨酶融合后仍具有单碱基编辑能力.2020年,David R. Liu团队[54]利用噬菌体辅助持续进化(phage-assisted continuous evolution,PACE)方法获得了三种识别非G PAM序列的SpCas9突变体,从而进一步提高了CRISPR/Cas9系统的靶向范围.Walton等[32]通过改造与PAM序列直接作用的氨基酸位点获得了能稳定识别NRN PAM序列的突变体蛋白SpRY,同时其能以较低效率识别NYN PAM序列,使其在靶点识别上几乎不受PAM限制.Hu等[20]应用PACE方法获得一种能够识别NG、GAA及GAT等PAM序列的SpCas9突变体xCas9,同时其能以优于SpCas9的特异性靶向NGG,且对各种PAM的靶向能力均在人类细胞中的基因编辑、转录调控以及胞嘧啶与腺嘌呤核苷酸的碱基修饰中得到了应用.2014年,Guilinger等[34]将FokⅠ核酸酶与核酸酶活性失活的Cas9蛋白(catalytically inactive Cas9,dCas9)融合后得到了一种融合蛋白fCas9,只有两个fCas9单体同时结合在靶基因附近时才能切割目的基因,使其在人类细胞中进行基因编辑的特异性比野生型Cas9提高了140倍.多种研究已报道了将SpCas9或sgRNA工程化后,通过质粒载体携带并表达CRISPR/Cas9系统或通过RNP递送的方式均能够在基因组范围上降低CRISPR/Cas9系统的脱靶效应,并显著提高该系统的保真性[33,55-58].2018年,Casini等[33]筛选并表征了能够提高CRISPR/Cas9系统编辑效率的Cas9 REC3结构域突变库并组合了四种有利突变后,获得了一种保真性比野生型SpCas9提高了79倍的突变体蛋白evoCas9,同时编辑效率接近野生型SpCas9.除了对Cas9蛋白进行优化,对gRNA不断进行工程化改造也能使CRISPR/Cas9系统的性能得到显著提升.2014年,Fu等[44]证明了间隔区截短后的gRNA的脱靶率明显低于全长的gRNA(20 nt),最大降低幅度达5000倍.2019年,Kocak等[21]在工程化设计的sgRNA上的间隔区加入一“发夹”形RNA二级结构,该结构能够在发生脱靶编辑时在脱靶位点处提高gRNA或crRNA链入侵的热力学屏障而并不影响中靶效率,使CRISPR系统的特异性增加了两个数量级.2020年,Rose等[59]基于过短的gRNA(间隔区小于16 nt)与Cas9形成的RNP只能结合却不能切割靶位点特性设计的失活RNA(dead-RNAs,dRNAs)可保护多个脱靶位点,从而使得SpCas9的靶向特异性提高了40倍.相较于工程化改造的高特异性Cas9突变体来说,通过缩短gRNA的间隔区来提高靶向特异性的方法在设计和应用方面均更易用和灵活,预计将成为未来的研究热点方向. ...
High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells
1
2013
... 尽管CRISPR/Cas9系统已在多种生物和细胞的基因组上得到了应用[46-51],Cas9对于PAM序列NGG的严格识别、复杂基因组中gRNA潜在的脱靶效应以及该系统作为医疗手段使用时的保真性仍大大限制其在更广范围下的应用与开发.如在人类基因组DNA上平均每8个碱基对存在一个NGG位点[52],Fu等[53]在人类细胞中表征了CRISPR/Cas9系统的脱靶效应,结果显示该系统的脱靶率高达66%.因此,研究人员通过工程化Cas9蛋白或gRNA等方式,使CRISPR/Cas9系统的靶向范围、靶向特异性和保真性均得到了拓展和提高[19-20].2018年,张锋团队和Osamu Nureki团队[31]通过对SpCas9进行理性设计获得了能够识别NG PAM序列的突变体蛋白SpCas9-NG,并证明了其在人类细胞中的基因组编辑能力以及与胞苷脱氨酶融合后仍具有单碱基编辑能力.2020年,David R. Liu团队[54]利用噬菌体辅助持续进化(phage-assisted continuous evolution,PACE)方法获得了三种识别非G PAM序列的SpCas9突变体,从而进一步提高了CRISPR/Cas9系统的靶向范围.Walton等[32]通过改造与PAM序列直接作用的氨基酸位点获得了能稳定识别NRN PAM序列的突变体蛋白SpRY,同时其能以较低效率识别NYN PAM序列,使其在靶点识别上几乎不受PAM限制.Hu等[20]应用PACE方法获得一种能够识别NG、GAA及GAT等PAM序列的SpCas9突变体xCas9,同时其能以优于SpCas9的特异性靶向NGG,且对各种PAM的靶向能力均在人类细胞中的基因编辑、转录调控以及胞嘧啶与腺嘌呤核苷酸的碱基修饰中得到了应用.2014年,Guilinger等[34]将FokⅠ核酸酶与核酸酶活性失活的Cas9蛋白(catalytically inactive Cas9,dCas9)融合后得到了一种融合蛋白fCas9,只有两个fCas9单体同时结合在靶基因附近时才能切割目的基因,使其在人类细胞中进行基因编辑的特异性比野生型Cas9提高了140倍.多种研究已报道了将SpCas9或sgRNA工程化后,通过质粒载体携带并表达CRISPR/Cas9系统或通过RNP递送的方式均能够在基因组范围上降低CRISPR/Cas9系统的脱靶效应,并显著提高该系统的保真性[33,55-58].2018年,Casini等[33]筛选并表征了能够提高CRISPR/Cas9系统编辑效率的Cas9 REC3结构域突变库并组合了四种有利突变后,获得了一种保真性比野生型SpCas9提高了79倍的突变体蛋白evoCas9,同时编辑效率接近野生型SpCas9.除了对Cas9蛋白进行优化,对gRNA不断进行工程化改造也能使CRISPR/Cas9系统的性能得到显著提升.2014年,Fu等[44]证明了间隔区截短后的gRNA的脱靶率明显低于全长的gRNA(20 nt),最大降低幅度达5000倍.2019年,Kocak等[21]在工程化设计的sgRNA上的间隔区加入一“发夹”形RNA二级结构,该结构能够在发生脱靶编辑时在脱靶位点处提高gRNA或crRNA链入侵的热力学屏障而并不影响中靶效率,使CRISPR系统的特异性增加了两个数量级.2020年,Rose等[59]基于过短的gRNA(间隔区小于16 nt)与Cas9形成的RNP只能结合却不能切割靶位点特性设计的失活RNA(dead-RNAs,dRNAs)可保护多个脱靶位点,从而使得SpCas9的靶向特异性提高了40倍.相较于工程化改造的高特异性Cas9突变体来说,通过缩短gRNA的间隔区来提高靶向特异性的方法在设计和应用方面均更易用和灵活,预计将成为未来的研究热点方向. ...
Continuous evolution of SpCas9 variants compatible with non-G PAMs
1
2020
... 尽管CRISPR/Cas9系统已在多种生物和细胞的基因组上得到了应用[46-51],Cas9对于PAM序列NGG的严格识别、复杂基因组中gRNA潜在的脱靶效应以及该系统作为医疗手段使用时的保真性仍大大限制其在更广范围下的应用与开发.如在人类基因组DNA上平均每8个碱基对存在一个NGG位点[52],Fu等[53]在人类细胞中表征了CRISPR/Cas9系统的脱靶效应,结果显示该系统的脱靶率高达66%.因此,研究人员通过工程化Cas9蛋白或gRNA等方式,使CRISPR/Cas9系统的靶向范围、靶向特异性和保真性均得到了拓展和提高[19-20].2018年,张锋团队和Osamu Nureki团队[31]通过对SpCas9进行理性设计获得了能够识别NG PAM序列的突变体蛋白SpCas9-NG,并证明了其在人类细胞中的基因组编辑能力以及与胞苷脱氨酶融合后仍具有单碱基编辑能力.2020年,David R. Liu团队[54]利用噬菌体辅助持续进化(phage-assisted continuous evolution,PACE)方法获得了三种识别非G PAM序列的SpCas9突变体,从而进一步提高了CRISPR/Cas9系统的靶向范围.Walton等[32]通过改造与PAM序列直接作用的氨基酸位点获得了能稳定识别NRN PAM序列的突变体蛋白SpRY,同时其能以较低效率识别NYN PAM序列,使其在靶点识别上几乎不受PAM限制.Hu等[20]应用PACE方法获得一种能够识别NG、GAA及GAT等PAM序列的SpCas9突变体xCas9,同时其能以优于SpCas9的特异性靶向NGG,且对各种PAM的靶向能力均在人类细胞中的基因编辑、转录调控以及胞嘧啶与腺嘌呤核苷酸的碱基修饰中得到了应用.2014年,Guilinger等[34]将FokⅠ核酸酶与核酸酶活性失活的Cas9蛋白(catalytically inactive Cas9,dCas9)融合后得到了一种融合蛋白fCas9,只有两个fCas9单体同时结合在靶基因附近时才能切割目的基因,使其在人类细胞中进行基因编辑的特异性比野生型Cas9提高了140倍.多种研究已报道了将SpCas9或sgRNA工程化后,通过质粒载体携带并表达CRISPR/Cas9系统或通过RNP递送的方式均能够在基因组范围上降低CRISPR/Cas9系统的脱靶效应,并显著提高该系统的保真性[33,55-58].2018年,Casini等[33]筛选并表征了能够提高CRISPR/Cas9系统编辑效率的Cas9 REC3结构域突变库并组合了四种有利突变后,获得了一种保真性比野生型SpCas9提高了79倍的突变体蛋白evoCas9,同时编辑效率接近野生型SpCas9.除了对Cas9蛋白进行优化,对gRNA不断进行工程化改造也能使CRISPR/Cas9系统的性能得到显著提升.2014年,Fu等[44]证明了间隔区截短后的gRNA的脱靶率明显低于全长的gRNA(20 nt),最大降低幅度达5000倍.2019年,Kocak等[21]在工程化设计的sgRNA上的间隔区加入一“发夹”形RNA二级结构,该结构能够在发生脱靶编辑时在脱靶位点处提高gRNA或crRNA链入侵的热力学屏障而并不影响中靶效率,使CRISPR系统的特异性增加了两个数量级.2020年,Rose等[59]基于过短的gRNA(间隔区小于16 nt)与Cas9形成的RNP只能结合却不能切割靶位点特性设计的失活RNA(dead-RNAs,dRNAs)可保护多个脱靶位点,从而使得SpCas9的靶向特异性提高了40倍.相较于工程化改造的高特异性Cas9突变体来说,通过缩短gRNA的间隔区来提高靶向特异性的方法在设计和应用方面均更易用和灵活,预计将成为未来的研究热点方向. ...
Rationally engineered Cas9 nucleases with improved specificity
1
2016
... 尽管CRISPR/Cas9系统已在多种生物和细胞的基因组上得到了应用[46-51],Cas9对于PAM序列NGG的严格识别、复杂基因组中gRNA潜在的脱靶效应以及该系统作为医疗手段使用时的保真性仍大大限制其在更广范围下的应用与开发.如在人类基因组DNA上平均每8个碱基对存在一个NGG位点[52],Fu等[53]在人类细胞中表征了CRISPR/Cas9系统的脱靶效应,结果显示该系统的脱靶率高达66%.因此,研究人员通过工程化Cas9蛋白或gRNA等方式,使CRISPR/Cas9系统的靶向范围、靶向特异性和保真性均得到了拓展和提高[19-20].2018年,张锋团队和Osamu Nureki团队[31]通过对SpCas9进行理性设计获得了能够识别NG PAM序列的突变体蛋白SpCas9-NG,并证明了其在人类细胞中的基因组编辑能力以及与胞苷脱氨酶融合后仍具有单碱基编辑能力.2020年,David R. Liu团队[54]利用噬菌体辅助持续进化(phage-assisted continuous evolution,PACE)方法获得了三种识别非G PAM序列的SpCas9突变体,从而进一步提高了CRISPR/Cas9系统的靶向范围.Walton等[32]通过改造与PAM序列直接作用的氨基酸位点获得了能稳定识别NRN PAM序列的突变体蛋白SpRY,同时其能以较低效率识别NYN PAM序列,使其在靶点识别上几乎不受PAM限制.Hu等[20]应用PACE方法获得一种能够识别NG、GAA及GAT等PAM序列的SpCas9突变体xCas9,同时其能以优于SpCas9的特异性靶向NGG,且对各种PAM的靶向能力均在人类细胞中的基因编辑、转录调控以及胞嘧啶与腺嘌呤核苷酸的碱基修饰中得到了应用.2014年,Guilinger等[34]将FokⅠ核酸酶与核酸酶活性失活的Cas9蛋白(catalytically inactive Cas9,dCas9)融合后得到了一种融合蛋白fCas9,只有两个fCas9单体同时结合在靶基因附近时才能切割目的基因,使其在人类细胞中进行基因编辑的特异性比野生型Cas9提高了140倍.多种研究已报道了将SpCas9或sgRNA工程化后,通过质粒载体携带并表达CRISPR/Cas9系统或通过RNP递送的方式均能够在基因组范围上降低CRISPR/Cas9系统的脱靶效应,并显著提高该系统的保真性[33,55-58].2018年,Casini等[33]筛选并表征了能够提高CRISPR/Cas9系统编辑效率的Cas9 REC3结构域突变库并组合了四种有利突变后,获得了一种保真性比野生型SpCas9提高了79倍的突变体蛋白evoCas9,同时编辑效率接近野生型SpCas9.除了对Cas9蛋白进行优化,对gRNA不断进行工程化改造也能使CRISPR/Cas9系统的性能得到显著提升.2014年,Fu等[44]证明了间隔区截短后的gRNA的脱靶率明显低于全长的gRNA(20 nt),最大降低幅度达5000倍.2019年,Kocak等[21]在工程化设计的sgRNA上的间隔区加入一“发夹”形RNA二级结构,该结构能够在发生脱靶编辑时在脱靶位点处提高gRNA或crRNA链入侵的热力学屏障而并不影响中靶效率,使CRISPR系统的特异性增加了两个数量级.2020年,Rose等[59]基于过短的gRNA(间隔区小于16 nt)与Cas9形成的RNP只能结合却不能切割靶位点特性设计的失活RNA(dead-RNAs,dRNAs)可保护多个脱靶位点,从而使得SpCas9的靶向特异性提高了40倍.相较于工程化改造的高特异性Cas9突变体来说,通过缩短gRNA的间隔区来提高靶向特异性的方法在设计和应用方面均更易用和灵活,预计将成为未来的研究热点方向. ...
High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects
0
2016
Enhanced proofreading governs CRISPR-Cas9 targeting accuracy
0
2017
A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells
1
2018
... 尽管CRISPR/Cas9系统已在多种生物和细胞的基因组上得到了应用[46-51],Cas9对于PAM序列NGG的严格识别、复杂基因组中gRNA潜在的脱靶效应以及该系统作为医疗手段使用时的保真性仍大大限制其在更广范围下的应用与开发.如在人类基因组DNA上平均每8个碱基对存在一个NGG位点[52],Fu等[53]在人类细胞中表征了CRISPR/Cas9系统的脱靶效应,结果显示该系统的脱靶率高达66%.因此,研究人员通过工程化Cas9蛋白或gRNA等方式,使CRISPR/Cas9系统的靶向范围、靶向特异性和保真性均得到了拓展和提高[19-20].2018年,张锋团队和Osamu Nureki团队[31]通过对SpCas9进行理性设计获得了能够识别NG PAM序列的突变体蛋白SpCas9-NG,并证明了其在人类细胞中的基因组编辑能力以及与胞苷脱氨酶融合后仍具有单碱基编辑能力.2020年,David R. Liu团队[54]利用噬菌体辅助持续进化(phage-assisted continuous evolution,PACE)方法获得了三种识别非G PAM序列的SpCas9突变体,从而进一步提高了CRISPR/Cas9系统的靶向范围.Walton等[32]通过改造与PAM序列直接作用的氨基酸位点获得了能稳定识别NRN PAM序列的突变体蛋白SpRY,同时其能以较低效率识别NYN PAM序列,使其在靶点识别上几乎不受PAM限制.Hu等[20]应用PACE方法获得一种能够识别NG、GAA及GAT等PAM序列的SpCas9突变体xCas9,同时其能以优于SpCas9的特异性靶向NGG,且对各种PAM的靶向能力均在人类细胞中的基因编辑、转录调控以及胞嘧啶与腺嘌呤核苷酸的碱基修饰中得到了应用.2014年,Guilinger等[34]将FokⅠ核酸酶与核酸酶活性失活的Cas9蛋白(catalytically inactive Cas9,dCas9)融合后得到了一种融合蛋白fCas9,只有两个fCas9单体同时结合在靶基因附近时才能切割目的基因,使其在人类细胞中进行基因编辑的特异性比野生型Cas9提高了140倍.多种研究已报道了将SpCas9或sgRNA工程化后,通过质粒载体携带并表达CRISPR/Cas9系统或通过RNP递送的方式均能够在基因组范围上降低CRISPR/Cas9系统的脱靶效应,并显著提高该系统的保真性[33,55-58].2018年,Casini等[33]筛选并表征了能够提高CRISPR/Cas9系统编辑效率的Cas9 REC3结构域突变库并组合了四种有利突变后,获得了一种保真性比野生型SpCas9提高了79倍的突变体蛋白evoCas9,同时编辑效率接近野生型SpCas9.除了对Cas9蛋白进行优化,对gRNA不断进行工程化改造也能使CRISPR/Cas9系统的性能得到显著提升.2014年,Fu等[44]证明了间隔区截短后的gRNA的脱靶率明显低于全长的gRNA(20 nt),最大降低幅度达5000倍.2019年,Kocak等[21]在工程化设计的sgRNA上的间隔区加入一“发夹”形RNA二级结构,该结构能够在发生脱靶编辑时在脱靶位点处提高gRNA或crRNA链入侵的热力学屏障而并不影响中靶效率,使CRISPR系统的特异性增加了两个数量级.2020年,Rose等[59]基于过短的gRNA(间隔区小于16 nt)与Cas9形成的RNP只能结合却不能切割靶位点特性设计的失活RNA(dead-RNAs,dRNAs)可保护多个脱靶位点,从而使得SpCas9的靶向特异性提高了40倍.相较于工程化改造的高特异性Cas9突变体来说,通过缩短gRNA的间隔区来提高靶向特异性的方法在设计和应用方面均更易用和灵活,预计将成为未来的研究热点方向. ...
Suppression of unwanted CRISPR-Cas9 editing by co-administration of catalytically inactivating truncated guide RNAs
1
2020
... 尽管CRISPR/Cas9系统已在多种生物和细胞的基因组上得到了应用[46-51],Cas9对于PAM序列NGG的严格识别、复杂基因组中gRNA潜在的脱靶效应以及该系统作为医疗手段使用时的保真性仍大大限制其在更广范围下的应用与开发.如在人类基因组DNA上平均每8个碱基对存在一个NGG位点[52],Fu等[53]在人类细胞中表征了CRISPR/Cas9系统的脱靶效应,结果显示该系统的脱靶率高达66%.因此,研究人员通过工程化Cas9蛋白或gRNA等方式,使CRISPR/Cas9系统的靶向范围、靶向特异性和保真性均得到了拓展和提高[19-20].2018年,张锋团队和Osamu Nureki团队[31]通过对SpCas9进行理性设计获得了能够识别NG PAM序列的突变体蛋白SpCas9-NG,并证明了其在人类细胞中的基因组编辑能力以及与胞苷脱氨酶融合后仍具有单碱基编辑能力.2020年,David R. Liu团队[54]利用噬菌体辅助持续进化(phage-assisted continuous evolution,PACE)方法获得了三种识别非G PAM序列的SpCas9突变体,从而进一步提高了CRISPR/Cas9系统的靶向范围.Walton等[32]通过改造与PAM序列直接作用的氨基酸位点获得了能稳定识别NRN PAM序列的突变体蛋白SpRY,同时其能以较低效率识别NYN PAM序列,使其在靶点识别上几乎不受PAM限制.Hu等[20]应用PACE方法获得一种能够识别NG、GAA及GAT等PAM序列的SpCas9突变体xCas9,同时其能以优于SpCas9的特异性靶向NGG,且对各种PAM的靶向能力均在人类细胞中的基因编辑、转录调控以及胞嘧啶与腺嘌呤核苷酸的碱基修饰中得到了应用.2014年,Guilinger等[34]将FokⅠ核酸酶与核酸酶活性失活的Cas9蛋白(catalytically inactive Cas9,dCas9)融合后得到了一种融合蛋白fCas9,只有两个fCas9单体同时结合在靶基因附近时才能切割目的基因,使其在人类细胞中进行基因编辑的特异性比野生型Cas9提高了140倍.多种研究已报道了将SpCas9或sgRNA工程化后,通过质粒载体携带并表达CRISPR/Cas9系统或通过RNP递送的方式均能够在基因组范围上降低CRISPR/Cas9系统的脱靶效应,并显著提高该系统的保真性[33,55-58].2018年,Casini等[33]筛选并表征了能够提高CRISPR/Cas9系统编辑效率的Cas9 REC3结构域突变库并组合了四种有利突变后,获得了一种保真性比野生型SpCas9提高了79倍的突变体蛋白evoCas9,同时编辑效率接近野生型SpCas9.除了对Cas9蛋白进行优化,对gRNA不断进行工程化改造也能使CRISPR/Cas9系统的性能得到显著提升.2014年,Fu等[44]证明了间隔区截短后的gRNA的脱靶率明显低于全长的gRNA(20 nt),最大降低幅度达5000倍.2019年,Kocak等[21]在工程化设计的sgRNA上的间隔区加入一“发夹”形RNA二级结构,该结构能够在发生脱靶编辑时在脱靶位点处提高gRNA或crRNA链入侵的热力学屏障而并不影响中靶效率,使CRISPR系统的特异性增加了两个数量级.2020年,Rose等[59]基于过短的gRNA(间隔区小于16 nt)与Cas9形成的RNP只能结合却不能切割靶位点特性设计的失活RNA(dead-RNAs,dRNAs)可保护多个脱靶位点,从而使得SpCas9的靶向特异性提高了40倍.相较于工程化改造的高特异性Cas9突变体来说,通过缩短gRNA的间隔区来提高靶向特异性的方法在设计和应用方面均更易用和灵活,预计将成为未来的研究热点方向. ...
Processing-independent CRISPR RNAs limit natural transformation in Neisseria meningitidis
1
2013
... 除了开发Cas9的突变体以弥补和拓展CRISPR/Cas9系统的不足与应用范围外,对Cas9 直系同源蛋白进行挖掘和开发也能够使该系统识别范围得到极大扩展.如来自嗜热链球菌(Streptococcus thermophilus)的StCas9能识别NNGAAW(其中W代表A或T)的PAM序列[4,35],来自脑膜炎奈瑟菌(Neisseria meningitidis)的NmCas9识别NNNNGATT序列[36,60-61],来自金黄色葡萄球菌(Staphylococcus aureus)的SaCas9识别NNGRRT序列(其中R代表A或G)[37],这些SpCas9同源蛋白或其组合应用均已在细菌和哺乳动物细胞中得到了开发.此外,SaCas9在肽链长度较小的条件下(1053个氨基酸,SpCas9含有1368个氨基酸)拥有与SpCas9相当的编辑效率,这使其在大小受限制的传递载体如腺相关病毒(adeno-associated virus,AAV)中得到广泛应用.来自曲状杆菌(Campylobacter jejuni)的CjCas9多肽链上仅含984个氨基酸,能识别PAM序列NNNVRYM,已被报道可以通过AAV递送至小鼠肌纤维和视网膜色素上皮细胞(retinal pigment epithelium,RPE)中并成功进行基因编辑[38].工程化的新型Cas9突变体、gRNA与Cas9直系同源的不断发掘为CRISPR/Cas9系统在各种背景下的应用提供了源源不断的新方法和工具,也证明了该系统作为第三代“基因剪刀”目前仍然处于高速发展阶段.该系统中存在的PAM依赖性、脱靶率较高等问题都需要利用进化工程或计算机辅助理性设计等方式对Cas9蛋白进行进一步探索和改造才能得到解决,使其能够在更多的场景下最大程度地发挥自身高灵活性、高精准性、易编程性的优势. ...
Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis
1
2013
... 除了开发Cas9的突变体以弥补和拓展CRISPR/Cas9系统的不足与应用范围外,对Cas9 直系同源蛋白进行挖掘和开发也能够使该系统识别范围得到极大扩展.如来自嗜热链球菌(Streptococcus thermophilus)的StCas9能识别NNGAAW(其中W代表A或T)的PAM序列[4,35],来自脑膜炎奈瑟菌(Neisseria meningitidis)的NmCas9识别NNNNGATT序列[36,60-61],来自金黄色葡萄球菌(Staphylococcus aureus)的SaCas9识别NNGRRT序列(其中R代表A或G)[37],这些SpCas9同源蛋白或其组合应用均已在细菌和哺乳动物细胞中得到了开发.此外,SaCas9在肽链长度较小的条件下(1053个氨基酸,SpCas9含有1368个氨基酸)拥有与SpCas9相当的编辑效率,这使其在大小受限制的传递载体如腺相关病毒(adeno-associated virus,AAV)中得到广泛应用.来自曲状杆菌(Campylobacter jejuni)的CjCas9多肽链上仅含984个氨基酸,能识别PAM序列NNNVRYM,已被报道可以通过AAV递送至小鼠肌纤维和视网膜色素上皮细胞(retinal pigment epithelium,RPE)中并成功进行基因编辑[38].工程化的新型Cas9突变体、gRNA与Cas9直系同源的不断发掘为CRISPR/Cas9系统在各种背景下的应用提供了源源不断的新方法和工具,也证明了该系统作为第三代“基因剪刀”目前仍然处于高速发展阶段.该系统中存在的PAM依赖性、脱靶率较高等问题都需要利用进化工程或计算机辅助理性设计等方式对Cas9蛋白进行进一步探索和改造才能得到解决,使其能够在更多的场景下最大程度地发挥自身高灵活性、高精准性、易编程性的优势. ...
CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes
8
2013
... Functional optimization through fusion expression of effector proteins with CRISPR/Cas9 system
Table 2| 功能 | 效应蛋白或gRNA工程 | 系统 | 特征 | 参考文献 |
|---|
| 表达调控 | 无 | dCas9 | 原核生物基因抑制表达,真核生物基因抑制效率低 | [22] |
| KRAB,MXI1,TUP1抑制结构域 | dCas9 | 可在真核生物中抑制基因表达 | [62-63] |
| KRAB-MeCP1融合抑制结构域 | dCas9 | 在哺乳动物细胞中高效抑制目的基因表达 | [64] |
| p65,VP64激活结构域 | dCas9 | 激活基因表达,效率较低 | [65-66] |
| VP64-p65-Rta(VPR)融合激活结构域 | dCas9 | 与VP64相比提高了22~320倍;多重基因激活表达;刺激iPSCs神经元分化 | [67] |
| gRNA工程;gRNA上携带携带两个MS2发夹二级结构 | dCas9 | 每个发夹结构招募一个MS2-p65-HSF1融合激活蛋白;与VP64相比,使10个靶基因上调2倍以上 | [68] |
| SPY系统(包括SpyTag和SpyCatcher)和Med2激活结构域 | dCas9 | 招募多拷贝SpyCatcher和Med2融合蛋白使酿酒酵母目的基因表达提高34.9倍 | [69] |
| SunTag系统(包括GCN4肽和抗GCN4抗体的单链可变片段scFv)和MIG1抑制结构域 | dCpf1 | 招募多拷贝ScFv与MIG1融合蛋白使酿酒酵母目的基因受到95%的抑制 | [69] |
| 表观基因组编辑 | KRAB-DNA甲基化酶催化结构域融合蛋白 | dCas9 | 融合抑制因子和甲基化酶;造成目的基因78%的稳定沉默 | [70] |
| 两侧分别融合KRAB和Dnmt3A-Dnmt3L融合DNA甲基化酶结构域 | dCas9 | 在干细胞分裂分化为神经细胞后仍维持DNA甲基化和基因抑制 | [71] |
| TET1甲基胞嘧啶双加氧酶1催化结构域 | dCas9 | dCas9- TET1靶向至BRCA1基因启动子后成功地上调其表达水平 | [72] |
| 人类组蛋白乙酰基转移酶p300催化核心 | dCas9 | 催化组蛋白H3 第27位赖氨酸残基的乙酰基化;基因表达上调 | [73] |
| 单碱基编辑 | 胞嘧啶脱氨酶,腺嘌呤脱氨酶 | nCas9 | 在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换 | [30,74-75] |
| CBE-UGI融合蛋白 | nCas9 | UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率 | [30] |
| 单链DNA结合结构域ssDBD融合到胞嘧啶脱氨酶与nCas9之间 | nCas9 | 通过延长ssDNA的暴露时间极大地提高CBEs的编辑效率;使CBEs的编辑窗口范围得到了提高 | [76] |
| 组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶 | nCas9 | 首次实现大肠杆菌中C到A 87.2%和哺乳动物细胞C到G 5.3%至53.0%的编辑效率 | [77] |
| 先导编辑 | 通过gRNA工程改造的pegRNA;与逆转录酶融合 | nCas9 | 改变pegRNA可实现精确的碱基替换、基因敲除和插入 | [27] |
| 通过gRNA工程改造的pegRNA;与逆转录酶和Rad51融合 | nCas9 | 融合单链DNA结合蛋白Rad51使PE2的编辑效率最高提升2.6倍 | [78] |
在CRISPR/Cas9的基因转录调控系统中,对Cas9蛋白工程化的dCas9分别在RuvC和HNH结构域发生突变使得RNP复合物仅能发挥靶序列识别功能,而无法切断核苷酸双链[29].nCas9(Cas9 nickase)是仅能使靶DNA发生单链断裂(single-strand break,SSB)的Cas9突变体,其中nCas9(H840A)和nCas9(D10A)分别是Cas9在HNH和RuvC结构域发生突变后的突变体蛋白,从而能够使该结构域失去内切酶活性,使DNA单链断裂,产生“缺口”[22,39].将dCas9或nCas9与各种效应蛋白如转录激活因子或抑制因子、表观遗传修饰因子、碱基编辑器甚至具有特定功能的酶等融合后,研究人员便能够借助CRISPR/Cas9系统对靶序列实现特定生物学功能,如表征转录基因乃至基因组上非编码区的功能、通过将碱基编辑器与nCas9融合能够在HDR活性低的细胞中进行精确高效的单碱基编辑等.因此本节将重点回顾与总结多种多样的效应蛋白为CRISPR/Cas9系统带来的多元化优化和发展,以及赋予该系统的多种新功能. ...
... 转录因子通过结合或识别基因上游特定区域,进而调节RNA聚合酶的招募,最终使靶基因表达上调或下调[79-80].其不仅可以与启动子区域结合,多个转录因子间也可结合形成转录因子复合体进而对靶基因进行更复杂、精细的调控,促进或抑制DNA上遗传信息向RNA转录的过程.在基因组范围上对靶基因进行调控是探究基因功能、重塑代谢调控网络的重要方法[62].自20世纪末以来,研究人员就已经将自然存在的或工程化后的DNA靶向蛋白如四环素阻遏蛋白[81-82]、Gal4蛋白[83]、锌指蛋白[84]以及TALE蛋白[85-87]等与转录因子融合进行靶基因调控.但由于依赖蛋白质靶向 DNA的特性,靶向不同的DNA序列时需要工程化设计不同的蛋白质氨基酸序列,使得这些蛋白普遍可编程性较差且分子庞大,因此开发用于基因组范围的转录调控蛋白文库相当耗时且价格昂贵[9].CRISPR基因调控系统包括CRISPR激活系统(CRISPR activation system,CRISPRa)和CRISPR干扰系统(CRISPR interference system,CRISPRi),该系统将转录激活或抑制因子与dCas9蛋白融合后,借助CRISPR系统精准的靶向性和灵活的可编程性将融合蛋白靶向至目的基因从而对该基因的转录进行激活或干扰(图2)[62,81-88].不同于其他DNA靶向蛋白,CRISPR激活系统的dCas9-effector融合蛋白利用gRNA与靶序列的碱基互补配对进行精确定位并最终实现目的基因调控,同时gRNA的易编程性和dCas9蛋白较小的体量使该系统在基因组范围上进行调控成为了可能. ...
... [62,81-88].不同于其他DNA靶向蛋白,CRISPR激活系统的dCas9-effector融合蛋白利用gRNA与靶序列的碱基互补配对进行精确定位并最终实现目的基因调控,同时gRNA的易编程性和dCas9蛋白较小的体量使该系统在基因组范围上进行调控成为了可能. ...
... 仅将dCas9蛋白靶向至目的基因上游时,dCas9蛋白对RNA聚合酶的空间位阻效应便可在一定程度上抑制基因表达[22],而该效应抑制表达的方式仅在原核细胞中有较高的效率,在真核细胞中由于其内部复杂的调控机制使得抑制效率较低[22,62,89].将dCas9蛋白与转录抑制结构域如KRAB[62]、MXI1[62]和TUP1[63]等蛋白融合表达则可提高CRISPRi系统在真核细胞中的抑制效率.其中KRAB锌指蛋白(KRAB-ZFP)属于哺乳动物细胞中最大的转录调节因子家族[90],该蛋白能够通过诱导靶序列的染色质结构变化从而抑制基因转录[91].KRAB与dCas9融合后被靶向至启动子区、5′非翻译区以及近端或远端增强子元件区后可以有效地沉默单个基因或非编码RNA[62,92-94].2018年,Yeo等[64]表征了20多种已知的能够诱导基因沉默的转录因子的抑制效率后,将获得的最高效的6种抑制因子——MeCP2、SIN3A、HDT1、MBD2B、NIPP1和HP1A分别与通用抑制因子KRAB融合形成双重抑制因子,再与dCas9蛋白C端融合表达并表征CRISPRi系统的抑制效率,最终获得了比单独dCas9抑制效率提高了60倍的双重抑制因子融合蛋白dCas9-KRAB-MeCP2.而之后研究人员继续表征的三重抑制因子融合蛋白可能由于功能冗余或蛋白体量较大从而影响抑制因子正常工作导致抑制效率不如双重抑制因子,证明了通过无限累加融合的方式提高融合蛋白的调控效率是不可行的[64]. ...
... [62]、MXI1[62]和TUP1[63]等蛋白融合表达则可提高CRISPRi系统在真核细胞中的抑制效率.其中KRAB锌指蛋白(KRAB-ZFP)属于哺乳动物细胞中最大的转录调节因子家族[90],该蛋白能够通过诱导靶序列的染色质结构变化从而抑制基因转录[91].KRAB与dCas9融合后被靶向至启动子区、5′非翻译区以及近端或远端增强子元件区后可以有效地沉默单个基因或非编码RNA[62,92-94].2018年,Yeo等[64]表征了20多种已知的能够诱导基因沉默的转录因子的抑制效率后,将获得的最高效的6种抑制因子——MeCP2、SIN3A、HDT1、MBD2B、NIPP1和HP1A分别与通用抑制因子KRAB融合形成双重抑制因子,再与dCas9蛋白C端融合表达并表征CRISPRi系统的抑制效率,最终获得了比单独dCas9抑制效率提高了60倍的双重抑制因子融合蛋白dCas9-KRAB-MeCP2.而之后研究人员继续表征的三重抑制因子融合蛋白可能由于功能冗余或蛋白体量较大从而影响抑制因子正常工作导致抑制效率不如双重抑制因子,证明了通过无限累加融合的方式提高融合蛋白的调控效率是不可行的[64]. ...
... [62]和TUP1[63]等蛋白融合表达则可提高CRISPRi系统在真核细胞中的抑制效率.其中KRAB锌指蛋白(KRAB-ZFP)属于哺乳动物细胞中最大的转录调节因子家族[90],该蛋白能够通过诱导靶序列的染色质结构变化从而抑制基因转录[91].KRAB与dCas9融合后被靶向至启动子区、5′非翻译区以及近端或远端增强子元件区后可以有效地沉默单个基因或非编码RNA[62,92-94].2018年,Yeo等[64]表征了20多种已知的能够诱导基因沉默的转录因子的抑制效率后,将获得的最高效的6种抑制因子——MeCP2、SIN3A、HDT1、MBD2B、NIPP1和HP1A分别与通用抑制因子KRAB融合形成双重抑制因子,再与dCas9蛋白C端融合表达并表征CRISPRi系统的抑制效率,最终获得了比单独dCas9抑制效率提高了60倍的双重抑制因子融合蛋白dCas9-KRAB-MeCP2.而之后研究人员继续表征的三重抑制因子融合蛋白可能由于功能冗余或蛋白体量较大从而影响抑制因子正常工作导致抑制效率不如双重抑制因子,证明了通过无限累加融合的方式提高融合蛋白的调控效率是不可行的[64]. ...
... [62,92-94].2018年,Yeo等[64]表征了20多种已知的能够诱导基因沉默的转录因子的抑制效率后,将获得的最高效的6种抑制因子——MeCP2、SIN3A、HDT1、MBD2B、NIPP1和HP1A分别与通用抑制因子KRAB融合形成双重抑制因子,再与dCas9蛋白C端融合表达并表征CRISPRi系统的抑制效率,最终获得了比单独dCas9抑制效率提高了60倍的双重抑制因子融合蛋白dCas9-KRAB-MeCP2.而之后研究人员继续表征的三重抑制因子融合蛋白可能由于功能冗余或蛋白体量较大从而影响抑制因子正常工作导致抑制效率不如双重抑制因子,证明了通过无限累加融合的方式提高融合蛋白的调控效率是不可行的[64]. ...
... 将dCas9与各种转录激活因子融合后,则可在gRNA的靶向作用下精确激活目的基因,该系统称为CRISPR激活系统或CRISPRa.最初的CRISPRa系统中dCas9仅融合了单个反式激活因子p65(也被称为RelA,是构成NF-κB转录因子家族的五种成员之一)或VP64 (由单纯疱疹病毒蛋白VP16的四个重复结构域构成),这些早期工作共同构成了最初的可编程dCas9基因激活工具箱,但效果差强人意[23-28,39-44,62,65-66].为了不断提高CRISPRa系统的激活效率,研究人员尝试了用各种方式将更复杂或多拷贝的激活结构域应用于CRISPRa.2015年,Chavez等[67]通过蛋白质理性设计,构建了三重融合激活因子VP64-p65-Rta(VPR)并应用于CRISPRa系统,其中Rta是来源于EB病毒(epstein-barr virus)的反式激活因子.与dCas9-VP64相比,dCas9-VPR使人胚胎肾细胞293(HEK 293T cells)的内源靶基因表达上调了22~320倍,同时证明了该系统也能够在多重基因的激活表达与刺激人诱导多功能干细胞(induced pluripotent stem cells,iPSCs)的神经元分化上发挥作用.同年,张锋研究团队[68]开发的基于CRISPRa的协同激活介导系统(synergistic activation mediator system, SAM)通过修饰sgRNA使其携带了两个MS2发夹二级结构,每个发夹结构都能招募一个MS2-p65-HSF1融合蛋白,其中包括反式激活因子p65、MS2结合蛋白以及热休克蛋白1(heat shock protein 1),最终证明该修饰后的gRNA与dCas9-VP64的协同作用能够使10个靶基因的表达均上调2倍以上.2022年,山东大学候进课题组[69]构建的CRISPR介导的蛋白质标签信号放大系统首次将SPY系统(包括SpyTag和SpyCatcher)和SunTag系统分别引入dCas9介导的CRISPRa和dCpf1(一种丧失核酸酶活性的Cas9同源蛋白)介导的CRISPRi系统并进行骨架长度和转录因子优化后,成功使酿酒酵母内目的基因转录水平分别提高了34.9倍以及受到几乎完全的抑制.其中,SunTag系统是由若干个拷贝的GCN4肽以及作为标签的抗GCN4抗体的单链可变片段scFv组成[95],与dCas9融合的多拷贝GCN4肽通过招募多拷贝的转录因子与GCN4抗体标签的融合蛋白从而进行转录调控,该系统除了与CRISPR系统结合后用于碱基编辑以及基因调控,还在哺乳动物和植物细胞的DNA的甲基化和去甲基化诱导中发挥作用[96-100];而SPY系统作为一种广泛使用的蛋白质组装系统首次在酿酒酵母的转录调控中得到了应用[69].可见在dCas9与转录因子融合的基础上,CRISPRa与CRISPRi系统能够利用融合多重结构域或融合蛋白质骨架系统的方式提高调控效率.而通过利用RNA与蛋白质间的相互作用,将多个适配子融合在gRNA中使其同时含有目的基因靶向序列和转录因子招募序列的方式能够产生与dCas9融合多个转录激活或抑制结构域的策略类似的效果[101].此外,针对不同细胞乃至不同dCas9同源蛋白,CRISPRa与CRISPRi系统均需对不同转录激活或抑制结构域乃至其组合进行优化才能发挥该系统的最大效能. ...
Repression domain of the yeast global repressor Tup1 interacts directly with histones H3 and H4
2
1996
... Functional optimization through fusion expression of effector proteins with CRISPR/Cas9 system
Table 2| 功能 | 效应蛋白或gRNA工程 | 系统 | 特征 | 参考文献 |
|---|
| 表达调控 | 无 | dCas9 | 原核生物基因抑制表达,真核生物基因抑制效率低 | [22] |
| KRAB,MXI1,TUP1抑制结构域 | dCas9 | 可在真核生物中抑制基因表达 | [62-63] |
| KRAB-MeCP1融合抑制结构域 | dCas9 | 在哺乳动物细胞中高效抑制目的基因表达 | [64] |
| p65,VP64激活结构域 | dCas9 | 激活基因表达,效率较低 | [65-66] |
| VP64-p65-Rta(VPR)融合激活结构域 | dCas9 | 与VP64相比提高了22~320倍;多重基因激活表达;刺激iPSCs神经元分化 | [67] |
| gRNA工程;gRNA上携带携带两个MS2发夹二级结构 | dCas9 | 每个发夹结构招募一个MS2-p65-HSF1融合激活蛋白;与VP64相比,使10个靶基因上调2倍以上 | [68] |
| SPY系统(包括SpyTag和SpyCatcher)和Med2激活结构域 | dCas9 | 招募多拷贝SpyCatcher和Med2融合蛋白使酿酒酵母目的基因表达提高34.9倍 | [69] |
| SunTag系统(包括GCN4肽和抗GCN4抗体的单链可变片段scFv)和MIG1抑制结构域 | dCpf1 | 招募多拷贝ScFv与MIG1融合蛋白使酿酒酵母目的基因受到95%的抑制 | [69] |
| 表观基因组编辑 | KRAB-DNA甲基化酶催化结构域融合蛋白 | dCas9 | 融合抑制因子和甲基化酶;造成目的基因78%的稳定沉默 | [70] |
| 两侧分别融合KRAB和Dnmt3A-Dnmt3L融合DNA甲基化酶结构域 | dCas9 | 在干细胞分裂分化为神经细胞后仍维持DNA甲基化和基因抑制 | [71] |
| TET1甲基胞嘧啶双加氧酶1催化结构域 | dCas9 | dCas9- TET1靶向至BRCA1基因启动子后成功地上调其表达水平 | [72] |
| 人类组蛋白乙酰基转移酶p300催化核心 | dCas9 | 催化组蛋白H3 第27位赖氨酸残基的乙酰基化;基因表达上调 | [73] |
| 单碱基编辑 | 胞嘧啶脱氨酶,腺嘌呤脱氨酶 | nCas9 | 在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换 | [30,74-75] |
| CBE-UGI融合蛋白 | nCas9 | UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率 | [30] |
| 单链DNA结合结构域ssDBD融合到胞嘧啶脱氨酶与nCas9之间 | nCas9 | 通过延长ssDNA的暴露时间极大地提高CBEs的编辑效率;使CBEs的编辑窗口范围得到了提高 | [76] |
| 组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶 | nCas9 | 首次实现大肠杆菌中C到A 87.2%和哺乳动物细胞C到G 5.3%至53.0%的编辑效率 | [77] |
| 先导编辑 | 通过gRNA工程改造的pegRNA;与逆转录酶融合 | nCas9 | 改变pegRNA可实现精确的碱基替换、基因敲除和插入 | [27] |
| 通过gRNA工程改造的pegRNA;与逆转录酶和Rad51融合 | nCas9 | 融合单链DNA结合蛋白Rad51使PE2的编辑效率最高提升2.6倍 | [78] |
在CRISPR/Cas9的基因转录调控系统中,对Cas9蛋白工程化的dCas9分别在RuvC和HNH结构域发生突变使得RNP复合物仅能发挥靶序列识别功能,而无法切断核苷酸双链[29].nCas9(Cas9 nickase)是仅能使靶DNA发生单链断裂(single-strand break,SSB)的Cas9突变体,其中nCas9(H840A)和nCas9(D10A)分别是Cas9在HNH和RuvC结构域发生突变后的突变体蛋白,从而能够使该结构域失去内切酶活性,使DNA单链断裂,产生“缺口”[22,39].将dCas9或nCas9与各种效应蛋白如转录激活因子或抑制因子、表观遗传修饰因子、碱基编辑器甚至具有特定功能的酶等融合后,研究人员便能够借助CRISPR/Cas9系统对靶序列实现特定生物学功能,如表征转录基因乃至基因组上非编码区的功能、通过将碱基编辑器与nCas9融合能够在HDR活性低的细胞中进行精确高效的单碱基编辑等.因此本节将重点回顾与总结多种多样的效应蛋白为CRISPR/Cas9系统带来的多元化优化和发展,以及赋予该系统的多种新功能. ...
... 仅将dCas9蛋白靶向至目的基因上游时,dCas9蛋白对RNA聚合酶的空间位阻效应便可在一定程度上抑制基因表达[22],而该效应抑制表达的方式仅在原核细胞中有较高的效率,在真核细胞中由于其内部复杂的调控机制使得抑制效率较低[22,62,89].将dCas9蛋白与转录抑制结构域如KRAB[62]、MXI1[62]和TUP1[63]等蛋白融合表达则可提高CRISPRi系统在真核细胞中的抑制效率.其中KRAB锌指蛋白(KRAB-ZFP)属于哺乳动物细胞中最大的转录调节因子家族[90],该蛋白能够通过诱导靶序列的染色质结构变化从而抑制基因转录[91].KRAB与dCas9融合后被靶向至启动子区、5′非翻译区以及近端或远端增强子元件区后可以有效地沉默单个基因或非编码RNA[62,92-94].2018年,Yeo等[64]表征了20多种已知的能够诱导基因沉默的转录因子的抑制效率后,将获得的最高效的6种抑制因子——MeCP2、SIN3A、HDT1、MBD2B、NIPP1和HP1A分别与通用抑制因子KRAB融合形成双重抑制因子,再与dCas9蛋白C端融合表达并表征CRISPRi系统的抑制效率,最终获得了比单独dCas9抑制效率提高了60倍的双重抑制因子融合蛋白dCas9-KRAB-MeCP2.而之后研究人员继续表征的三重抑制因子融合蛋白可能由于功能冗余或蛋白体量较大从而影响抑制因子正常工作导致抑制效率不如双重抑制因子,证明了通过无限累加融合的方式提高融合蛋白的调控效率是不可行的[64]. ...
An enhanced CRISPR repressor for targeted mammalian gene regulation
3
2018
... Functional optimization through fusion expression of effector proteins with CRISPR/Cas9 system
Table 2| 功能 | 效应蛋白或gRNA工程 | 系统 | 特征 | 参考文献 |
|---|
| 表达调控 | 无 | dCas9 | 原核生物基因抑制表达,真核生物基因抑制效率低 | [22] |
| KRAB,MXI1,TUP1抑制结构域 | dCas9 | 可在真核生物中抑制基因表达 | [62-63] |
| KRAB-MeCP1融合抑制结构域 | dCas9 | 在哺乳动物细胞中高效抑制目的基因表达 | [64] |
| p65,VP64激活结构域 | dCas9 | 激活基因表达,效率较低 | [65-66] |
| VP64-p65-Rta(VPR)融合激活结构域 | dCas9 | 与VP64相比提高了22~320倍;多重基因激活表达;刺激iPSCs神经元分化 | [67] |
| gRNA工程;gRNA上携带携带两个MS2发夹二级结构 | dCas9 | 每个发夹结构招募一个MS2-p65-HSF1融合激活蛋白;与VP64相比,使10个靶基因上调2倍以上 | [68] |
| SPY系统(包括SpyTag和SpyCatcher)和Med2激活结构域 | dCas9 | 招募多拷贝SpyCatcher和Med2融合蛋白使酿酒酵母目的基因表达提高34.9倍 | [69] |
| SunTag系统(包括GCN4肽和抗GCN4抗体的单链可变片段scFv)和MIG1抑制结构域 | dCpf1 | 招募多拷贝ScFv与MIG1融合蛋白使酿酒酵母目的基因受到95%的抑制 | [69] |
| 表观基因组编辑 | KRAB-DNA甲基化酶催化结构域融合蛋白 | dCas9 | 融合抑制因子和甲基化酶;造成目的基因78%的稳定沉默 | [70] |
| 两侧分别融合KRAB和Dnmt3A-Dnmt3L融合DNA甲基化酶结构域 | dCas9 | 在干细胞分裂分化为神经细胞后仍维持DNA甲基化和基因抑制 | [71] |
| TET1甲基胞嘧啶双加氧酶1催化结构域 | dCas9 | dCas9- TET1靶向至BRCA1基因启动子后成功地上调其表达水平 | [72] |
| 人类组蛋白乙酰基转移酶p300催化核心 | dCas9 | 催化组蛋白H3 第27位赖氨酸残基的乙酰基化;基因表达上调 | [73] |
| 单碱基编辑 | 胞嘧啶脱氨酶,腺嘌呤脱氨酶 | nCas9 | 在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换 | [30,74-75] |
| CBE-UGI融合蛋白 | nCas9 | UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率 | [30] |
| 单链DNA结合结构域ssDBD融合到胞嘧啶脱氨酶与nCas9之间 | nCas9 | 通过延长ssDNA的暴露时间极大地提高CBEs的编辑效率;使CBEs的编辑窗口范围得到了提高 | [76] |
| 组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶 | nCas9 | 首次实现大肠杆菌中C到A 87.2%和哺乳动物细胞C到G 5.3%至53.0%的编辑效率 | [77] |
| 先导编辑 | 通过gRNA工程改造的pegRNA;与逆转录酶融合 | nCas9 | 改变pegRNA可实现精确的碱基替换、基因敲除和插入 | [27] |
| 通过gRNA工程改造的pegRNA;与逆转录酶和Rad51融合 | nCas9 | 融合单链DNA结合蛋白Rad51使PE2的编辑效率最高提升2.6倍 | [78] |
在CRISPR/Cas9的基因转录调控系统中,对Cas9蛋白工程化的dCas9分别在RuvC和HNH结构域发生突变使得RNP复合物仅能发挥靶序列识别功能,而无法切断核苷酸双链[29].nCas9(Cas9 nickase)是仅能使靶DNA发生单链断裂(single-strand break,SSB)的Cas9突变体,其中nCas9(H840A)和nCas9(D10A)分别是Cas9在HNH和RuvC结构域发生突变后的突变体蛋白,从而能够使该结构域失去内切酶活性,使DNA单链断裂,产生“缺口”[22,39].将dCas9或nCas9与各种效应蛋白如转录激活因子或抑制因子、表观遗传修饰因子、碱基编辑器甚至具有特定功能的酶等融合后,研究人员便能够借助CRISPR/Cas9系统对靶序列实现特定生物学功能,如表征转录基因乃至基因组上非编码区的功能、通过将碱基编辑器与nCas9融合能够在HDR活性低的细胞中进行精确高效的单碱基编辑等.因此本节将重点回顾与总结多种多样的效应蛋白为CRISPR/Cas9系统带来的多元化优化和发展,以及赋予该系统的多种新功能. ...
... 仅将dCas9蛋白靶向至目的基因上游时,dCas9蛋白对RNA聚合酶的空间位阻效应便可在一定程度上抑制基因表达[22],而该效应抑制表达的方式仅在原核细胞中有较高的效率,在真核细胞中由于其内部复杂的调控机制使得抑制效率较低[22,62,89].将dCas9蛋白与转录抑制结构域如KRAB[62]、MXI1[62]和TUP1[63]等蛋白融合表达则可提高CRISPRi系统在真核细胞中的抑制效率.其中KRAB锌指蛋白(KRAB-ZFP)属于哺乳动物细胞中最大的转录调节因子家族[90],该蛋白能够通过诱导靶序列的染色质结构变化从而抑制基因转录[91].KRAB与dCas9融合后被靶向至启动子区、5′非翻译区以及近端或远端增强子元件区后可以有效地沉默单个基因或非编码RNA[62,92-94].2018年,Yeo等[64]表征了20多种已知的能够诱导基因沉默的转录因子的抑制效率后,将获得的最高效的6种抑制因子——MeCP2、SIN3A、HDT1、MBD2B、NIPP1和HP1A分别与通用抑制因子KRAB融合形成双重抑制因子,再与dCas9蛋白C端融合表达并表征CRISPRi系统的抑制效率,最终获得了比单独dCas9抑制效率提高了60倍的双重抑制因子融合蛋白dCas9-KRAB-MeCP2.而之后研究人员继续表征的三重抑制因子融合蛋白可能由于功能冗余或蛋白体量较大从而影响抑制因子正常工作导致抑制效率不如双重抑制因子,证明了通过无限累加融合的方式提高融合蛋白的调控效率是不可行的[64]. ...
... [64]. ...
Tunable and multifunctional eukaryotic transcription factors based on CRISPR/Cas
2
2013
... Functional optimization through fusion expression of effector proteins with CRISPR/Cas9 system
Table 2| 功能 | 效应蛋白或gRNA工程 | 系统 | 特征 | 参考文献 |
|---|
| 表达调控 | 无 | dCas9 | 原核生物基因抑制表达,真核生物基因抑制效率低 | [22] |
| KRAB,MXI1,TUP1抑制结构域 | dCas9 | 可在真核生物中抑制基因表达 | [62-63] |
| KRAB-MeCP1融合抑制结构域 | dCas9 | 在哺乳动物细胞中高效抑制目的基因表达 | [64] |
| p65,VP64激活结构域 | dCas9 | 激活基因表达,效率较低 | [65-66] |
| VP64-p65-Rta(VPR)融合激活结构域 | dCas9 | 与VP64相比提高了22~320倍;多重基因激活表达;刺激iPSCs神经元分化 | [67] |
| gRNA工程;gRNA上携带携带两个MS2发夹二级结构 | dCas9 | 每个发夹结构招募一个MS2-p65-HSF1融合激活蛋白;与VP64相比,使10个靶基因上调2倍以上 | [68] |
| SPY系统(包括SpyTag和SpyCatcher)和Med2激活结构域 | dCas9 | 招募多拷贝SpyCatcher和Med2融合蛋白使酿酒酵母目的基因表达提高34.9倍 | [69] |
| SunTag系统(包括GCN4肽和抗GCN4抗体的单链可变片段scFv)和MIG1抑制结构域 | dCpf1 | 招募多拷贝ScFv与MIG1融合蛋白使酿酒酵母目的基因受到95%的抑制 | [69] |
| 表观基因组编辑 | KRAB-DNA甲基化酶催化结构域融合蛋白 | dCas9 | 融合抑制因子和甲基化酶;造成目的基因78%的稳定沉默 | [70] |
| 两侧分别融合KRAB和Dnmt3A-Dnmt3L融合DNA甲基化酶结构域 | dCas9 | 在干细胞分裂分化为神经细胞后仍维持DNA甲基化和基因抑制 | [71] |
| TET1甲基胞嘧啶双加氧酶1催化结构域 | dCas9 | dCas9- TET1靶向至BRCA1基因启动子后成功地上调其表达水平 | [72] |
| 人类组蛋白乙酰基转移酶p300催化核心 | dCas9 | 催化组蛋白H3 第27位赖氨酸残基的乙酰基化;基因表达上调 | [73] |
| 单碱基编辑 | 胞嘧啶脱氨酶,腺嘌呤脱氨酶 | nCas9 | 在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换 | [30,74-75] |
| CBE-UGI融合蛋白 | nCas9 | UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率 | [30] |
| 单链DNA结合结构域ssDBD融合到胞嘧啶脱氨酶与nCas9之间 | nCas9 | 通过延长ssDNA的暴露时间极大地提高CBEs的编辑效率;使CBEs的编辑窗口范围得到了提高 | [76] |
| 组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶 | nCas9 | 首次实现大肠杆菌中C到A 87.2%和哺乳动物细胞C到G 5.3%至53.0%的编辑效率 | [77] |
| 先导编辑 | 通过gRNA工程改造的pegRNA;与逆转录酶融合 | nCas9 | 改变pegRNA可实现精确的碱基替换、基因敲除和插入 | [27] |
| 通过gRNA工程改造的pegRNA;与逆转录酶和Rad51融合 | nCas9 | 融合单链DNA结合蛋白Rad51使PE2的编辑效率最高提升2.6倍 | [78] |
在CRISPR/Cas9的基因转录调控系统中,对Cas9蛋白工程化的dCas9分别在RuvC和HNH结构域发生突变使得RNP复合物仅能发挥靶序列识别功能,而无法切断核苷酸双链[29].nCas9(Cas9 nickase)是仅能使靶DNA发生单链断裂(single-strand break,SSB)的Cas9突变体,其中nCas9(H840A)和nCas9(D10A)分别是Cas9在HNH和RuvC结构域发生突变后的突变体蛋白,从而能够使该结构域失去内切酶活性,使DNA单链断裂,产生“缺口”[22,39].将dCas9或nCas9与各种效应蛋白如转录激活因子或抑制因子、表观遗传修饰因子、碱基编辑器甚至具有特定功能的酶等融合后,研究人员便能够借助CRISPR/Cas9系统对靶序列实现特定生物学功能,如表征转录基因乃至基因组上非编码区的功能、通过将碱基编辑器与nCas9融合能够在HDR活性低的细胞中进行精确高效的单碱基编辑等.因此本节将重点回顾与总结多种多样的效应蛋白为CRISPR/Cas9系统带来的多元化优化和发展,以及赋予该系统的多种新功能. ...
... 将dCas9与各种转录激活因子融合后,则可在gRNA的靶向作用下精确激活目的基因,该系统称为CRISPR激活系统或CRISPRa.最初的CRISPRa系统中dCas9仅融合了单个反式激活因子p65(也被称为RelA,是构成NF-κB转录因子家族的五种成员之一)或VP64 (由单纯疱疹病毒蛋白VP16的四个重复结构域构成),这些早期工作共同构成了最初的可编程dCas9基因激活工具箱,但效果差强人意[23-28,39-44,62,65-66].为了不断提高CRISPRa系统的激活效率,研究人员尝试了用各种方式将更复杂或多拷贝的激活结构域应用于CRISPRa.2015年,Chavez等[67]通过蛋白质理性设计,构建了三重融合激活因子VP64-p65-Rta(VPR)并应用于CRISPRa系统,其中Rta是来源于EB病毒(epstein-barr virus)的反式激活因子.与dCas9-VP64相比,dCas9-VPR使人胚胎肾细胞293(HEK 293T cells)的内源靶基因表达上调了22~320倍,同时证明了该系统也能够在多重基因的激活表达与刺激人诱导多功能干细胞(induced pluripotent stem cells,iPSCs)的神经元分化上发挥作用.同年,张锋研究团队[68]开发的基于CRISPRa的协同激活介导系统(synergistic activation mediator system, SAM)通过修饰sgRNA使其携带了两个MS2发夹二级结构,每个发夹结构都能招募一个MS2-p65-HSF1融合蛋白,其中包括反式激活因子p65、MS2结合蛋白以及热休克蛋白1(heat shock protein 1),最终证明该修饰后的gRNA与dCas9-VP64的协同作用能够使10个靶基因的表达均上调2倍以上.2022年,山东大学候进课题组[69]构建的CRISPR介导的蛋白质标签信号放大系统首次将SPY系统(包括SpyTag和SpyCatcher)和SunTag系统分别引入dCas9介导的CRISPRa和dCpf1(一种丧失核酸酶活性的Cas9同源蛋白)介导的CRISPRi系统并进行骨架长度和转录因子优化后,成功使酿酒酵母内目的基因转录水平分别提高了34.9倍以及受到几乎完全的抑制.其中,SunTag系统是由若干个拷贝的GCN4肽以及作为标签的抗GCN4抗体的单链可变片段scFv组成[95],与dCas9融合的多拷贝GCN4肽通过招募多拷贝的转录因子与GCN4抗体标签的融合蛋白从而进行转录调控,该系统除了与CRISPR系统结合后用于碱基编辑以及基因调控,还在哺乳动物和植物细胞的DNA的甲基化和去甲基化诱导中发挥作用[96-100];而SPY系统作为一种广泛使用的蛋白质组装系统首次在酿酒酵母的转录调控中得到了应用[69].可见在dCas9与转录因子融合的基础上,CRISPRa与CRISPRi系统能够利用融合多重结构域或融合蛋白质骨架系统的方式提高调控效率.而通过利用RNA与蛋白质间的相互作用,将多个适配子融合在gRNA中使其同时含有目的基因靶向序列和转录因子招募序列的方式能够产生与dCas9融合多个转录激活或抑制结构域的策略类似的效果[101].此外,针对不同细胞乃至不同dCas9同源蛋白,CRISPRa与CRISPRi系统均需对不同转录激活或抑制结构域乃至其组合进行优化才能发挥该系统的最大效能. ...
A CRISPR-dCas toolbox for genetic engineering and synthetic biology
2
2019
... Functional optimization through fusion expression of effector proteins with CRISPR/Cas9 system
Table 2| 功能 | 效应蛋白或gRNA工程 | 系统 | 特征 | 参考文献 |
|---|
| 表达调控 | 无 | dCas9 | 原核生物基因抑制表达,真核生物基因抑制效率低 | [22] |
| KRAB,MXI1,TUP1抑制结构域 | dCas9 | 可在真核生物中抑制基因表达 | [62-63] |
| KRAB-MeCP1融合抑制结构域 | dCas9 | 在哺乳动物细胞中高效抑制目的基因表达 | [64] |
| p65,VP64激活结构域 | dCas9 | 激活基因表达,效率较低 | [65-66] |
| VP64-p65-Rta(VPR)融合激活结构域 | dCas9 | 与VP64相比提高了22~320倍;多重基因激活表达;刺激iPSCs神经元分化 | [67] |
| gRNA工程;gRNA上携带携带两个MS2发夹二级结构 | dCas9 | 每个发夹结构招募一个MS2-p65-HSF1融合激活蛋白;与VP64相比,使10个靶基因上调2倍以上 | [68] |
| SPY系统(包括SpyTag和SpyCatcher)和Med2激活结构域 | dCas9 | 招募多拷贝SpyCatcher和Med2融合蛋白使酿酒酵母目的基因表达提高34.9倍 | [69] |
| SunTag系统(包括GCN4肽和抗GCN4抗体的单链可变片段scFv)和MIG1抑制结构域 | dCpf1 | 招募多拷贝ScFv与MIG1融合蛋白使酿酒酵母目的基因受到95%的抑制 | [69] |
| 表观基因组编辑 | KRAB-DNA甲基化酶催化结构域融合蛋白 | dCas9 | 融合抑制因子和甲基化酶;造成目的基因78%的稳定沉默 | [70] |
| 两侧分别融合KRAB和Dnmt3A-Dnmt3L融合DNA甲基化酶结构域 | dCas9 | 在干细胞分裂分化为神经细胞后仍维持DNA甲基化和基因抑制 | [71] |
| TET1甲基胞嘧啶双加氧酶1催化结构域 | dCas9 | dCas9- TET1靶向至BRCA1基因启动子后成功地上调其表达水平 | [72] |
| 人类组蛋白乙酰基转移酶p300催化核心 | dCas9 | 催化组蛋白H3 第27位赖氨酸残基的乙酰基化;基因表达上调 | [73] |
| 单碱基编辑 | 胞嘧啶脱氨酶,腺嘌呤脱氨酶 | nCas9 | 在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换 | [30,74-75] |
| CBE-UGI融合蛋白 | nCas9 | UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率 | [30] |
| 单链DNA结合结构域ssDBD融合到胞嘧啶脱氨酶与nCas9之间 | nCas9 | 通过延长ssDNA的暴露时间极大地提高CBEs的编辑效率;使CBEs的编辑窗口范围得到了提高 | [76] |
| 组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶 | nCas9 | 首次实现大肠杆菌中C到A 87.2%和哺乳动物细胞C到G 5.3%至53.0%的编辑效率 | [77] |
| 先导编辑 | 通过gRNA工程改造的pegRNA;与逆转录酶融合 | nCas9 | 改变pegRNA可实现精确的碱基替换、基因敲除和插入 | [27] |
| 通过gRNA工程改造的pegRNA;与逆转录酶和Rad51融合 | nCas9 | 融合单链DNA结合蛋白Rad51使PE2的编辑效率最高提升2.6倍 | [78] |
在CRISPR/Cas9的基因转录调控系统中,对Cas9蛋白工程化的dCas9分别在RuvC和HNH结构域发生突变使得RNP复合物仅能发挥靶序列识别功能,而无法切断核苷酸双链[29].nCas9(Cas9 nickase)是仅能使靶DNA发生单链断裂(single-strand break,SSB)的Cas9突变体,其中nCas9(H840A)和nCas9(D10A)分别是Cas9在HNH和RuvC结构域发生突变后的突变体蛋白,从而能够使该结构域失去内切酶活性,使DNA单链断裂,产生“缺口”[22,39].将dCas9或nCas9与各种效应蛋白如转录激活因子或抑制因子、表观遗传修饰因子、碱基编辑器甚至具有特定功能的酶等融合后,研究人员便能够借助CRISPR/Cas9系统对靶序列实现特定生物学功能,如表征转录基因乃至基因组上非编码区的功能、通过将碱基编辑器与nCas9融合能够在HDR活性低的细胞中进行精确高效的单碱基编辑等.因此本节将重点回顾与总结多种多样的效应蛋白为CRISPR/Cas9系统带来的多元化优化和发展,以及赋予该系统的多种新功能. ...
... 将dCas9与各种转录激活因子融合后,则可在gRNA的靶向作用下精确激活目的基因,该系统称为CRISPR激活系统或CRISPRa.最初的CRISPRa系统中dCas9仅融合了单个反式激活因子p65(也被称为RelA,是构成NF-κB转录因子家族的五种成员之一)或VP64 (由单纯疱疹病毒蛋白VP16的四个重复结构域构成),这些早期工作共同构成了最初的可编程dCas9基因激活工具箱,但效果差强人意[23-28,39-44,62,65-66].为了不断提高CRISPRa系统的激活效率,研究人员尝试了用各种方式将更复杂或多拷贝的激活结构域应用于CRISPRa.2015年,Chavez等[67]通过蛋白质理性设计,构建了三重融合激活因子VP64-p65-Rta(VPR)并应用于CRISPRa系统,其中Rta是来源于EB病毒(epstein-barr virus)的反式激活因子.与dCas9-VP64相比,dCas9-VPR使人胚胎肾细胞293(HEK 293T cells)的内源靶基因表达上调了22~320倍,同时证明了该系统也能够在多重基因的激活表达与刺激人诱导多功能干细胞(induced pluripotent stem cells,iPSCs)的神经元分化上发挥作用.同年,张锋研究团队[68]开发的基于CRISPRa的协同激活介导系统(synergistic activation mediator system, SAM)通过修饰sgRNA使其携带了两个MS2发夹二级结构,每个发夹结构都能招募一个MS2-p65-HSF1融合蛋白,其中包括反式激活因子p65、MS2结合蛋白以及热休克蛋白1(heat shock protein 1),最终证明该修饰后的gRNA与dCas9-VP64的协同作用能够使10个靶基因的表达均上调2倍以上.2022年,山东大学候进课题组[69]构建的CRISPR介导的蛋白质标签信号放大系统首次将SPY系统(包括SpyTag和SpyCatcher)和SunTag系统分别引入dCas9介导的CRISPRa和dCpf1(一种丧失核酸酶活性的Cas9同源蛋白)介导的CRISPRi系统并进行骨架长度和转录因子优化后,成功使酿酒酵母内目的基因转录水平分别提高了34.9倍以及受到几乎完全的抑制.其中,SunTag系统是由若干个拷贝的GCN4肽以及作为标签的抗GCN4抗体的单链可变片段scFv组成[95],与dCas9融合的多拷贝GCN4肽通过招募多拷贝的转录因子与GCN4抗体标签的融合蛋白从而进行转录调控,该系统除了与CRISPR系统结合后用于碱基编辑以及基因调控,还在哺乳动物和植物细胞的DNA的甲基化和去甲基化诱导中发挥作用[96-100];而SPY系统作为一种广泛使用的蛋白质组装系统首次在酿酒酵母的转录调控中得到了应用[69].可见在dCas9与转录因子融合的基础上,CRISPRa与CRISPRi系统能够利用融合多重结构域或融合蛋白质骨架系统的方式提高调控效率.而通过利用RNA与蛋白质间的相互作用,将多个适配子融合在gRNA中使其同时含有目的基因靶向序列和转录因子招募序列的方式能够产生与dCas9融合多个转录激活或抑制结构域的策略类似的效果[101].此外,针对不同细胞乃至不同dCas9同源蛋白,CRISPRa与CRISPRi系统均需对不同转录激活或抑制结构域乃至其组合进行优化才能发挥该系统的最大效能. ...
Highly efficient Cas9-mediated transcriptional programming
2
2015
... Functional optimization through fusion expression of effector proteins with CRISPR/Cas9 system
Table 2| 功能 | 效应蛋白或gRNA工程 | 系统 | 特征 | 参考文献 |
|---|
| 表达调控 | 无 | dCas9 | 原核生物基因抑制表达,真核生物基因抑制效率低 | [22] |
| KRAB,MXI1,TUP1抑制结构域 | dCas9 | 可在真核生物中抑制基因表达 | [62-63] |
| KRAB-MeCP1融合抑制结构域 | dCas9 | 在哺乳动物细胞中高效抑制目的基因表达 | [64] |
| p65,VP64激活结构域 | dCas9 | 激活基因表达,效率较低 | [65-66] |
| VP64-p65-Rta(VPR)融合激活结构域 | dCas9 | 与VP64相比提高了22~320倍;多重基因激活表达;刺激iPSCs神经元分化 | [67] |
| gRNA工程;gRNA上携带携带两个MS2发夹二级结构 | dCas9 | 每个发夹结构招募一个MS2-p65-HSF1融合激活蛋白;与VP64相比,使10个靶基因上调2倍以上 | [68] |
| SPY系统(包括SpyTag和SpyCatcher)和Med2激活结构域 | dCas9 | 招募多拷贝SpyCatcher和Med2融合蛋白使酿酒酵母目的基因表达提高34.9倍 | [69] |
| SunTag系统(包括GCN4肽和抗GCN4抗体的单链可变片段scFv)和MIG1抑制结构域 | dCpf1 | 招募多拷贝ScFv与MIG1融合蛋白使酿酒酵母目的基因受到95%的抑制 | [69] |
| 表观基因组编辑 | KRAB-DNA甲基化酶催化结构域融合蛋白 | dCas9 | 融合抑制因子和甲基化酶;造成目的基因78%的稳定沉默 | [70] |
| 两侧分别融合KRAB和Dnmt3A-Dnmt3L融合DNA甲基化酶结构域 | dCas9 | 在干细胞分裂分化为神经细胞后仍维持DNA甲基化和基因抑制 | [71] |
| TET1甲基胞嘧啶双加氧酶1催化结构域 | dCas9 | dCas9- TET1靶向至BRCA1基因启动子后成功地上调其表达水平 | [72] |
| 人类组蛋白乙酰基转移酶p300催化核心 | dCas9 | 催化组蛋白H3 第27位赖氨酸残基的乙酰基化;基因表达上调 | [73] |
| 单碱基编辑 | 胞嘧啶脱氨酶,腺嘌呤脱氨酶 | nCas9 | 在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换 | [30,74-75] |
| CBE-UGI融合蛋白 | nCas9 | UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率 | [30] |
| 单链DNA结合结构域ssDBD融合到胞嘧啶脱氨酶与nCas9之间 | nCas9 | 通过延长ssDNA的暴露时间极大地提高CBEs的编辑效率;使CBEs的编辑窗口范围得到了提高 | [76] |
| 组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶 | nCas9 | 首次实现大肠杆菌中C到A 87.2%和哺乳动物细胞C到G 5.3%至53.0%的编辑效率 | [77] |
| 先导编辑 | 通过gRNA工程改造的pegRNA;与逆转录酶融合 | nCas9 | 改变pegRNA可实现精确的碱基替换、基因敲除和插入 | [27] |
| 通过gRNA工程改造的pegRNA;与逆转录酶和Rad51融合 | nCas9 | 融合单链DNA结合蛋白Rad51使PE2的编辑效率最高提升2.6倍 | [78] |
在CRISPR/Cas9的基因转录调控系统中,对Cas9蛋白工程化的dCas9分别在RuvC和HNH结构域发生突变使得RNP复合物仅能发挥靶序列识别功能,而无法切断核苷酸双链[29].nCas9(Cas9 nickase)是仅能使靶DNA发生单链断裂(single-strand break,SSB)的Cas9突变体,其中nCas9(H840A)和nCas9(D10A)分别是Cas9在HNH和RuvC结构域发生突变后的突变体蛋白,从而能够使该结构域失去内切酶活性,使DNA单链断裂,产生“缺口”[22,39].将dCas9或nCas9与各种效应蛋白如转录激活因子或抑制因子、表观遗传修饰因子、碱基编辑器甚至具有特定功能的酶等融合后,研究人员便能够借助CRISPR/Cas9系统对靶序列实现特定生物学功能,如表征转录基因乃至基因组上非编码区的功能、通过将碱基编辑器与nCas9融合能够在HDR活性低的细胞中进行精确高效的单碱基编辑等.因此本节将重点回顾与总结多种多样的效应蛋白为CRISPR/Cas9系统带来的多元化优化和发展,以及赋予该系统的多种新功能. ...
... 将dCas9与各种转录激活因子融合后,则可在gRNA的靶向作用下精确激活目的基因,该系统称为CRISPR激活系统或CRISPRa.最初的CRISPRa系统中dCas9仅融合了单个反式激活因子p65(也被称为RelA,是构成NF-κB转录因子家族的五种成员之一)或VP64 (由单纯疱疹病毒蛋白VP16的四个重复结构域构成),这些早期工作共同构成了最初的可编程dCas9基因激活工具箱,但效果差强人意[23-28,39-44,62,65-66].为了不断提高CRISPRa系统的激活效率,研究人员尝试了用各种方式将更复杂或多拷贝的激活结构域应用于CRISPRa.2015年,Chavez等[67]通过蛋白质理性设计,构建了三重融合激活因子VP64-p65-Rta(VPR)并应用于CRISPRa系统,其中Rta是来源于EB病毒(epstein-barr virus)的反式激活因子.与dCas9-VP64相比,dCas9-VPR使人胚胎肾细胞293(HEK 293T cells)的内源靶基因表达上调了22~320倍,同时证明了该系统也能够在多重基因的激活表达与刺激人诱导多功能干细胞(induced pluripotent stem cells,iPSCs)的神经元分化上发挥作用.同年,张锋研究团队[68]开发的基于CRISPRa的协同激活介导系统(synergistic activation mediator system, SAM)通过修饰sgRNA使其携带了两个MS2发夹二级结构,每个发夹结构都能招募一个MS2-p65-HSF1融合蛋白,其中包括反式激活因子p65、MS2结合蛋白以及热休克蛋白1(heat shock protein 1),最终证明该修饰后的gRNA与dCas9-VP64的协同作用能够使10个靶基因的表达均上调2倍以上.2022年,山东大学候进课题组[69]构建的CRISPR介导的蛋白质标签信号放大系统首次将SPY系统(包括SpyTag和SpyCatcher)和SunTag系统分别引入dCas9介导的CRISPRa和dCpf1(一种丧失核酸酶活性的Cas9同源蛋白)介导的CRISPRi系统并进行骨架长度和转录因子优化后,成功使酿酒酵母内目的基因转录水平分别提高了34.9倍以及受到几乎完全的抑制.其中,SunTag系统是由若干个拷贝的GCN4肽以及作为标签的抗GCN4抗体的单链可变片段scFv组成[95],与dCas9融合的多拷贝GCN4肽通过招募多拷贝的转录因子与GCN4抗体标签的融合蛋白从而进行转录调控,该系统除了与CRISPR系统结合后用于碱基编辑以及基因调控,还在哺乳动物和植物细胞的DNA的甲基化和去甲基化诱导中发挥作用[96-100];而SPY系统作为一种广泛使用的蛋白质组装系统首次在酿酒酵母的转录调控中得到了应用[69].可见在dCas9与转录因子融合的基础上,CRISPRa与CRISPRi系统能够利用融合多重结构域或融合蛋白质骨架系统的方式提高调控效率.而通过利用RNA与蛋白质间的相互作用,将多个适配子融合在gRNA中使其同时含有目的基因靶向序列和转录因子招募序列的方式能够产生与dCas9融合多个转录激活或抑制结构域的策略类似的效果[101].此外,针对不同细胞乃至不同dCas9同源蛋白,CRISPRa与CRISPRi系统均需对不同转录激活或抑制结构域乃至其组合进行优化才能发挥该系统的最大效能. ...
Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex
2
2015
... Functional optimization through fusion expression of effector proteins with CRISPR/Cas9 system
Table 2| 功能 | 效应蛋白或gRNA工程 | 系统 | 特征 | 参考文献 |
|---|
| 表达调控 | 无 | dCas9 | 原核生物基因抑制表达,真核生物基因抑制效率低 | [22] |
| KRAB,MXI1,TUP1抑制结构域 | dCas9 | 可在真核生物中抑制基因表达 | [62-63] |
| KRAB-MeCP1融合抑制结构域 | dCas9 | 在哺乳动物细胞中高效抑制目的基因表达 | [64] |
| p65,VP64激活结构域 | dCas9 | 激活基因表达,效率较低 | [65-66] |
| VP64-p65-Rta(VPR)融合激活结构域 | dCas9 | 与VP64相比提高了22~320倍;多重基因激活表达;刺激iPSCs神经元分化 | [67] |
| gRNA工程;gRNA上携带携带两个MS2发夹二级结构 | dCas9 | 每个发夹结构招募一个MS2-p65-HSF1融合激活蛋白;与VP64相比,使10个靶基因上调2倍以上 | [68] |
| SPY系统(包括SpyTag和SpyCatcher)和Med2激活结构域 | dCas9 | 招募多拷贝SpyCatcher和Med2融合蛋白使酿酒酵母目的基因表达提高34.9倍 | [69] |
| SunTag系统(包括GCN4肽和抗GCN4抗体的单链可变片段scFv)和MIG1抑制结构域 | dCpf1 | 招募多拷贝ScFv与MIG1融合蛋白使酿酒酵母目的基因受到95%的抑制 | [69] |
| 表观基因组编辑 | KRAB-DNA甲基化酶催化结构域融合蛋白 | dCas9 | 融合抑制因子和甲基化酶;造成目的基因78%的稳定沉默 | [70] |
| 两侧分别融合KRAB和Dnmt3A-Dnmt3L融合DNA甲基化酶结构域 | dCas9 | 在干细胞分裂分化为神经细胞后仍维持DNA甲基化和基因抑制 | [71] |
| TET1甲基胞嘧啶双加氧酶1催化结构域 | dCas9 | dCas9- TET1靶向至BRCA1基因启动子后成功地上调其表达水平 | [72] |
| 人类组蛋白乙酰基转移酶p300催化核心 | dCas9 | 催化组蛋白H3 第27位赖氨酸残基的乙酰基化;基因表达上调 | [73] |
| 单碱基编辑 | 胞嘧啶脱氨酶,腺嘌呤脱氨酶 | nCas9 | 在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换 | [30,74-75] |
| CBE-UGI融合蛋白 | nCas9 | UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率 | [30] |
| 单链DNA结合结构域ssDBD融合到胞嘧啶脱氨酶与nCas9之间 | nCas9 | 通过延长ssDNA的暴露时间极大地提高CBEs的编辑效率;使CBEs的编辑窗口范围得到了提高 | [76] |
| 组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶 | nCas9 | 首次实现大肠杆菌中C到A 87.2%和哺乳动物细胞C到G 5.3%至53.0%的编辑效率 | [77] |
| 先导编辑 | 通过gRNA工程改造的pegRNA;与逆转录酶融合 | nCas9 | 改变pegRNA可实现精确的碱基替换、基因敲除和插入 | [27] |
| 通过gRNA工程改造的pegRNA;与逆转录酶和Rad51融合 | nCas9 | 融合单链DNA结合蛋白Rad51使PE2的编辑效率最高提升2.6倍 | [78] |
在CRISPR/Cas9的基因转录调控系统中,对Cas9蛋白工程化的dCas9分别在RuvC和HNH结构域发生突变使得RNP复合物仅能发挥靶序列识别功能,而无法切断核苷酸双链[29].nCas9(Cas9 nickase)是仅能使靶DNA发生单链断裂(single-strand break,SSB)的Cas9突变体,其中nCas9(H840A)和nCas9(D10A)分别是Cas9在HNH和RuvC结构域发生突变后的突变体蛋白,从而能够使该结构域失去内切酶活性,使DNA单链断裂,产生“缺口”[22,39].将dCas9或nCas9与各种效应蛋白如转录激活因子或抑制因子、表观遗传修饰因子、碱基编辑器甚至具有特定功能的酶等融合后,研究人员便能够借助CRISPR/Cas9系统对靶序列实现特定生物学功能,如表征转录基因乃至基因组上非编码区的功能、通过将碱基编辑器与nCas9融合能够在HDR活性低的细胞中进行精确高效的单碱基编辑等.因此本节将重点回顾与总结多种多样的效应蛋白为CRISPR/Cas9系统带来的多元化优化和发展,以及赋予该系统的多种新功能. ...
... 将dCas9与各种转录激活因子融合后,则可在gRNA的靶向作用下精确激活目的基因,该系统称为CRISPR激活系统或CRISPRa.最初的CRISPRa系统中dCas9仅融合了单个反式激活因子p65(也被称为RelA,是构成NF-κB转录因子家族的五种成员之一)或VP64 (由单纯疱疹病毒蛋白VP16的四个重复结构域构成),这些早期工作共同构成了最初的可编程dCas9基因激活工具箱,但效果差强人意[23-28,39-44,62,65-66].为了不断提高CRISPRa系统的激活效率,研究人员尝试了用各种方式将更复杂或多拷贝的激活结构域应用于CRISPRa.2015年,Chavez等[67]通过蛋白质理性设计,构建了三重融合激活因子VP64-p65-Rta(VPR)并应用于CRISPRa系统,其中Rta是来源于EB病毒(epstein-barr virus)的反式激活因子.与dCas9-VP64相比,dCas9-VPR使人胚胎肾细胞293(HEK 293T cells)的内源靶基因表达上调了22~320倍,同时证明了该系统也能够在多重基因的激活表达与刺激人诱导多功能干细胞(induced pluripotent stem cells,iPSCs)的神经元分化上发挥作用.同年,张锋研究团队[68]开发的基于CRISPRa的协同激活介导系统(synergistic activation mediator system, SAM)通过修饰sgRNA使其携带了两个MS2发夹二级结构,每个发夹结构都能招募一个MS2-p65-HSF1融合蛋白,其中包括反式激活因子p65、MS2结合蛋白以及热休克蛋白1(heat shock protein 1),最终证明该修饰后的gRNA与dCas9-VP64的协同作用能够使10个靶基因的表达均上调2倍以上.2022年,山东大学候进课题组[69]构建的CRISPR介导的蛋白质标签信号放大系统首次将SPY系统(包括SpyTag和SpyCatcher)和SunTag系统分别引入dCas9介导的CRISPRa和dCpf1(一种丧失核酸酶活性的Cas9同源蛋白)介导的CRISPRi系统并进行骨架长度和转录因子优化后,成功使酿酒酵母内目的基因转录水平分别提高了34.9倍以及受到几乎完全的抑制.其中,SunTag系统是由若干个拷贝的GCN4肽以及作为标签的抗GCN4抗体的单链可变片段scFv组成[95],与dCas9融合的多拷贝GCN4肽通过招募多拷贝的转录因子与GCN4抗体标签的融合蛋白从而进行转录调控,该系统除了与CRISPR系统结合后用于碱基编辑以及基因调控,还在哺乳动物和植物细胞的DNA的甲基化和去甲基化诱导中发挥作用[96-100];而SPY系统作为一种广泛使用的蛋白质组装系统首次在酿酒酵母的转录调控中得到了应用[69].可见在dCas9与转录因子融合的基础上,CRISPRa与CRISPRi系统能够利用融合多重结构域或融合蛋白质骨架系统的方式提高调控效率.而通过利用RNA与蛋白质间的相互作用,将多个适配子融合在gRNA中使其同时含有目的基因靶向序列和转录因子招募序列的方式能够产生与dCas9融合多个转录激活或抑制结构域的策略类似的效果[101].此外,针对不同细胞乃至不同dCas9同源蛋白,CRISPRa与CRISPRi系统均需对不同转录激活或抑制结构域乃至其组合进行优化才能发挥该系统的最大效能. ...
CRISPR-mediated protein-tagging signal amplification systems for efficient transcriptional activation and repression in Saccharomyces cerevisiae
4
2022
... Functional optimization through fusion expression of effector proteins with CRISPR/Cas9 system
Table 2| 功能 | 效应蛋白或gRNA工程 | 系统 | 特征 | 参考文献 |
|---|
| 表达调控 | 无 | dCas9 | 原核生物基因抑制表达,真核生物基因抑制效率低 | [22] |
| KRAB,MXI1,TUP1抑制结构域 | dCas9 | 可在真核生物中抑制基因表达 | [62-63] |
| KRAB-MeCP1融合抑制结构域 | dCas9 | 在哺乳动物细胞中高效抑制目的基因表达 | [64] |
| p65,VP64激活结构域 | dCas9 | 激活基因表达,效率较低 | [65-66] |
| VP64-p65-Rta(VPR)融合激活结构域 | dCas9 | 与VP64相比提高了22~320倍;多重基因激活表达;刺激iPSCs神经元分化 | [67] |
| gRNA工程;gRNA上携带携带两个MS2发夹二级结构 | dCas9 | 每个发夹结构招募一个MS2-p65-HSF1融合激活蛋白;与VP64相比,使10个靶基因上调2倍以上 | [68] |
| SPY系统(包括SpyTag和SpyCatcher)和Med2激活结构域 | dCas9 | 招募多拷贝SpyCatcher和Med2融合蛋白使酿酒酵母目的基因表达提高34.9倍 | [69] |
| SunTag系统(包括GCN4肽和抗GCN4抗体的单链可变片段scFv)和MIG1抑制结构域 | dCpf1 | 招募多拷贝ScFv与MIG1融合蛋白使酿酒酵母目的基因受到95%的抑制 | [69] |
| 表观基因组编辑 | KRAB-DNA甲基化酶催化结构域融合蛋白 | dCas9 | 融合抑制因子和甲基化酶;造成目的基因78%的稳定沉默 | [70] |
| 两侧分别融合KRAB和Dnmt3A-Dnmt3L融合DNA甲基化酶结构域 | dCas9 | 在干细胞分裂分化为神经细胞后仍维持DNA甲基化和基因抑制 | [71] |
| TET1甲基胞嘧啶双加氧酶1催化结构域 | dCas9 | dCas9- TET1靶向至BRCA1基因启动子后成功地上调其表达水平 | [72] |
| 人类组蛋白乙酰基转移酶p300催化核心 | dCas9 | 催化组蛋白H3 第27位赖氨酸残基的乙酰基化;基因表达上调 | [73] |
| 单碱基编辑 | 胞嘧啶脱氨酶,腺嘌呤脱氨酶 | nCas9 | 在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换 | [30,74-75] |
| CBE-UGI融合蛋白 | nCas9 | UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率 | [30] |
| 单链DNA结合结构域ssDBD融合到胞嘧啶脱氨酶与nCas9之间 | nCas9 | 通过延长ssDNA的暴露时间极大地提高CBEs的编辑效率;使CBEs的编辑窗口范围得到了提高 | [76] |
| 组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶 | nCas9 | 首次实现大肠杆菌中C到A 87.2%和哺乳动物细胞C到G 5.3%至53.0%的编辑效率 | [77] |
| 先导编辑 | 通过gRNA工程改造的pegRNA;与逆转录酶融合 | nCas9 | 改变pegRNA可实现精确的碱基替换、基因敲除和插入 | [27] |
| 通过gRNA工程改造的pegRNA;与逆转录酶和Rad51融合 | nCas9 | 融合单链DNA结合蛋白Rad51使PE2的编辑效率最高提升2.6倍 | [78] |
在CRISPR/Cas9的基因转录调控系统中,对Cas9蛋白工程化的dCas9分别在RuvC和HNH结构域发生突变使得RNP复合物仅能发挥靶序列识别功能,而无法切断核苷酸双链[29].nCas9(Cas9 nickase)是仅能使靶DNA发生单链断裂(single-strand break,SSB)的Cas9突变体,其中nCas9(H840A)和nCas9(D10A)分别是Cas9在HNH和RuvC结构域发生突变后的突变体蛋白,从而能够使该结构域失去内切酶活性,使DNA单链断裂,产生“缺口”[22,39].将dCas9或nCas9与各种效应蛋白如转录激活因子或抑制因子、表观遗传修饰因子、碱基编辑器甚至具有特定功能的酶等融合后,研究人员便能够借助CRISPR/Cas9系统对靶序列实现特定生物学功能,如表征转录基因乃至基因组上非编码区的功能、通过将碱基编辑器与nCas9融合能够在HDR活性低的细胞中进行精确高效的单碱基编辑等.因此本节将重点回顾与总结多种多样的效应蛋白为CRISPR/Cas9系统带来的多元化优化和发展,以及赋予该系统的多种新功能. ...
... [
69]
| 表观基因组编辑 | KRAB-DNA甲基化酶催化结构域融合蛋白 | dCas9 | 融合抑制因子和甲基化酶;造成目的基因78%的稳定沉默 | [70] |
| 两侧分别融合KRAB和Dnmt3A-Dnmt3L融合DNA甲基化酶结构域 | dCas9 | 在干细胞分裂分化为神经细胞后仍维持DNA甲基化和基因抑制 | [71] |
| TET1甲基胞嘧啶双加氧酶1催化结构域 | dCas9 | dCas9- TET1靶向至BRCA1基因启动子后成功地上调其表达水平 | [72] |
| 人类组蛋白乙酰基转移酶p300催化核心 | dCas9 | 催化组蛋白H3 第27位赖氨酸残基的乙酰基化;基因表达上调 | [73] |
| 单碱基编辑 | 胞嘧啶脱氨酶,腺嘌呤脱氨酶 | nCas9 | 在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换 | [30,74-75] |
| CBE-UGI融合蛋白 | nCas9 | UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率 | [30] |
| 单链DNA结合结构域ssDBD融合到胞嘧啶脱氨酶与nCas9之间 | nCas9 | 通过延长ssDNA的暴露时间极大地提高CBEs的编辑效率;使CBEs的编辑窗口范围得到了提高 | [76] |
| 组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶 | nCas9 | 首次实现大肠杆菌中C到A 87.2%和哺乳动物细胞C到G 5.3%至53.0%的编辑效率 | [77] |
| 先导编辑 | 通过gRNA工程改造的pegRNA;与逆转录酶融合 | nCas9 | 改变pegRNA可实现精确的碱基替换、基因敲除和插入 | [27] |
| 通过gRNA工程改造的pegRNA;与逆转录酶和Rad51融合 | nCas9 | 融合单链DNA结合蛋白Rad51使PE2的编辑效率最高提升2.6倍 | [78] |
在CRISPR/Cas9的基因转录调控系统中,对Cas9蛋白工程化的dCas9分别在RuvC和HNH结构域发生突变使得RNP复合物仅能发挥靶序列识别功能,而无法切断核苷酸双链[29].nCas9(Cas9 nickase)是仅能使靶DNA发生单链断裂(single-strand break,SSB)的Cas9突变体,其中nCas9(H840A)和nCas9(D10A)分别是Cas9在HNH和RuvC结构域发生突变后的突变体蛋白,从而能够使该结构域失去内切酶活性,使DNA单链断裂,产生“缺口”[22,39].将dCas9或nCas9与各种效应蛋白如转录激活因子或抑制因子、表观遗传修饰因子、碱基编辑器甚至具有特定功能的酶等融合后,研究人员便能够借助CRISPR/Cas9系统对靶序列实现特定生物学功能,如表征转录基因乃至基因组上非编码区的功能、通过将碱基编辑器与nCas9融合能够在HDR活性低的细胞中进行精确高效的单碱基编辑等.因此本节将重点回顾与总结多种多样的效应蛋白为CRISPR/Cas9系统带来的多元化优化和发展,以及赋予该系统的多种新功能. ...
... 将dCas9与各种转录激活因子融合后,则可在gRNA的靶向作用下精确激活目的基因,该系统称为CRISPR激活系统或CRISPRa.最初的CRISPRa系统中dCas9仅融合了单个反式激活因子p65(也被称为RelA,是构成NF-κB转录因子家族的五种成员之一)或VP64 (由单纯疱疹病毒蛋白VP16的四个重复结构域构成),这些早期工作共同构成了最初的可编程dCas9基因激活工具箱,但效果差强人意[23-28,39-44,62,65-66].为了不断提高CRISPRa系统的激活效率,研究人员尝试了用各种方式将更复杂或多拷贝的激活结构域应用于CRISPRa.2015年,Chavez等[67]通过蛋白质理性设计,构建了三重融合激活因子VP64-p65-Rta(VPR)并应用于CRISPRa系统,其中Rta是来源于EB病毒(epstein-barr virus)的反式激活因子.与dCas9-VP64相比,dCas9-VPR使人胚胎肾细胞293(HEK 293T cells)的内源靶基因表达上调了22~320倍,同时证明了该系统也能够在多重基因的激活表达与刺激人诱导多功能干细胞(induced pluripotent stem cells,iPSCs)的神经元分化上发挥作用.同年,张锋研究团队[68]开发的基于CRISPRa的协同激活介导系统(synergistic activation mediator system, SAM)通过修饰sgRNA使其携带了两个MS2发夹二级结构,每个发夹结构都能招募一个MS2-p65-HSF1融合蛋白,其中包括反式激活因子p65、MS2结合蛋白以及热休克蛋白1(heat shock protein 1),最终证明该修饰后的gRNA与dCas9-VP64的协同作用能够使10个靶基因的表达均上调2倍以上.2022年,山东大学候进课题组[69]构建的CRISPR介导的蛋白质标签信号放大系统首次将SPY系统(包括SpyTag和SpyCatcher)和SunTag系统分别引入dCas9介导的CRISPRa和dCpf1(一种丧失核酸酶活性的Cas9同源蛋白)介导的CRISPRi系统并进行骨架长度和转录因子优化后,成功使酿酒酵母内目的基因转录水平分别提高了34.9倍以及受到几乎完全的抑制.其中,SunTag系统是由若干个拷贝的GCN4肽以及作为标签的抗GCN4抗体的单链可变片段scFv组成[95],与dCas9融合的多拷贝GCN4肽通过招募多拷贝的转录因子与GCN4抗体标签的融合蛋白从而进行转录调控,该系统除了与CRISPR系统结合后用于碱基编辑以及基因调控,还在哺乳动物和植物细胞的DNA的甲基化和去甲基化诱导中发挥作用[96-100];而SPY系统作为一种广泛使用的蛋白质组装系统首次在酿酒酵母的转录调控中得到了应用[69].可见在dCas9与转录因子融合的基础上,CRISPRa与CRISPRi系统能够利用融合多重结构域或融合蛋白质骨架系统的方式提高调控效率.而通过利用RNA与蛋白质间的相互作用,将多个适配子融合在gRNA中使其同时含有目的基因靶向序列和转录因子招募序列的方式能够产生与dCas9融合多个转录激活或抑制结构域的策略类似的效果[101].此外,针对不同细胞乃至不同dCas9同源蛋白,CRISPRa与CRISPRi系统均需对不同转录激活或抑制结构域乃至其组合进行优化才能发挥该系统的最大效能. ...
... [69].可见在dCas9与转录因子融合的基础上,CRISPRa与CRISPRi系统能够利用融合多重结构域或融合蛋白质骨架系统的方式提高调控效率.而通过利用RNA与蛋白质间的相互作用,将多个适配子融合在gRNA中使其同时含有目的基因靶向序列和转录因子招募序列的方式能够产生与dCas9融合多个转录激活或抑制结构域的策略类似的效果[101].此外,针对不同细胞乃至不同dCas9同源蛋白,CRISPRa与CRISPRi系统均需对不同转录激活或抑制结构域乃至其组合进行优化才能发挥该系统的最大效能. ...
Inheritable silencing of endogenous genes by hit-and-run targeted epigenetic editing
2
2016
... Functional optimization through fusion expression of effector proteins with CRISPR/Cas9 system
Table 2| 功能 | 效应蛋白或gRNA工程 | 系统 | 特征 | 参考文献 |
|---|
| 表达调控 | 无 | dCas9 | 原核生物基因抑制表达,真核生物基因抑制效率低 | [22] |
| KRAB,MXI1,TUP1抑制结构域 | dCas9 | 可在真核生物中抑制基因表达 | [62-63] |
| KRAB-MeCP1融合抑制结构域 | dCas9 | 在哺乳动物细胞中高效抑制目的基因表达 | [64] |
| p65,VP64激活结构域 | dCas9 | 激活基因表达,效率较低 | [65-66] |
| VP64-p65-Rta(VPR)融合激活结构域 | dCas9 | 与VP64相比提高了22~320倍;多重基因激活表达;刺激iPSCs神经元分化 | [67] |
| gRNA工程;gRNA上携带携带两个MS2发夹二级结构 | dCas9 | 每个发夹结构招募一个MS2-p65-HSF1融合激活蛋白;与VP64相比,使10个靶基因上调2倍以上 | [68] |
| SPY系统(包括SpyTag和SpyCatcher)和Med2激活结构域 | dCas9 | 招募多拷贝SpyCatcher和Med2融合蛋白使酿酒酵母目的基因表达提高34.9倍 | [69] |
| SunTag系统(包括GCN4肽和抗GCN4抗体的单链可变片段scFv)和MIG1抑制结构域 | dCpf1 | 招募多拷贝ScFv与MIG1融合蛋白使酿酒酵母目的基因受到95%的抑制 | [69] |
| 表观基因组编辑 | KRAB-DNA甲基化酶催化结构域融合蛋白 | dCas9 | 融合抑制因子和甲基化酶;造成目的基因78%的稳定沉默 | [70] |
| 两侧分别融合KRAB和Dnmt3A-Dnmt3L融合DNA甲基化酶结构域 | dCas9 | 在干细胞分裂分化为神经细胞后仍维持DNA甲基化和基因抑制 | [71] |
| TET1甲基胞嘧啶双加氧酶1催化结构域 | dCas9 | dCas9- TET1靶向至BRCA1基因启动子后成功地上调其表达水平 | [72] |
| 人类组蛋白乙酰基转移酶p300催化核心 | dCas9 | 催化组蛋白H3 第27位赖氨酸残基的乙酰基化;基因表达上调 | [73] |
| 单碱基编辑 | 胞嘧啶脱氨酶,腺嘌呤脱氨酶 | nCas9 | 在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换 | [30,74-75] |
| CBE-UGI融合蛋白 | nCas9 | UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率 | [30] |
| 单链DNA结合结构域ssDBD融合到胞嘧啶脱氨酶与nCas9之间 | nCas9 | 通过延长ssDNA的暴露时间极大地提高CBEs的编辑效率;使CBEs的编辑窗口范围得到了提高 | [76] |
| 组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶 | nCas9 | 首次实现大肠杆菌中C到A 87.2%和哺乳动物细胞C到G 5.3%至53.0%的编辑效率 | [77] |
| 先导编辑 | 通过gRNA工程改造的pegRNA;与逆转录酶融合 | nCas9 | 改变pegRNA可实现精确的碱基替换、基因敲除和插入 | [27] |
| 通过gRNA工程改造的pegRNA;与逆转录酶和Rad51融合 | nCas9 | 融合单链DNA结合蛋白Rad51使PE2的编辑效率最高提升2.6倍 | [78] |
在CRISPR/Cas9的基因转录调控系统中,对Cas9蛋白工程化的dCas9分别在RuvC和HNH结构域发生突变使得RNP复合物仅能发挥靶序列识别功能,而无法切断核苷酸双链[29].nCas9(Cas9 nickase)是仅能使靶DNA发生单链断裂(single-strand break,SSB)的Cas9突变体,其中nCas9(H840A)和nCas9(D10A)分别是Cas9在HNH和RuvC结构域发生突变后的突变体蛋白,从而能够使该结构域失去内切酶活性,使DNA单链断裂,产生“缺口”[22,39].将dCas9或nCas9与各种效应蛋白如转录激活因子或抑制因子、表观遗传修饰因子、碱基编辑器甚至具有特定功能的酶等融合后,研究人员便能够借助CRISPR/Cas9系统对靶序列实现特定生物学功能,如表征转录基因乃至基因组上非编码区的功能、通过将碱基编辑器与nCas9融合能够在HDR活性低的细胞中进行精确高效的单碱基编辑等.因此本节将重点回顾与总结多种多样的效应蛋白为CRISPR/Cas9系统带来的多元化优化和发展,以及赋予该系统的多种新功能. ...
... 通过使用基于CRISPR/dCas9系统的一系列融合蛋白工具可以实现诸多靶向的表观遗传修饰,如DNA甲基化和去甲基化、组蛋白乙酰基化/去乙酰化和甲基化/去甲基化等,从而调控基因的表达并在许多细胞过程、癌症治疗及细胞工厂中发挥关键作用(图2)[30].其中DNA甲基化如5′胞嘧啶甲基化(5mC)是一种稳定的抑制性化学修饰调控.2016年,研究人员将dCas9、KRAB转录抑制因子以及DNA甲基化酶(DNA methyltransferase)的催化结构域融合蛋白基因与7个gRNA在K562细胞中共表达后获得了β2-微球蛋白启动子-增强子78%的稳定沉默[70].2021年,Nunez等[71]开发的CRISPRoff甲基化修饰工具能够在细胞分裂及分化过程中稳定维持靶基因的甲基化并抑制其表达水平.DNA去甲基化是将甲基胞嘧啶双加氧酶1(methylcytosine dioxygenase 1,TET1)的肽链C端催化结构域与dCas9融合后,再靶向至目的基因启动子或增强子区,从而实现目的基因的表达上调.2016年,Choudhury等[72]将dCas9- TET1靶向至乳腺癌基因1(breast cancer 1,BRCA1)启动子后成功地上调其表达水平.BRCA1作为抑制恶性肿瘤发生的关键基因之一,其成功的靶向上调为研究癌变新疗法带来新的思路,证明了通过去甲基化靶向激活人类肿瘤抑制基因的疗法是可行的.同样的,将dCas9与人类组蛋白乙酰基转移酶p300的催化核心融合后靶向至启动子和增强子区从而催化组蛋白H3 第27位赖氨酸残基的乙酰基化也会造成较强的基因上调[73].2019年,Chen等[102]通过提高麦角生物碱合成基因簇(ergot alkaloid synthesis gene cluster)的组蛋白乙酰化水平,使麦角菌Cp-1菌株(Claviceps pururea Cp-1 strain)中的麦角生物碱含量提高了一倍.通过融合dCas9与适当的表观遗传效应蛋白,CRISPR表观遗传工程工具则可利用CRISPR系统稳健地靶向性实现各种所需的表观遗传修饰,而不同工具间的适当组合,理论上讲可以为表观遗传学工具箱带来更多新型和功能复杂的应用策略,如利用与CRISPRa和CRISPRi优化策略类似的方式优化CRISPR表观遗传工程工具箱的特异性、稳定性和性能[103]. ...
Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing
2
2021
... Functional optimization through fusion expression of effector proteins with CRISPR/Cas9 system
Table 2| 功能 | 效应蛋白或gRNA工程 | 系统 | 特征 | 参考文献 |
|---|
| 表达调控 | 无 | dCas9 | 原核生物基因抑制表达,真核生物基因抑制效率低 | [22] |
| KRAB,MXI1,TUP1抑制结构域 | dCas9 | 可在真核生物中抑制基因表达 | [62-63] |
| KRAB-MeCP1融合抑制结构域 | dCas9 | 在哺乳动物细胞中高效抑制目的基因表达 | [64] |
| p65,VP64激活结构域 | dCas9 | 激活基因表达,效率较低 | [65-66] |
| VP64-p65-Rta(VPR)融合激活结构域 | dCas9 | 与VP64相比提高了22~320倍;多重基因激活表达;刺激iPSCs神经元分化 | [67] |
| gRNA工程;gRNA上携带携带两个MS2发夹二级结构 | dCas9 | 每个发夹结构招募一个MS2-p65-HSF1融合激活蛋白;与VP64相比,使10个靶基因上调2倍以上 | [68] |
| SPY系统(包括SpyTag和SpyCatcher)和Med2激活结构域 | dCas9 | 招募多拷贝SpyCatcher和Med2融合蛋白使酿酒酵母目的基因表达提高34.9倍 | [69] |
| SunTag系统(包括GCN4肽和抗GCN4抗体的单链可变片段scFv)和MIG1抑制结构域 | dCpf1 | 招募多拷贝ScFv与MIG1融合蛋白使酿酒酵母目的基因受到95%的抑制 | [69] |
| 表观基因组编辑 | KRAB-DNA甲基化酶催化结构域融合蛋白 | dCas9 | 融合抑制因子和甲基化酶;造成目的基因78%的稳定沉默 | [70] |
| 两侧分别融合KRAB和Dnmt3A-Dnmt3L融合DNA甲基化酶结构域 | dCas9 | 在干细胞分裂分化为神经细胞后仍维持DNA甲基化和基因抑制 | [71] |
| TET1甲基胞嘧啶双加氧酶1催化结构域 | dCas9 | dCas9- TET1靶向至BRCA1基因启动子后成功地上调其表达水平 | [72] |
| 人类组蛋白乙酰基转移酶p300催化核心 | dCas9 | 催化组蛋白H3 第27位赖氨酸残基的乙酰基化;基因表达上调 | [73] |
| 单碱基编辑 | 胞嘧啶脱氨酶,腺嘌呤脱氨酶 | nCas9 | 在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换 | [30,74-75] |
| CBE-UGI融合蛋白 | nCas9 | UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率 | [30] |
| 单链DNA结合结构域ssDBD融合到胞嘧啶脱氨酶与nCas9之间 | nCas9 | 通过延长ssDNA的暴露时间极大地提高CBEs的编辑效率;使CBEs的编辑窗口范围得到了提高 | [76] |
| 组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶 | nCas9 | 首次实现大肠杆菌中C到A 87.2%和哺乳动物细胞C到G 5.3%至53.0%的编辑效率 | [77] |
| 先导编辑 | 通过gRNA工程改造的pegRNA;与逆转录酶融合 | nCas9 | 改变pegRNA可实现精确的碱基替换、基因敲除和插入 | [27] |
| 通过gRNA工程改造的pegRNA;与逆转录酶和Rad51融合 | nCas9 | 融合单链DNA结合蛋白Rad51使PE2的编辑效率最高提升2.6倍 | [78] |
在CRISPR/Cas9的基因转录调控系统中,对Cas9蛋白工程化的dCas9分别在RuvC和HNH结构域发生突变使得RNP复合物仅能发挥靶序列识别功能,而无法切断核苷酸双链[29].nCas9(Cas9 nickase)是仅能使靶DNA发生单链断裂(single-strand break,SSB)的Cas9突变体,其中nCas9(H840A)和nCas9(D10A)分别是Cas9在HNH和RuvC结构域发生突变后的突变体蛋白,从而能够使该结构域失去内切酶活性,使DNA单链断裂,产生“缺口”[22,39].将dCas9或nCas9与各种效应蛋白如转录激活因子或抑制因子、表观遗传修饰因子、碱基编辑器甚至具有特定功能的酶等融合后,研究人员便能够借助CRISPR/Cas9系统对靶序列实现特定生物学功能,如表征转录基因乃至基因组上非编码区的功能、通过将碱基编辑器与nCas9融合能够在HDR活性低的细胞中进行精确高效的单碱基编辑等.因此本节将重点回顾与总结多种多样的效应蛋白为CRISPR/Cas9系统带来的多元化优化和发展,以及赋予该系统的多种新功能. ...
... 通过使用基于CRISPR/dCas9系统的一系列融合蛋白工具可以实现诸多靶向的表观遗传修饰,如DNA甲基化和去甲基化、组蛋白乙酰基化/去乙酰化和甲基化/去甲基化等,从而调控基因的表达并在许多细胞过程、癌症治疗及细胞工厂中发挥关键作用(图2)[30].其中DNA甲基化如5′胞嘧啶甲基化(5mC)是一种稳定的抑制性化学修饰调控.2016年,研究人员将dCas9、KRAB转录抑制因子以及DNA甲基化酶(DNA methyltransferase)的催化结构域融合蛋白基因与7个gRNA在K562细胞中共表达后获得了β2-微球蛋白启动子-增强子78%的稳定沉默[70].2021年,Nunez等[71]开发的CRISPRoff甲基化修饰工具能够在细胞分裂及分化过程中稳定维持靶基因的甲基化并抑制其表达水平.DNA去甲基化是将甲基胞嘧啶双加氧酶1(methylcytosine dioxygenase 1,TET1)的肽链C端催化结构域与dCas9融合后,再靶向至目的基因启动子或增强子区,从而实现目的基因的表达上调.2016年,Choudhury等[72]将dCas9- TET1靶向至乳腺癌基因1(breast cancer 1,BRCA1)启动子后成功地上调其表达水平.BRCA1作为抑制恶性肿瘤发生的关键基因之一,其成功的靶向上调为研究癌变新疗法带来新的思路,证明了通过去甲基化靶向激活人类肿瘤抑制基因的疗法是可行的.同样的,将dCas9与人类组蛋白乙酰基转移酶p300的催化核心融合后靶向至启动子和增强子区从而催化组蛋白H3 第27位赖氨酸残基的乙酰基化也会造成较强的基因上调[73].2019年,Chen等[102]通过提高麦角生物碱合成基因簇(ergot alkaloid synthesis gene cluster)的组蛋白乙酰化水平,使麦角菌Cp-1菌株(Claviceps pururea Cp-1 strain)中的麦角生物碱含量提高了一倍.通过融合dCas9与适当的表观遗传效应蛋白,CRISPR表观遗传工程工具则可利用CRISPR系统稳健地靶向性实现各种所需的表观遗传修饰,而不同工具间的适当组合,理论上讲可以为表观遗传学工具箱带来更多新型和功能复杂的应用策略,如利用与CRISPRa和CRISPRi优化策略类似的方式优化CRISPR表观遗传工程工具箱的特异性、稳定性和性能[103]. ...
CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter
2
2016
... Functional optimization through fusion expression of effector proteins with CRISPR/Cas9 system
Table 2| 功能 | 效应蛋白或gRNA工程 | 系统 | 特征 | 参考文献 |
|---|
| 表达调控 | 无 | dCas9 | 原核生物基因抑制表达,真核生物基因抑制效率低 | [22] |
| KRAB,MXI1,TUP1抑制结构域 | dCas9 | 可在真核生物中抑制基因表达 | [62-63] |
| KRAB-MeCP1融合抑制结构域 | dCas9 | 在哺乳动物细胞中高效抑制目的基因表达 | [64] |
| p65,VP64激活结构域 | dCas9 | 激活基因表达,效率较低 | [65-66] |
| VP64-p65-Rta(VPR)融合激活结构域 | dCas9 | 与VP64相比提高了22~320倍;多重基因激活表达;刺激iPSCs神经元分化 | [67] |
| gRNA工程;gRNA上携带携带两个MS2发夹二级结构 | dCas9 | 每个发夹结构招募一个MS2-p65-HSF1融合激活蛋白;与VP64相比,使10个靶基因上调2倍以上 | [68] |
| SPY系统(包括SpyTag和SpyCatcher)和Med2激活结构域 | dCas9 | 招募多拷贝SpyCatcher和Med2融合蛋白使酿酒酵母目的基因表达提高34.9倍 | [69] |
| SunTag系统(包括GCN4肽和抗GCN4抗体的单链可变片段scFv)和MIG1抑制结构域 | dCpf1 | 招募多拷贝ScFv与MIG1融合蛋白使酿酒酵母目的基因受到95%的抑制 | [69] |
| 表观基因组编辑 | KRAB-DNA甲基化酶催化结构域融合蛋白 | dCas9 | 融合抑制因子和甲基化酶;造成目的基因78%的稳定沉默 | [70] |
| 两侧分别融合KRAB和Dnmt3A-Dnmt3L融合DNA甲基化酶结构域 | dCas9 | 在干细胞分裂分化为神经细胞后仍维持DNA甲基化和基因抑制 | [71] |
| TET1甲基胞嘧啶双加氧酶1催化结构域 | dCas9 | dCas9- TET1靶向至BRCA1基因启动子后成功地上调其表达水平 | [72] |
| 人类组蛋白乙酰基转移酶p300催化核心 | dCas9 | 催化组蛋白H3 第27位赖氨酸残基的乙酰基化;基因表达上调 | [73] |
| 单碱基编辑 | 胞嘧啶脱氨酶,腺嘌呤脱氨酶 | nCas9 | 在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换 | [30,74-75] |
| CBE-UGI融合蛋白 | nCas9 | UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率 | [30] |
| 单链DNA结合结构域ssDBD融合到胞嘧啶脱氨酶与nCas9之间 | nCas9 | 通过延长ssDNA的暴露时间极大地提高CBEs的编辑效率;使CBEs的编辑窗口范围得到了提高 | [76] |
| 组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶 | nCas9 | 首次实现大肠杆菌中C到A 87.2%和哺乳动物细胞C到G 5.3%至53.0%的编辑效率 | [77] |
| 先导编辑 | 通过gRNA工程改造的pegRNA;与逆转录酶融合 | nCas9 | 改变pegRNA可实现精确的碱基替换、基因敲除和插入 | [27] |
| 通过gRNA工程改造的pegRNA;与逆转录酶和Rad51融合 | nCas9 | 融合单链DNA结合蛋白Rad51使PE2的编辑效率最高提升2.6倍 | [78] |
在CRISPR/Cas9的基因转录调控系统中,对Cas9蛋白工程化的dCas9分别在RuvC和HNH结构域发生突变使得RNP复合物仅能发挥靶序列识别功能,而无法切断核苷酸双链[29].nCas9(Cas9 nickase)是仅能使靶DNA发生单链断裂(single-strand break,SSB)的Cas9突变体,其中nCas9(H840A)和nCas9(D10A)分别是Cas9在HNH和RuvC结构域发生突变后的突变体蛋白,从而能够使该结构域失去内切酶活性,使DNA单链断裂,产生“缺口”[22,39].将dCas9或nCas9与各种效应蛋白如转录激活因子或抑制因子、表观遗传修饰因子、碱基编辑器甚至具有特定功能的酶等融合后,研究人员便能够借助CRISPR/Cas9系统对靶序列实现特定生物学功能,如表征转录基因乃至基因组上非编码区的功能、通过将碱基编辑器与nCas9融合能够在HDR活性低的细胞中进行精确高效的单碱基编辑等.因此本节将重点回顾与总结多种多样的效应蛋白为CRISPR/Cas9系统带来的多元化优化和发展,以及赋予该系统的多种新功能. ...
... 通过使用基于CRISPR/dCas9系统的一系列融合蛋白工具可以实现诸多靶向的表观遗传修饰,如DNA甲基化和去甲基化、组蛋白乙酰基化/去乙酰化和甲基化/去甲基化等,从而调控基因的表达并在许多细胞过程、癌症治疗及细胞工厂中发挥关键作用(图2)[30].其中DNA甲基化如5′胞嘧啶甲基化(5mC)是一种稳定的抑制性化学修饰调控.2016年,研究人员将dCas9、KRAB转录抑制因子以及DNA甲基化酶(DNA methyltransferase)的催化结构域融合蛋白基因与7个gRNA在K562细胞中共表达后获得了β2-微球蛋白启动子-增强子78%的稳定沉默[70].2021年,Nunez等[71]开发的CRISPRoff甲基化修饰工具能够在细胞分裂及分化过程中稳定维持靶基因的甲基化并抑制其表达水平.DNA去甲基化是将甲基胞嘧啶双加氧酶1(methylcytosine dioxygenase 1,TET1)的肽链C端催化结构域与dCas9融合后,再靶向至目的基因启动子或增强子区,从而实现目的基因的表达上调.2016年,Choudhury等[72]将dCas9- TET1靶向至乳腺癌基因1(breast cancer 1,BRCA1)启动子后成功地上调其表达水平.BRCA1作为抑制恶性肿瘤发生的关键基因之一,其成功的靶向上调为研究癌变新疗法带来新的思路,证明了通过去甲基化靶向激活人类肿瘤抑制基因的疗法是可行的.同样的,将dCas9与人类组蛋白乙酰基转移酶p300的催化核心融合后靶向至启动子和增强子区从而催化组蛋白H3 第27位赖氨酸残基的乙酰基化也会造成较强的基因上调[73].2019年,Chen等[102]通过提高麦角生物碱合成基因簇(ergot alkaloid synthesis gene cluster)的组蛋白乙酰化水平,使麦角菌Cp-1菌株(Claviceps pururea Cp-1 strain)中的麦角生物碱含量提高了一倍.通过融合dCas9与适当的表观遗传效应蛋白,CRISPR表观遗传工程工具则可利用CRISPR系统稳健地靶向性实现各种所需的表观遗传修饰,而不同工具间的适当组合,理论上讲可以为表观遗传学工具箱带来更多新型和功能复杂的应用策略,如利用与CRISPRa和CRISPRi优化策略类似的方式优化CRISPR表观遗传工程工具箱的特异性、稳定性和性能[103]. ...
Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers
2
2015
... Functional optimization through fusion expression of effector proteins with CRISPR/Cas9 system
Table 2| 功能 | 效应蛋白或gRNA工程 | 系统 | 特征 | 参考文献 |
|---|
| 表达调控 | 无 | dCas9 | 原核生物基因抑制表达,真核生物基因抑制效率低 | [22] |
| KRAB,MXI1,TUP1抑制结构域 | dCas9 | 可在真核生物中抑制基因表达 | [62-63] |
| KRAB-MeCP1融合抑制结构域 | dCas9 | 在哺乳动物细胞中高效抑制目的基因表达 | [64] |
| p65,VP64激活结构域 | dCas9 | 激活基因表达,效率较低 | [65-66] |
| VP64-p65-Rta(VPR)融合激活结构域 | dCas9 | 与VP64相比提高了22~320倍;多重基因激活表达;刺激iPSCs神经元分化 | [67] |
| gRNA工程;gRNA上携带携带两个MS2发夹二级结构 | dCas9 | 每个发夹结构招募一个MS2-p65-HSF1融合激活蛋白;与VP64相比,使10个靶基因上调2倍以上 | [68] |
| SPY系统(包括SpyTag和SpyCatcher)和Med2激活结构域 | dCas9 | 招募多拷贝SpyCatcher和Med2融合蛋白使酿酒酵母目的基因表达提高34.9倍 | [69] |
| SunTag系统(包括GCN4肽和抗GCN4抗体的单链可变片段scFv)和MIG1抑制结构域 | dCpf1 | 招募多拷贝ScFv与MIG1融合蛋白使酿酒酵母目的基因受到95%的抑制 | [69] |
| 表观基因组编辑 | KRAB-DNA甲基化酶催化结构域融合蛋白 | dCas9 | 融合抑制因子和甲基化酶;造成目的基因78%的稳定沉默 | [70] |
| 两侧分别融合KRAB和Dnmt3A-Dnmt3L融合DNA甲基化酶结构域 | dCas9 | 在干细胞分裂分化为神经细胞后仍维持DNA甲基化和基因抑制 | [71] |
| TET1甲基胞嘧啶双加氧酶1催化结构域 | dCas9 | dCas9- TET1靶向至BRCA1基因启动子后成功地上调其表达水平 | [72] |
| 人类组蛋白乙酰基转移酶p300催化核心 | dCas9 | 催化组蛋白H3 第27位赖氨酸残基的乙酰基化;基因表达上调 | [73] |
| 单碱基编辑 | 胞嘧啶脱氨酶,腺嘌呤脱氨酶 | nCas9 | 在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换 | [30,74-75] |
| CBE-UGI融合蛋白 | nCas9 | UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率 | [30] |
| 单链DNA结合结构域ssDBD融合到胞嘧啶脱氨酶与nCas9之间 | nCas9 | 通过延长ssDNA的暴露时间极大地提高CBEs的编辑效率;使CBEs的编辑窗口范围得到了提高 | [76] |
| 组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶 | nCas9 | 首次实现大肠杆菌中C到A 87.2%和哺乳动物细胞C到G 5.3%至53.0%的编辑效率 | [77] |
| 先导编辑 | 通过gRNA工程改造的pegRNA;与逆转录酶融合 | nCas9 | 改变pegRNA可实现精确的碱基替换、基因敲除和插入 | [27] |
| 通过gRNA工程改造的pegRNA;与逆转录酶和Rad51融合 | nCas9 | 融合单链DNA结合蛋白Rad51使PE2的编辑效率最高提升2.6倍 | [78] |
在CRISPR/Cas9的基因转录调控系统中,对Cas9蛋白工程化的dCas9分别在RuvC和HNH结构域发生突变使得RNP复合物仅能发挥靶序列识别功能,而无法切断核苷酸双链[29].nCas9(Cas9 nickase)是仅能使靶DNA发生单链断裂(single-strand break,SSB)的Cas9突变体,其中nCas9(H840A)和nCas9(D10A)分别是Cas9在HNH和RuvC结构域发生突变后的突变体蛋白,从而能够使该结构域失去内切酶活性,使DNA单链断裂,产生“缺口”[22,39].将dCas9或nCas9与各种效应蛋白如转录激活因子或抑制因子、表观遗传修饰因子、碱基编辑器甚至具有特定功能的酶等融合后,研究人员便能够借助CRISPR/Cas9系统对靶序列实现特定生物学功能,如表征转录基因乃至基因组上非编码区的功能、通过将碱基编辑器与nCas9融合能够在HDR活性低的细胞中进行精确高效的单碱基编辑等.因此本节将重点回顾与总结多种多样的效应蛋白为CRISPR/Cas9系统带来的多元化优化和发展,以及赋予该系统的多种新功能. ...
... 通过使用基于CRISPR/dCas9系统的一系列融合蛋白工具可以实现诸多靶向的表观遗传修饰,如DNA甲基化和去甲基化、组蛋白乙酰基化/去乙酰化和甲基化/去甲基化等,从而调控基因的表达并在许多细胞过程、癌症治疗及细胞工厂中发挥关键作用(图2)[30].其中DNA甲基化如5′胞嘧啶甲基化(5mC)是一种稳定的抑制性化学修饰调控.2016年,研究人员将dCas9、KRAB转录抑制因子以及DNA甲基化酶(DNA methyltransferase)的催化结构域融合蛋白基因与7个gRNA在K562细胞中共表达后获得了β2-微球蛋白启动子-增强子78%的稳定沉默[70].2021年,Nunez等[71]开发的CRISPRoff甲基化修饰工具能够在细胞分裂及分化过程中稳定维持靶基因的甲基化并抑制其表达水平.DNA去甲基化是将甲基胞嘧啶双加氧酶1(methylcytosine dioxygenase 1,TET1)的肽链C端催化结构域与dCas9融合后,再靶向至目的基因启动子或增强子区,从而实现目的基因的表达上调.2016年,Choudhury等[72]将dCas9- TET1靶向至乳腺癌基因1(breast cancer 1,BRCA1)启动子后成功地上调其表达水平.BRCA1作为抑制恶性肿瘤发生的关键基因之一,其成功的靶向上调为研究癌变新疗法带来新的思路,证明了通过去甲基化靶向激活人类肿瘤抑制基因的疗法是可行的.同样的,将dCas9与人类组蛋白乙酰基转移酶p300的催化核心融合后靶向至启动子和增强子区从而催化组蛋白H3 第27位赖氨酸残基的乙酰基化也会造成较强的基因上调[73].2019年,Chen等[102]通过提高麦角生物碱合成基因簇(ergot alkaloid synthesis gene cluster)的组蛋白乙酰化水平,使麦角菌Cp-1菌株(Claviceps pururea Cp-1 strain)中的麦角生物碱含量提高了一倍.通过融合dCas9与适当的表观遗传效应蛋白,CRISPR表观遗传工程工具则可利用CRISPR系统稳健地靶向性实现各种所需的表观遗传修饰,而不同工具间的适当组合,理论上讲可以为表观遗传学工具箱带来更多新型和功能复杂的应用策略,如利用与CRISPRa和CRISPRi优化策略类似的方式优化CRISPR表观遗传工程工具箱的特异性、稳定性和性能[103]. ...
Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells
2
2016
... Functional optimization through fusion expression of effector proteins with CRISPR/Cas9 system
Table 2| 功能 | 效应蛋白或gRNA工程 | 系统 | 特征 | 参考文献 |
|---|
| 表达调控 | 无 | dCas9 | 原核生物基因抑制表达,真核生物基因抑制效率低 | [22] |
| KRAB,MXI1,TUP1抑制结构域 | dCas9 | 可在真核生物中抑制基因表达 | [62-63] |
| KRAB-MeCP1融合抑制结构域 | dCas9 | 在哺乳动物细胞中高效抑制目的基因表达 | [64] |
| p65,VP64激活结构域 | dCas9 | 激活基因表达,效率较低 | [65-66] |
| VP64-p65-Rta(VPR)融合激活结构域 | dCas9 | 与VP64相比提高了22~320倍;多重基因激活表达;刺激iPSCs神经元分化 | [67] |
| gRNA工程;gRNA上携带携带两个MS2发夹二级结构 | dCas9 | 每个发夹结构招募一个MS2-p65-HSF1融合激活蛋白;与VP64相比,使10个靶基因上调2倍以上 | [68] |
| SPY系统(包括SpyTag和SpyCatcher)和Med2激活结构域 | dCas9 | 招募多拷贝SpyCatcher和Med2融合蛋白使酿酒酵母目的基因表达提高34.9倍 | [69] |
| SunTag系统(包括GCN4肽和抗GCN4抗体的单链可变片段scFv)和MIG1抑制结构域 | dCpf1 | 招募多拷贝ScFv与MIG1融合蛋白使酿酒酵母目的基因受到95%的抑制 | [69] |
| 表观基因组编辑 | KRAB-DNA甲基化酶催化结构域融合蛋白 | dCas9 | 融合抑制因子和甲基化酶;造成目的基因78%的稳定沉默 | [70] |
| 两侧分别融合KRAB和Dnmt3A-Dnmt3L融合DNA甲基化酶结构域 | dCas9 | 在干细胞分裂分化为神经细胞后仍维持DNA甲基化和基因抑制 | [71] |
| TET1甲基胞嘧啶双加氧酶1催化结构域 | dCas9 | dCas9- TET1靶向至BRCA1基因启动子后成功地上调其表达水平 | [72] |
| 人类组蛋白乙酰基转移酶p300催化核心 | dCas9 | 催化组蛋白H3 第27位赖氨酸残基的乙酰基化;基因表达上调 | [73] |
| 单碱基编辑 | 胞嘧啶脱氨酶,腺嘌呤脱氨酶 | nCas9 | 在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换 | [30,74-75] |
| CBE-UGI融合蛋白 | nCas9 | UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率 | [30] |
| 单链DNA结合结构域ssDBD融合到胞嘧啶脱氨酶与nCas9之间 | nCas9 | 通过延长ssDNA的暴露时间极大地提高CBEs的编辑效率;使CBEs的编辑窗口范围得到了提高 | [76] |
| 组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶 | nCas9 | 首次实现大肠杆菌中C到A 87.2%和哺乳动物细胞C到G 5.3%至53.0%的编辑效率 | [77] |
| 先导编辑 | 通过gRNA工程改造的pegRNA;与逆转录酶融合 | nCas9 | 改变pegRNA可实现精确的碱基替换、基因敲除和插入 | [27] |
| 通过gRNA工程改造的pegRNA;与逆转录酶和Rad51融合 | nCas9 | 融合单链DNA结合蛋白Rad51使PE2的编辑效率最高提升2.6倍 | [78] |
在CRISPR/Cas9的基因转录调控系统中,对Cas9蛋白工程化的dCas9分别在RuvC和HNH结构域发生突变使得RNP复合物仅能发挥靶序列识别功能,而无法切断核苷酸双链[29].nCas9(Cas9 nickase)是仅能使靶DNA发生单链断裂(single-strand break,SSB)的Cas9突变体,其中nCas9(H840A)和nCas9(D10A)分别是Cas9在HNH和RuvC结构域发生突变后的突变体蛋白,从而能够使该结构域失去内切酶活性,使DNA单链断裂,产生“缺口”[22,39].将dCas9或nCas9与各种效应蛋白如转录激活因子或抑制因子、表观遗传修饰因子、碱基编辑器甚至具有特定功能的酶等融合后,研究人员便能够借助CRISPR/Cas9系统对靶序列实现特定生物学功能,如表征转录基因乃至基因组上非编码区的功能、通过将碱基编辑器与nCas9融合能够在HDR活性低的细胞中进行精确高效的单碱基编辑等.因此本节将重点回顾与总结多种多样的效应蛋白为CRISPR/Cas9系统带来的多元化优化和发展,以及赋予该系统的多种新功能. ...
... 为了改善CRIPSR/Cas9系统的碱基编辑工具箱,研究人员通过将nCas9与胞嘧啶脱氨酶(cytosine deaminase)或腺嘌呤脱氨酶(adenine deaminase)融合的方式在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换(图2),其中相应的融合蛋白分别被称为胞嘧啶碱基编辑器(cytosine base editors,CBEs)[30,74]和腺嘌呤碱基编辑器(adenine base editors,ABEs)[75].CBEs中的胞嘧啶脱氨酶能使被nCas9释放的单链DNA(single strand DNA,ssDNA)上的胞嘧啶C转变为尿嘧啶U,随后该位置产生的U:G错配碱基对被修复为T:A碱基对;相应的,ABEs中的腺嘌呤脱氨酶使腺嘌呤A水解脱氨为次黄嘌呤I,随后DNA聚合酶能将次黄嘌呤I识别为鸟嘌呤G进行复制,互补链上原来与腺嘌呤A配对的胸腺嘧啶T则会相应变成胞嘧啶C,至此完成碱基置换全过程[107].由于不存在天然的ssDNA腺嘌呤脱氨酶[75],Kim等[108]通过定向进化改造得到一种大肠杆菌(Escherichia coli)tRNA来源的腺嘌呤脱氨酶ecTadA(E.coli TadA)突变体TadA*.由于在天然环境(大肠杆菌)中TadA是以同型二聚体的方式发挥作用,所以最初的TadA*-nCas9融合蛋白在哺乳动物细胞中的编辑效率并不高,直到研究人员将野生型TadA单体、进化的突变体TadA*及nCas9整合到一起(TadA- TadA*- nCas9),才使ABE在哺乳动物中的编辑效率大大提高.2018年,David R. Liu团队[109]工程化改造并优化了ABE7.10后获得的ABEmax最高可获得前者7.9倍的编辑效率,是目前为止在哺乳动物中编辑效率最高的ABEs.为了方便体内编辑,该团队[110]在2022年7月发表的紧凑型ABEs递送系统通过将小型腺嘌呤编辑器SaABE8e及其PAM变体SaKKH-ABE8e的基因长度最小化以及AAV组件基因长度最小化后使得ABEs能够进行单AAV递送,且在肝脏(66%)、心脏(33%)和肌肉细胞(22%)中的编辑效率均高于双AAV递送系统.同年,吉林大学李占军团队和赖学良团队[111]合作开发的紧凑型的Cas9(Cje2Cas9和Cje3Cas9)能够与sgRNA一起包装到单个AAV中,并在成年小鼠肝脏中以12%的效率成功编辑Pcsk9基因,最终有效降低了小鼠体内的胆固醇水平.高效的单AAV递送系统的开发不仅确立了其相对于双AAV递送系统的优势,还促进了CRISPR/Cas9系统作为体内碱基编辑工具时的研究和治疗应用,具有巨大的临床治疗价值.为了改善编辑效率,Zhang等[76]通过将单链DNA结合结构域(single-stranded DNA-binding domain,ssDBD)融合到胞嘧啶脱氨酶与nCas9之间从而延长ssDNA的暴露时间,可在不增加插入缺失突变(indels)、细胞毒性或脱靶效应等负面效应概率的条件下极大地提高CBEs的编辑效率.ssDBD的融合还使CBEs的编辑窗口范围得到了提高,从而拓宽了其靶向作用范围及用于疾病建模和基因治疗的适用范围.David R. Liu团队[30]在CBE的基础上融合了尿嘧啶糖基化酶抑制剂(uracil glycosylase inhibitor,UGI),作为一种体量较小的蛋白(9.5kDa),UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率.该团队还发现通过招募多拷贝的脱氨酶或更改其空间位置以优化编辑窗口能有效提高编辑效率和特异性.此外,与nCas9融合的脱氨酶若工程化改造后具有序列偏好性,则可通过降低旁观者编辑效应从而提高编辑精度.尽管ABEs和CBEs碱基编辑器的性能不断提升,A:T到G:C和C:G到T:A碱基对的单向固定替换仍限制着BEs作为单碱基改变工具使用时的灵活性.2020年,Zhao等[77]通过组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶(uracil-DNA glycosylase,Ung)实现了大肠杆菌中C到A 87.2%的编辑效率和哺乳动物细胞C到G 5.3%~53.0%的编辑效率.先导编辑作为一种多功能编辑手段,虽然同时具备可编程性和高精确度,但对于单碱基编辑的效率仍与BEs有较大差距[28].2021年,Song等[78]构建的hyPE2在PE2的基础上融合加入了一个ssDBD结构域Rad51,使得PE2的编辑效率最高提升了2.6倍,同时该团队还通过深度学习开发了能够预测不同的hyPE2与PE2相比编辑效率提升倍数的计算模型PEselector,该工具的网址为http://deepcrispr.info/PEselector.2022年,Velimirovic等[112]通过高通量肽链自编辑测序(peptide self-editing sequencing assay,PepSEq)测出12000个85个氨基酸的肽段对PE效率的影响,从而通过肽链融合提高PE的效率.综上所述,基于CRIPSR/Cas9系统的碱基编辑器编辑效率、编辑精度的不断提高以及编辑窗口乃至碱基固定替换模式的扩展相对于大量已知且不断增加的遗传病相关点突变数量仍捉襟见肘,各种脱氨酶赋予CRIPSR/Cas9系统的碱基编辑能力在其他结构域如UGI和ssDBD加持下不仅能够发挥CRIPSR系统高效率、高灵活、易编程的优势,也能够充分发挥脱氨酶的效能,但仍需进一步优化它们以有效地转换碱基和在无旁观者编辑效应的条件下进行目的碱基替换. ...
Programmable base editing of A·T to G·C in genomic DNA without DNA cleavage
3
2017
... Functional optimization through fusion expression of effector proteins with CRISPR/Cas9 system
Table 2| 功能 | 效应蛋白或gRNA工程 | 系统 | 特征 | 参考文献 |
|---|
| 表达调控 | 无 | dCas9 | 原核生物基因抑制表达,真核生物基因抑制效率低 | [22] |
| KRAB,MXI1,TUP1抑制结构域 | dCas9 | 可在真核生物中抑制基因表达 | [62-63] |
| KRAB-MeCP1融合抑制结构域 | dCas9 | 在哺乳动物细胞中高效抑制目的基因表达 | [64] |
| p65,VP64激活结构域 | dCas9 | 激活基因表达,效率较低 | [65-66] |
| VP64-p65-Rta(VPR)融合激活结构域 | dCas9 | 与VP64相比提高了22~320倍;多重基因激活表达;刺激iPSCs神经元分化 | [67] |
| gRNA工程;gRNA上携带携带两个MS2发夹二级结构 | dCas9 | 每个发夹结构招募一个MS2-p65-HSF1融合激活蛋白;与VP64相比,使10个靶基因上调2倍以上 | [68] |
| SPY系统(包括SpyTag和SpyCatcher)和Med2激活结构域 | dCas9 | 招募多拷贝SpyCatcher和Med2融合蛋白使酿酒酵母目的基因表达提高34.9倍 | [69] |
| SunTag系统(包括GCN4肽和抗GCN4抗体的单链可变片段scFv)和MIG1抑制结构域 | dCpf1 | 招募多拷贝ScFv与MIG1融合蛋白使酿酒酵母目的基因受到95%的抑制 | [69] |
| 表观基因组编辑 | KRAB-DNA甲基化酶催化结构域融合蛋白 | dCas9 | 融合抑制因子和甲基化酶;造成目的基因78%的稳定沉默 | [70] |
| 两侧分别融合KRAB和Dnmt3A-Dnmt3L融合DNA甲基化酶结构域 | dCas9 | 在干细胞分裂分化为神经细胞后仍维持DNA甲基化和基因抑制 | [71] |
| TET1甲基胞嘧啶双加氧酶1催化结构域 | dCas9 | dCas9- TET1靶向至BRCA1基因启动子后成功地上调其表达水平 | [72] |
| 人类组蛋白乙酰基转移酶p300催化核心 | dCas9 | 催化组蛋白H3 第27位赖氨酸残基的乙酰基化;基因表达上调 | [73] |
| 单碱基编辑 | 胞嘧啶脱氨酶,腺嘌呤脱氨酶 | nCas9 | 在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换 | [30,74-75] |
| CBE-UGI融合蛋白 | nCas9 | UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率 | [30] |
| 单链DNA结合结构域ssDBD融合到胞嘧啶脱氨酶与nCas9之间 | nCas9 | 通过延长ssDNA的暴露时间极大地提高CBEs的编辑效率;使CBEs的编辑窗口范围得到了提高 | [76] |
| 组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶 | nCas9 | 首次实现大肠杆菌中C到A 87.2%和哺乳动物细胞C到G 5.3%至53.0%的编辑效率 | [77] |
| 先导编辑 | 通过gRNA工程改造的pegRNA;与逆转录酶融合 | nCas9 | 改变pegRNA可实现精确的碱基替换、基因敲除和插入 | [27] |
| 通过gRNA工程改造的pegRNA;与逆转录酶和Rad51融合 | nCas9 | 融合单链DNA结合蛋白Rad51使PE2的编辑效率最高提升2.6倍 | [78] |
在CRISPR/Cas9的基因转录调控系统中,对Cas9蛋白工程化的dCas9分别在RuvC和HNH结构域发生突变使得RNP复合物仅能发挥靶序列识别功能,而无法切断核苷酸双链[29].nCas9(Cas9 nickase)是仅能使靶DNA发生单链断裂(single-strand break,SSB)的Cas9突变体,其中nCas9(H840A)和nCas9(D10A)分别是Cas9在HNH和RuvC结构域发生突变后的突变体蛋白,从而能够使该结构域失去内切酶活性,使DNA单链断裂,产生“缺口”[22,39].将dCas9或nCas9与各种效应蛋白如转录激活因子或抑制因子、表观遗传修饰因子、碱基编辑器甚至具有特定功能的酶等融合后,研究人员便能够借助CRISPR/Cas9系统对靶序列实现特定生物学功能,如表征转录基因乃至基因组上非编码区的功能、通过将碱基编辑器与nCas9融合能够在HDR活性低的细胞中进行精确高效的单碱基编辑等.因此本节将重点回顾与总结多种多样的效应蛋白为CRISPR/Cas9系统带来的多元化优化和发展,以及赋予该系统的多种新功能. ...
... 为了改善CRIPSR/Cas9系统的碱基编辑工具箱,研究人员通过将nCas9与胞嘧啶脱氨酶(cytosine deaminase)或腺嘌呤脱氨酶(adenine deaminase)融合的方式在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换(图2),其中相应的融合蛋白分别被称为胞嘧啶碱基编辑器(cytosine base editors,CBEs)[30,74]和腺嘌呤碱基编辑器(adenine base editors,ABEs)[75].CBEs中的胞嘧啶脱氨酶能使被nCas9释放的单链DNA(single strand DNA,ssDNA)上的胞嘧啶C转变为尿嘧啶U,随后该位置产生的U:G错配碱基对被修复为T:A碱基对;相应的,ABEs中的腺嘌呤脱氨酶使腺嘌呤A水解脱氨为次黄嘌呤I,随后DNA聚合酶能将次黄嘌呤I识别为鸟嘌呤G进行复制,互补链上原来与腺嘌呤A配对的胸腺嘧啶T则会相应变成胞嘧啶C,至此完成碱基置换全过程[107].由于不存在天然的ssDNA腺嘌呤脱氨酶[75],Kim等[108]通过定向进化改造得到一种大肠杆菌(Escherichia coli)tRNA来源的腺嘌呤脱氨酶ecTadA(E.coli TadA)突变体TadA*.由于在天然环境(大肠杆菌)中TadA是以同型二聚体的方式发挥作用,所以最初的TadA*-nCas9融合蛋白在哺乳动物细胞中的编辑效率并不高,直到研究人员将野生型TadA单体、进化的突变体TadA*及nCas9整合到一起(TadA- TadA*- nCas9),才使ABE在哺乳动物中的编辑效率大大提高.2018年,David R. Liu团队[109]工程化改造并优化了ABE7.10后获得的ABEmax最高可获得前者7.9倍的编辑效率,是目前为止在哺乳动物中编辑效率最高的ABEs.为了方便体内编辑,该团队[110]在2022年7月发表的紧凑型ABEs递送系统通过将小型腺嘌呤编辑器SaABE8e及其PAM变体SaKKH-ABE8e的基因长度最小化以及AAV组件基因长度最小化后使得ABEs能够进行单AAV递送,且在肝脏(66%)、心脏(33%)和肌肉细胞(22%)中的编辑效率均高于双AAV递送系统.同年,吉林大学李占军团队和赖学良团队[111]合作开发的紧凑型的Cas9(Cje2Cas9和Cje3Cas9)能够与sgRNA一起包装到单个AAV中,并在成年小鼠肝脏中以12%的效率成功编辑Pcsk9基因,最终有效降低了小鼠体内的胆固醇水平.高效的单AAV递送系统的开发不仅确立了其相对于双AAV递送系统的优势,还促进了CRISPR/Cas9系统作为体内碱基编辑工具时的研究和治疗应用,具有巨大的临床治疗价值.为了改善编辑效率,Zhang等[76]通过将单链DNA结合结构域(single-stranded DNA-binding domain,ssDBD)融合到胞嘧啶脱氨酶与nCas9之间从而延长ssDNA的暴露时间,可在不增加插入缺失突变(indels)、细胞毒性或脱靶效应等负面效应概率的条件下极大地提高CBEs的编辑效率.ssDBD的融合还使CBEs的编辑窗口范围得到了提高,从而拓宽了其靶向作用范围及用于疾病建模和基因治疗的适用范围.David R. Liu团队[30]在CBE的基础上融合了尿嘧啶糖基化酶抑制剂(uracil glycosylase inhibitor,UGI),作为一种体量较小的蛋白(9.5kDa),UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率.该团队还发现通过招募多拷贝的脱氨酶或更改其空间位置以优化编辑窗口能有效提高编辑效率和特异性.此外,与nCas9融合的脱氨酶若工程化改造后具有序列偏好性,则可通过降低旁观者编辑效应从而提高编辑精度.尽管ABEs和CBEs碱基编辑器的性能不断提升,A:T到G:C和C:G到T:A碱基对的单向固定替换仍限制着BEs作为单碱基改变工具使用时的灵活性.2020年,Zhao等[77]通过组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶(uracil-DNA glycosylase,Ung)实现了大肠杆菌中C到A 87.2%的编辑效率和哺乳动物细胞C到G 5.3%~53.0%的编辑效率.先导编辑作为一种多功能编辑手段,虽然同时具备可编程性和高精确度,但对于单碱基编辑的效率仍与BEs有较大差距[28].2021年,Song等[78]构建的hyPE2在PE2的基础上融合加入了一个ssDBD结构域Rad51,使得PE2的编辑效率最高提升了2.6倍,同时该团队还通过深度学习开发了能够预测不同的hyPE2与PE2相比编辑效率提升倍数的计算模型PEselector,该工具的网址为http://deepcrispr.info/PEselector.2022年,Velimirovic等[112]通过高通量肽链自编辑测序(peptide self-editing sequencing assay,PepSEq)测出12000个85个氨基酸的肽段对PE效率的影响,从而通过肽链融合提高PE的效率.综上所述,基于CRIPSR/Cas9系统的碱基编辑器编辑效率、编辑精度的不断提高以及编辑窗口乃至碱基固定替换模式的扩展相对于大量已知且不断增加的遗传病相关点突变数量仍捉襟见肘,各种脱氨酶赋予CRIPSR/Cas9系统的碱基编辑能力在其他结构域如UGI和ssDBD加持下不仅能够发挥CRIPSR系统高效率、高灵活、易编程的优势,也能够充分发挥脱氨酶的效能,但仍需进一步优化它们以有效地转换碱基和在无旁观者编辑效应的条件下进行目的碱基替换. ...
... [75],Kim等[108]通过定向进化改造得到一种大肠杆菌(Escherichia coli)tRNA来源的腺嘌呤脱氨酶ecTadA(E.coli TadA)突变体TadA*.由于在天然环境(大肠杆菌)中TadA是以同型二聚体的方式发挥作用,所以最初的TadA*-nCas9融合蛋白在哺乳动物细胞中的编辑效率并不高,直到研究人员将野生型TadA单体、进化的突变体TadA*及nCas9整合到一起(TadA- TadA*- nCas9),才使ABE在哺乳动物中的编辑效率大大提高.2018年,David R. Liu团队[109]工程化改造并优化了ABE7.10后获得的ABEmax最高可获得前者7.9倍的编辑效率,是目前为止在哺乳动物中编辑效率最高的ABEs.为了方便体内编辑,该团队[110]在2022年7月发表的紧凑型ABEs递送系统通过将小型腺嘌呤编辑器SaABE8e及其PAM变体SaKKH-ABE8e的基因长度最小化以及AAV组件基因长度最小化后使得ABEs能够进行单AAV递送,且在肝脏(66%)、心脏(33%)和肌肉细胞(22%)中的编辑效率均高于双AAV递送系统.同年,吉林大学李占军团队和赖学良团队[111]合作开发的紧凑型的Cas9(Cje2Cas9和Cje3Cas9)能够与sgRNA一起包装到单个AAV中,并在成年小鼠肝脏中以12%的效率成功编辑Pcsk9基因,最终有效降低了小鼠体内的胆固醇水平.高效的单AAV递送系统的开发不仅确立了其相对于双AAV递送系统的优势,还促进了CRISPR/Cas9系统作为体内碱基编辑工具时的研究和治疗应用,具有巨大的临床治疗价值.为了改善编辑效率,Zhang等[76]通过将单链DNA结合结构域(single-stranded DNA-binding domain,ssDBD)融合到胞嘧啶脱氨酶与nCas9之间从而延长ssDNA的暴露时间,可在不增加插入缺失突变(indels)、细胞毒性或脱靶效应等负面效应概率的条件下极大地提高CBEs的编辑效率.ssDBD的融合还使CBEs的编辑窗口范围得到了提高,从而拓宽了其靶向作用范围及用于疾病建模和基因治疗的适用范围.David R. Liu团队[30]在CBE的基础上融合了尿嘧啶糖基化酶抑制剂(uracil glycosylase inhibitor,UGI),作为一种体量较小的蛋白(9.5kDa),UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率.该团队还发现通过招募多拷贝的脱氨酶或更改其空间位置以优化编辑窗口能有效提高编辑效率和特异性.此外,与nCas9融合的脱氨酶若工程化改造后具有序列偏好性,则可通过降低旁观者编辑效应从而提高编辑精度.尽管ABEs和CBEs碱基编辑器的性能不断提升,A:T到G:C和C:G到T:A碱基对的单向固定替换仍限制着BEs作为单碱基改变工具使用时的灵活性.2020年,Zhao等[77]通过组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶(uracil-DNA glycosylase,Ung)实现了大肠杆菌中C到A 87.2%的编辑效率和哺乳动物细胞C到G 5.3%~53.0%的编辑效率.先导编辑作为一种多功能编辑手段,虽然同时具备可编程性和高精确度,但对于单碱基编辑的效率仍与BEs有较大差距[28].2021年,Song等[78]构建的hyPE2在PE2的基础上融合加入了一个ssDBD结构域Rad51,使得PE2的编辑效率最高提升了2.6倍,同时该团队还通过深度学习开发了能够预测不同的hyPE2与PE2相比编辑效率提升倍数的计算模型PEselector,该工具的网址为http://deepcrispr.info/PEselector.2022年,Velimirovic等[112]通过高通量肽链自编辑测序(peptide self-editing sequencing assay,PepSEq)测出12000个85个氨基酸的肽段对PE效率的影响,从而通过肽链融合提高PE的效率.综上所述,基于CRIPSR/Cas9系统的碱基编辑器编辑效率、编辑精度的不断提高以及编辑窗口乃至碱基固定替换模式的扩展相对于大量已知且不断增加的遗传病相关点突变数量仍捉襟见肘,各种脱氨酶赋予CRIPSR/Cas9系统的碱基编辑能力在其他结构域如UGI和ssDBD加持下不仅能够发挥CRIPSR系统高效率、高灵活、易编程的优势,也能够充分发挥脱氨酶的效能,但仍需进一步优化它们以有效地转换碱基和在无旁观者编辑效应的条件下进行目的碱基替换. ...
Increasing the efficiency and targeting range of cytidine base editors through fusion of a single-stranded DNA-binding protein domain
2
2020
... Functional optimization through fusion expression of effector proteins with CRISPR/Cas9 system
Table 2| 功能 | 效应蛋白或gRNA工程 | 系统 | 特征 | 参考文献 |
|---|
| 表达调控 | 无 | dCas9 | 原核生物基因抑制表达,真核生物基因抑制效率低 | [22] |
| KRAB,MXI1,TUP1抑制结构域 | dCas9 | 可在真核生物中抑制基因表达 | [62-63] |
| KRAB-MeCP1融合抑制结构域 | dCas9 | 在哺乳动物细胞中高效抑制目的基因表达 | [64] |
| p65,VP64激活结构域 | dCas9 | 激活基因表达,效率较低 | [65-66] |
| VP64-p65-Rta(VPR)融合激活结构域 | dCas9 | 与VP64相比提高了22~320倍;多重基因激活表达;刺激iPSCs神经元分化 | [67] |
| gRNA工程;gRNA上携带携带两个MS2发夹二级结构 | dCas9 | 每个发夹结构招募一个MS2-p65-HSF1融合激活蛋白;与VP64相比,使10个靶基因上调2倍以上 | [68] |
| SPY系统(包括SpyTag和SpyCatcher)和Med2激活结构域 | dCas9 | 招募多拷贝SpyCatcher和Med2融合蛋白使酿酒酵母目的基因表达提高34.9倍 | [69] |
| SunTag系统(包括GCN4肽和抗GCN4抗体的单链可变片段scFv)和MIG1抑制结构域 | dCpf1 | 招募多拷贝ScFv与MIG1融合蛋白使酿酒酵母目的基因受到95%的抑制 | [69] |
| 表观基因组编辑 | KRAB-DNA甲基化酶催化结构域融合蛋白 | dCas9 | 融合抑制因子和甲基化酶;造成目的基因78%的稳定沉默 | [70] |
| 两侧分别融合KRAB和Dnmt3A-Dnmt3L融合DNA甲基化酶结构域 | dCas9 | 在干细胞分裂分化为神经细胞后仍维持DNA甲基化和基因抑制 | [71] |
| TET1甲基胞嘧啶双加氧酶1催化结构域 | dCas9 | dCas9- TET1靶向至BRCA1基因启动子后成功地上调其表达水平 | [72] |
| 人类组蛋白乙酰基转移酶p300催化核心 | dCas9 | 催化组蛋白H3 第27位赖氨酸残基的乙酰基化;基因表达上调 | [73] |
| 单碱基编辑 | 胞嘧啶脱氨酶,腺嘌呤脱氨酶 | nCas9 | 在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换 | [30,74-75] |
| CBE-UGI融合蛋白 | nCas9 | UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率 | [30] |
| 单链DNA结合结构域ssDBD融合到胞嘧啶脱氨酶与nCas9之间 | nCas9 | 通过延长ssDNA的暴露时间极大地提高CBEs的编辑效率;使CBEs的编辑窗口范围得到了提高 | [76] |
| 组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶 | nCas9 | 首次实现大肠杆菌中C到A 87.2%和哺乳动物细胞C到G 5.3%至53.0%的编辑效率 | [77] |
| 先导编辑 | 通过gRNA工程改造的pegRNA;与逆转录酶融合 | nCas9 | 改变pegRNA可实现精确的碱基替换、基因敲除和插入 | [27] |
| 通过gRNA工程改造的pegRNA;与逆转录酶和Rad51融合 | nCas9 | 融合单链DNA结合蛋白Rad51使PE2的编辑效率最高提升2.6倍 | [78] |
在CRISPR/Cas9的基因转录调控系统中,对Cas9蛋白工程化的dCas9分别在RuvC和HNH结构域发生突变使得RNP复合物仅能发挥靶序列识别功能,而无法切断核苷酸双链[29].nCas9(Cas9 nickase)是仅能使靶DNA发生单链断裂(single-strand break,SSB)的Cas9突变体,其中nCas9(H840A)和nCas9(D10A)分别是Cas9在HNH和RuvC结构域发生突变后的突变体蛋白,从而能够使该结构域失去内切酶活性,使DNA单链断裂,产生“缺口”[22,39].将dCas9或nCas9与各种效应蛋白如转录激活因子或抑制因子、表观遗传修饰因子、碱基编辑器甚至具有特定功能的酶等融合后,研究人员便能够借助CRISPR/Cas9系统对靶序列实现特定生物学功能,如表征转录基因乃至基因组上非编码区的功能、通过将碱基编辑器与nCas9融合能够在HDR活性低的细胞中进行精确高效的单碱基编辑等.因此本节将重点回顾与总结多种多样的效应蛋白为CRISPR/Cas9系统带来的多元化优化和发展,以及赋予该系统的多种新功能. ...
... 为了改善CRIPSR/Cas9系统的碱基编辑工具箱,研究人员通过将nCas9与胞嘧啶脱氨酶(cytosine deaminase)或腺嘌呤脱氨酶(adenine deaminase)融合的方式在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换(图2),其中相应的融合蛋白分别被称为胞嘧啶碱基编辑器(cytosine base editors,CBEs)[30,74]和腺嘌呤碱基编辑器(adenine base editors,ABEs)[75].CBEs中的胞嘧啶脱氨酶能使被nCas9释放的单链DNA(single strand DNA,ssDNA)上的胞嘧啶C转变为尿嘧啶U,随后该位置产生的U:G错配碱基对被修复为T:A碱基对;相应的,ABEs中的腺嘌呤脱氨酶使腺嘌呤A水解脱氨为次黄嘌呤I,随后DNA聚合酶能将次黄嘌呤I识别为鸟嘌呤G进行复制,互补链上原来与腺嘌呤A配对的胸腺嘧啶T则会相应变成胞嘧啶C,至此完成碱基置换全过程[107].由于不存在天然的ssDNA腺嘌呤脱氨酶[75],Kim等[108]通过定向进化改造得到一种大肠杆菌(Escherichia coli)tRNA来源的腺嘌呤脱氨酶ecTadA(E.coli TadA)突变体TadA*.由于在天然环境(大肠杆菌)中TadA是以同型二聚体的方式发挥作用,所以最初的TadA*-nCas9融合蛋白在哺乳动物细胞中的编辑效率并不高,直到研究人员将野生型TadA单体、进化的突变体TadA*及nCas9整合到一起(TadA- TadA*- nCas9),才使ABE在哺乳动物中的编辑效率大大提高.2018年,David R. Liu团队[109]工程化改造并优化了ABE7.10后获得的ABEmax最高可获得前者7.9倍的编辑效率,是目前为止在哺乳动物中编辑效率最高的ABEs.为了方便体内编辑,该团队[110]在2022年7月发表的紧凑型ABEs递送系统通过将小型腺嘌呤编辑器SaABE8e及其PAM变体SaKKH-ABE8e的基因长度最小化以及AAV组件基因长度最小化后使得ABEs能够进行单AAV递送,且在肝脏(66%)、心脏(33%)和肌肉细胞(22%)中的编辑效率均高于双AAV递送系统.同年,吉林大学李占军团队和赖学良团队[111]合作开发的紧凑型的Cas9(Cje2Cas9和Cje3Cas9)能够与sgRNA一起包装到单个AAV中,并在成年小鼠肝脏中以12%的效率成功编辑Pcsk9基因,最终有效降低了小鼠体内的胆固醇水平.高效的单AAV递送系统的开发不仅确立了其相对于双AAV递送系统的优势,还促进了CRISPR/Cas9系统作为体内碱基编辑工具时的研究和治疗应用,具有巨大的临床治疗价值.为了改善编辑效率,Zhang等[76]通过将单链DNA结合结构域(single-stranded DNA-binding domain,ssDBD)融合到胞嘧啶脱氨酶与nCas9之间从而延长ssDNA的暴露时间,可在不增加插入缺失突变(indels)、细胞毒性或脱靶效应等负面效应概率的条件下极大地提高CBEs的编辑效率.ssDBD的融合还使CBEs的编辑窗口范围得到了提高,从而拓宽了其靶向作用范围及用于疾病建模和基因治疗的适用范围.David R. Liu团队[30]在CBE的基础上融合了尿嘧啶糖基化酶抑制剂(uracil glycosylase inhibitor,UGI),作为一种体量较小的蛋白(9.5kDa),UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率.该团队还发现通过招募多拷贝的脱氨酶或更改其空间位置以优化编辑窗口能有效提高编辑效率和特异性.此外,与nCas9融合的脱氨酶若工程化改造后具有序列偏好性,则可通过降低旁观者编辑效应从而提高编辑精度.尽管ABEs和CBEs碱基编辑器的性能不断提升,A:T到G:C和C:G到T:A碱基对的单向固定替换仍限制着BEs作为单碱基改变工具使用时的灵活性.2020年,Zhao等[77]通过组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶(uracil-DNA glycosylase,Ung)实现了大肠杆菌中C到A 87.2%的编辑效率和哺乳动物细胞C到G 5.3%~53.0%的编辑效率.先导编辑作为一种多功能编辑手段,虽然同时具备可编程性和高精确度,但对于单碱基编辑的效率仍与BEs有较大差距[28].2021年,Song等[78]构建的hyPE2在PE2的基础上融合加入了一个ssDBD结构域Rad51,使得PE2的编辑效率最高提升了2.6倍,同时该团队还通过深度学习开发了能够预测不同的hyPE2与PE2相比编辑效率提升倍数的计算模型PEselector,该工具的网址为http://deepcrispr.info/PEselector.2022年,Velimirovic等[112]通过高通量肽链自编辑测序(peptide self-editing sequencing assay,PepSEq)测出12000个85个氨基酸的肽段对PE效率的影响,从而通过肽链融合提高PE的效率.综上所述,基于CRIPSR/Cas9系统的碱基编辑器编辑效率、编辑精度的不断提高以及编辑窗口乃至碱基固定替换模式的扩展相对于大量已知且不断增加的遗传病相关点突变数量仍捉襟见肘,各种脱氨酶赋予CRIPSR/Cas9系统的碱基编辑能力在其他结构域如UGI和ssDBD加持下不仅能够发挥CRIPSR系统高效率、高灵活、易编程的优势,也能够充分发挥脱氨酶的效能,但仍需进一步优化它们以有效地转换碱基和在无旁观者编辑效应的条件下进行目的碱基替换. ...
Glycosylase base editors enable C-to-A and C-to-G base changes
2
2021
... Functional optimization through fusion expression of effector proteins with CRISPR/Cas9 system
Table 2| 功能 | 效应蛋白或gRNA工程 | 系统 | 特征 | 参考文献 |
|---|
| 表达调控 | 无 | dCas9 | 原核生物基因抑制表达,真核生物基因抑制效率低 | [22] |
| KRAB,MXI1,TUP1抑制结构域 | dCas9 | 可在真核生物中抑制基因表达 | [62-63] |
| KRAB-MeCP1融合抑制结构域 | dCas9 | 在哺乳动物细胞中高效抑制目的基因表达 | [64] |
| p65,VP64激活结构域 | dCas9 | 激活基因表达,效率较低 | [65-66] |
| VP64-p65-Rta(VPR)融合激活结构域 | dCas9 | 与VP64相比提高了22~320倍;多重基因激活表达;刺激iPSCs神经元分化 | [67] |
| gRNA工程;gRNA上携带携带两个MS2发夹二级结构 | dCas9 | 每个发夹结构招募一个MS2-p65-HSF1融合激活蛋白;与VP64相比,使10个靶基因上调2倍以上 | [68] |
| SPY系统(包括SpyTag和SpyCatcher)和Med2激活结构域 | dCas9 | 招募多拷贝SpyCatcher和Med2融合蛋白使酿酒酵母目的基因表达提高34.9倍 | [69] |
| SunTag系统(包括GCN4肽和抗GCN4抗体的单链可变片段scFv)和MIG1抑制结构域 | dCpf1 | 招募多拷贝ScFv与MIG1融合蛋白使酿酒酵母目的基因受到95%的抑制 | [69] |
| 表观基因组编辑 | KRAB-DNA甲基化酶催化结构域融合蛋白 | dCas9 | 融合抑制因子和甲基化酶;造成目的基因78%的稳定沉默 | [70] |
| 两侧分别融合KRAB和Dnmt3A-Dnmt3L融合DNA甲基化酶结构域 | dCas9 | 在干细胞分裂分化为神经细胞后仍维持DNA甲基化和基因抑制 | [71] |
| TET1甲基胞嘧啶双加氧酶1催化结构域 | dCas9 | dCas9- TET1靶向至BRCA1基因启动子后成功地上调其表达水平 | [72] |
| 人类组蛋白乙酰基转移酶p300催化核心 | dCas9 | 催化组蛋白H3 第27位赖氨酸残基的乙酰基化;基因表达上调 | [73] |
| 单碱基编辑 | 胞嘧啶脱氨酶,腺嘌呤脱氨酶 | nCas9 | 在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换 | [30,74-75] |
| CBE-UGI融合蛋白 | nCas9 | UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率 | [30] |
| 单链DNA结合结构域ssDBD融合到胞嘧啶脱氨酶与nCas9之间 | nCas9 | 通过延长ssDNA的暴露时间极大地提高CBEs的编辑效率;使CBEs的编辑窗口范围得到了提高 | [76] |
| 组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶 | nCas9 | 首次实现大肠杆菌中C到A 87.2%和哺乳动物细胞C到G 5.3%至53.0%的编辑效率 | [77] |
| 先导编辑 | 通过gRNA工程改造的pegRNA;与逆转录酶融合 | nCas9 | 改变pegRNA可实现精确的碱基替换、基因敲除和插入 | [27] |
| 通过gRNA工程改造的pegRNA;与逆转录酶和Rad51融合 | nCas9 | 融合单链DNA结合蛋白Rad51使PE2的编辑效率最高提升2.6倍 | [78] |
在CRISPR/Cas9的基因转录调控系统中,对Cas9蛋白工程化的dCas9分别在RuvC和HNH结构域发生突变使得RNP复合物仅能发挥靶序列识别功能,而无法切断核苷酸双链[29].nCas9(Cas9 nickase)是仅能使靶DNA发生单链断裂(single-strand break,SSB)的Cas9突变体,其中nCas9(H840A)和nCas9(D10A)分别是Cas9在HNH和RuvC结构域发生突变后的突变体蛋白,从而能够使该结构域失去内切酶活性,使DNA单链断裂,产生“缺口”[22,39].将dCas9或nCas9与各种效应蛋白如转录激活因子或抑制因子、表观遗传修饰因子、碱基编辑器甚至具有特定功能的酶等融合后,研究人员便能够借助CRISPR/Cas9系统对靶序列实现特定生物学功能,如表征转录基因乃至基因组上非编码区的功能、通过将碱基编辑器与nCas9融合能够在HDR活性低的细胞中进行精确高效的单碱基编辑等.因此本节将重点回顾与总结多种多样的效应蛋白为CRISPR/Cas9系统带来的多元化优化和发展,以及赋予该系统的多种新功能. ...
... 为了改善CRIPSR/Cas9系统的碱基编辑工具箱,研究人员通过将nCas9与胞嘧啶脱氨酶(cytosine deaminase)或腺嘌呤脱氨酶(adenine deaminase)融合的方式在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换(图2),其中相应的融合蛋白分别被称为胞嘧啶碱基编辑器(cytosine base editors,CBEs)[30,74]和腺嘌呤碱基编辑器(adenine base editors,ABEs)[75].CBEs中的胞嘧啶脱氨酶能使被nCas9释放的单链DNA(single strand DNA,ssDNA)上的胞嘧啶C转变为尿嘧啶U,随后该位置产生的U:G错配碱基对被修复为T:A碱基对;相应的,ABEs中的腺嘌呤脱氨酶使腺嘌呤A水解脱氨为次黄嘌呤I,随后DNA聚合酶能将次黄嘌呤I识别为鸟嘌呤G进行复制,互补链上原来与腺嘌呤A配对的胸腺嘧啶T则会相应变成胞嘧啶C,至此完成碱基置换全过程[107].由于不存在天然的ssDNA腺嘌呤脱氨酶[75],Kim等[108]通过定向进化改造得到一种大肠杆菌(Escherichia coli)tRNA来源的腺嘌呤脱氨酶ecTadA(E.coli TadA)突变体TadA*.由于在天然环境(大肠杆菌)中TadA是以同型二聚体的方式发挥作用,所以最初的TadA*-nCas9融合蛋白在哺乳动物细胞中的编辑效率并不高,直到研究人员将野生型TadA单体、进化的突变体TadA*及nCas9整合到一起(TadA- TadA*- nCas9),才使ABE在哺乳动物中的编辑效率大大提高.2018年,David R. Liu团队[109]工程化改造并优化了ABE7.10后获得的ABEmax最高可获得前者7.9倍的编辑效率,是目前为止在哺乳动物中编辑效率最高的ABEs.为了方便体内编辑,该团队[110]在2022年7月发表的紧凑型ABEs递送系统通过将小型腺嘌呤编辑器SaABE8e及其PAM变体SaKKH-ABE8e的基因长度最小化以及AAV组件基因长度最小化后使得ABEs能够进行单AAV递送,且在肝脏(66%)、心脏(33%)和肌肉细胞(22%)中的编辑效率均高于双AAV递送系统.同年,吉林大学李占军团队和赖学良团队[111]合作开发的紧凑型的Cas9(Cje2Cas9和Cje3Cas9)能够与sgRNA一起包装到单个AAV中,并在成年小鼠肝脏中以12%的效率成功编辑Pcsk9基因,最终有效降低了小鼠体内的胆固醇水平.高效的单AAV递送系统的开发不仅确立了其相对于双AAV递送系统的优势,还促进了CRISPR/Cas9系统作为体内碱基编辑工具时的研究和治疗应用,具有巨大的临床治疗价值.为了改善编辑效率,Zhang等[76]通过将单链DNA结合结构域(single-stranded DNA-binding domain,ssDBD)融合到胞嘧啶脱氨酶与nCas9之间从而延长ssDNA的暴露时间,可在不增加插入缺失突变(indels)、细胞毒性或脱靶效应等负面效应概率的条件下极大地提高CBEs的编辑效率.ssDBD的融合还使CBEs的编辑窗口范围得到了提高,从而拓宽了其靶向作用范围及用于疾病建模和基因治疗的适用范围.David R. Liu团队[30]在CBE的基础上融合了尿嘧啶糖基化酶抑制剂(uracil glycosylase inhibitor,UGI),作为一种体量较小的蛋白(9.5kDa),UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率.该团队还发现通过招募多拷贝的脱氨酶或更改其空间位置以优化编辑窗口能有效提高编辑效率和特异性.此外,与nCas9融合的脱氨酶若工程化改造后具有序列偏好性,则可通过降低旁观者编辑效应从而提高编辑精度.尽管ABEs和CBEs碱基编辑器的性能不断提升,A:T到G:C和C:G到T:A碱基对的单向固定替换仍限制着BEs作为单碱基改变工具使用时的灵活性.2020年,Zhao等[77]通过组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶(uracil-DNA glycosylase,Ung)实现了大肠杆菌中C到A 87.2%的编辑效率和哺乳动物细胞C到G 5.3%~53.0%的编辑效率.先导编辑作为一种多功能编辑手段,虽然同时具备可编程性和高精确度,但对于单碱基编辑的效率仍与BEs有较大差距[28].2021年,Song等[78]构建的hyPE2在PE2的基础上融合加入了一个ssDBD结构域Rad51,使得PE2的编辑效率最高提升了2.6倍,同时该团队还通过深度学习开发了能够预测不同的hyPE2与PE2相比编辑效率提升倍数的计算模型PEselector,该工具的网址为http://deepcrispr.info/PEselector.2022年,Velimirovic等[112]通过高通量肽链自编辑测序(peptide self-editing sequencing assay,PepSEq)测出12000个85个氨基酸的肽段对PE效率的影响,从而通过肽链融合提高PE的效率.综上所述,基于CRIPSR/Cas9系统的碱基编辑器编辑效率、编辑精度的不断提高以及编辑窗口乃至碱基固定替换模式的扩展相对于大量已知且不断增加的遗传病相关点突变数量仍捉襟见肘,各种脱氨酶赋予CRIPSR/Cas9系统的碱基编辑能力在其他结构域如UGI和ssDBD加持下不仅能够发挥CRIPSR系统高效率、高灵活、易编程的优势,也能够充分发挥脱氨酶的效能,但仍需进一步优化它们以有效地转换碱基和在无旁观者编辑效应的条件下进行目的碱基替换. ...
Generation of a more efficient prime editor 2 by addition of the Rad51 DNA-binding domain
2
2021
... Functional optimization through fusion expression of effector proteins with CRISPR/Cas9 system
Table 2| 功能 | 效应蛋白或gRNA工程 | 系统 | 特征 | 参考文献 |
|---|
| 表达调控 | 无 | dCas9 | 原核生物基因抑制表达,真核生物基因抑制效率低 | [22] |
| KRAB,MXI1,TUP1抑制结构域 | dCas9 | 可在真核生物中抑制基因表达 | [62-63] |
| KRAB-MeCP1融合抑制结构域 | dCas9 | 在哺乳动物细胞中高效抑制目的基因表达 | [64] |
| p65,VP64激活结构域 | dCas9 | 激活基因表达,效率较低 | [65-66] |
| VP64-p65-Rta(VPR)融合激活结构域 | dCas9 | 与VP64相比提高了22~320倍;多重基因激活表达;刺激iPSCs神经元分化 | [67] |
| gRNA工程;gRNA上携带携带两个MS2发夹二级结构 | dCas9 | 每个发夹结构招募一个MS2-p65-HSF1融合激活蛋白;与VP64相比,使10个靶基因上调2倍以上 | [68] |
| SPY系统(包括SpyTag和SpyCatcher)和Med2激活结构域 | dCas9 | 招募多拷贝SpyCatcher和Med2融合蛋白使酿酒酵母目的基因表达提高34.9倍 | [69] |
| SunTag系统(包括GCN4肽和抗GCN4抗体的单链可变片段scFv)和MIG1抑制结构域 | dCpf1 | 招募多拷贝ScFv与MIG1融合蛋白使酿酒酵母目的基因受到95%的抑制 | [69] |
| 表观基因组编辑 | KRAB-DNA甲基化酶催化结构域融合蛋白 | dCas9 | 融合抑制因子和甲基化酶;造成目的基因78%的稳定沉默 | [70] |
| 两侧分别融合KRAB和Dnmt3A-Dnmt3L融合DNA甲基化酶结构域 | dCas9 | 在干细胞分裂分化为神经细胞后仍维持DNA甲基化和基因抑制 | [71] |
| TET1甲基胞嘧啶双加氧酶1催化结构域 | dCas9 | dCas9- TET1靶向至BRCA1基因启动子后成功地上调其表达水平 | [72] |
| 人类组蛋白乙酰基转移酶p300催化核心 | dCas9 | 催化组蛋白H3 第27位赖氨酸残基的乙酰基化;基因表达上调 | [73] |
| 单碱基编辑 | 胞嘧啶脱氨酶,腺嘌呤脱氨酶 | nCas9 | 在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换 | [30,74-75] |
| CBE-UGI融合蛋白 | nCas9 | UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率 | [30] |
| 单链DNA结合结构域ssDBD融合到胞嘧啶脱氨酶与nCas9之间 | nCas9 | 通过延长ssDNA的暴露时间极大地提高CBEs的编辑效率;使CBEs的编辑窗口范围得到了提高 | [76] |
| 组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶 | nCas9 | 首次实现大肠杆菌中C到A 87.2%和哺乳动物细胞C到G 5.3%至53.0%的编辑效率 | [77] |
| 先导编辑 | 通过gRNA工程改造的pegRNA;与逆转录酶融合 | nCas9 | 改变pegRNA可实现精确的碱基替换、基因敲除和插入 | [27] |
| 通过gRNA工程改造的pegRNA;与逆转录酶和Rad51融合 | nCas9 | 融合单链DNA结合蛋白Rad51使PE2的编辑效率最高提升2.6倍 | [78] |
在CRISPR/Cas9的基因转录调控系统中,对Cas9蛋白工程化的dCas9分别在RuvC和HNH结构域发生突变使得RNP复合物仅能发挥靶序列识别功能,而无法切断核苷酸双链[29].nCas9(Cas9 nickase)是仅能使靶DNA发生单链断裂(single-strand break,SSB)的Cas9突变体,其中nCas9(H840A)和nCas9(D10A)分别是Cas9在HNH和RuvC结构域发生突变后的突变体蛋白,从而能够使该结构域失去内切酶活性,使DNA单链断裂,产生“缺口”[22,39].将dCas9或nCas9与各种效应蛋白如转录激活因子或抑制因子、表观遗传修饰因子、碱基编辑器甚至具有特定功能的酶等融合后,研究人员便能够借助CRISPR/Cas9系统对靶序列实现特定生物学功能,如表征转录基因乃至基因组上非编码区的功能、通过将碱基编辑器与nCas9融合能够在HDR活性低的细胞中进行精确高效的单碱基编辑等.因此本节将重点回顾与总结多种多样的效应蛋白为CRISPR/Cas9系统带来的多元化优化和发展,以及赋予该系统的多种新功能. ...
... 为了改善CRIPSR/Cas9系统的碱基编辑工具箱,研究人员通过将nCas9与胞嘧啶脱氨酶(cytosine deaminase)或腺嘌呤脱氨酶(adenine deaminase)融合的方式在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换(图2),其中相应的融合蛋白分别被称为胞嘧啶碱基编辑器(cytosine base editors,CBEs)[30,74]和腺嘌呤碱基编辑器(adenine base editors,ABEs)[75].CBEs中的胞嘧啶脱氨酶能使被nCas9释放的单链DNA(single strand DNA,ssDNA)上的胞嘧啶C转变为尿嘧啶U,随后该位置产生的U:G错配碱基对被修复为T:A碱基对;相应的,ABEs中的腺嘌呤脱氨酶使腺嘌呤A水解脱氨为次黄嘌呤I,随后DNA聚合酶能将次黄嘌呤I识别为鸟嘌呤G进行复制,互补链上原来与腺嘌呤A配对的胸腺嘧啶T则会相应变成胞嘧啶C,至此完成碱基置换全过程[107].由于不存在天然的ssDNA腺嘌呤脱氨酶[75],Kim等[108]通过定向进化改造得到一种大肠杆菌(Escherichia coli)tRNA来源的腺嘌呤脱氨酶ecTadA(E.coli TadA)突变体TadA*.由于在天然环境(大肠杆菌)中TadA是以同型二聚体的方式发挥作用,所以最初的TadA*-nCas9融合蛋白在哺乳动物细胞中的编辑效率并不高,直到研究人员将野生型TadA单体、进化的突变体TadA*及nCas9整合到一起(TadA- TadA*- nCas9),才使ABE在哺乳动物中的编辑效率大大提高.2018年,David R. Liu团队[109]工程化改造并优化了ABE7.10后获得的ABEmax最高可获得前者7.9倍的编辑效率,是目前为止在哺乳动物中编辑效率最高的ABEs.为了方便体内编辑,该团队[110]在2022年7月发表的紧凑型ABEs递送系统通过将小型腺嘌呤编辑器SaABE8e及其PAM变体SaKKH-ABE8e的基因长度最小化以及AAV组件基因长度最小化后使得ABEs能够进行单AAV递送,且在肝脏(66%)、心脏(33%)和肌肉细胞(22%)中的编辑效率均高于双AAV递送系统.同年,吉林大学李占军团队和赖学良团队[111]合作开发的紧凑型的Cas9(Cje2Cas9和Cje3Cas9)能够与sgRNA一起包装到单个AAV中,并在成年小鼠肝脏中以12%的效率成功编辑Pcsk9基因,最终有效降低了小鼠体内的胆固醇水平.高效的单AAV递送系统的开发不仅确立了其相对于双AAV递送系统的优势,还促进了CRISPR/Cas9系统作为体内碱基编辑工具时的研究和治疗应用,具有巨大的临床治疗价值.为了改善编辑效率,Zhang等[76]通过将单链DNA结合结构域(single-stranded DNA-binding domain,ssDBD)融合到胞嘧啶脱氨酶与nCas9之间从而延长ssDNA的暴露时间,可在不增加插入缺失突变(indels)、细胞毒性或脱靶效应等负面效应概率的条件下极大地提高CBEs的编辑效率.ssDBD的融合还使CBEs的编辑窗口范围得到了提高,从而拓宽了其靶向作用范围及用于疾病建模和基因治疗的适用范围.David R. Liu团队[30]在CBE的基础上融合了尿嘧啶糖基化酶抑制剂(uracil glycosylase inhibitor,UGI),作为一种体量较小的蛋白(9.5kDa),UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率.该团队还发现通过招募多拷贝的脱氨酶或更改其空间位置以优化编辑窗口能有效提高编辑效率和特异性.此外,与nCas9融合的脱氨酶若工程化改造后具有序列偏好性,则可通过降低旁观者编辑效应从而提高编辑精度.尽管ABEs和CBEs碱基编辑器的性能不断提升,A:T到G:C和C:G到T:A碱基对的单向固定替换仍限制着BEs作为单碱基改变工具使用时的灵活性.2020年,Zhao等[77]通过组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶(uracil-DNA glycosylase,Ung)实现了大肠杆菌中C到A 87.2%的编辑效率和哺乳动物细胞C到G 5.3%~53.0%的编辑效率.先导编辑作为一种多功能编辑手段,虽然同时具备可编程性和高精确度,但对于单碱基编辑的效率仍与BEs有较大差距[28].2021年,Song等[78]构建的hyPE2在PE2的基础上融合加入了一个ssDBD结构域Rad51,使得PE2的编辑效率最高提升了2.6倍,同时该团队还通过深度学习开发了能够预测不同的hyPE2与PE2相比编辑效率提升倍数的计算模型PEselector,该工具的网址为http://deepcrispr.info/PEselector.2022年,Velimirovic等[112]通过高通量肽链自编辑测序(peptide self-editing sequencing assay,PepSEq)测出12000个85个氨基酸的肽段对PE效率的影响,从而通过肽链融合提高PE的效率.综上所述,基于CRIPSR/Cas9系统的碱基编辑器编辑效率、编辑精度的不断提高以及编辑窗口乃至碱基固定替换模式的扩展相对于大量已知且不断增加的遗传病相关点突变数量仍捉襟见肘,各种脱氨酶赋予CRIPSR/Cas9系统的碱基编辑能力在其他结构域如UGI和ssDBD加持下不仅能够发挥CRIPSR系统高效率、高灵活、易编程的优势,也能够充分发挥脱氨酶的效能,但仍需进一步优化它们以有效地转换碱基和在无旁观者编辑效应的条件下进行目的碱基替换. ...
Transcription factors: an overview
1
1997
... 转录因子通过结合或识别基因上游特定区域,进而调节RNA聚合酶的招募,最终使靶基因表达上调或下调[79-80].其不仅可以与启动子区域结合,多个转录因子间也可结合形成转录因子复合体进而对靶基因进行更复杂、精细的调控,促进或抑制DNA上遗传信息向RNA转录的过程.在基因组范围上对靶基因进行调控是探究基因功能、重塑代谢调控网络的重要方法[62].自20世纪末以来,研究人员就已经将自然存在的或工程化后的DNA靶向蛋白如四环素阻遏蛋白[81-82]、Gal4蛋白[83]、锌指蛋白[84]以及TALE蛋白[85-87]等与转录因子融合进行靶基因调控.但由于依赖蛋白质靶向 DNA的特性,靶向不同的DNA序列时需要工程化设计不同的蛋白质氨基酸序列,使得这些蛋白普遍可编程性较差且分子庞大,因此开发用于基因组范围的转录调控蛋白文库相当耗时且价格昂贵[9].CRISPR基因调控系统包括CRISPR激活系统(CRISPR activation system,CRISPRa)和CRISPR干扰系统(CRISPR interference system,CRISPRi),该系统将转录激活或抑制因子与dCas9蛋白融合后,借助CRISPR系统精准的靶向性和灵活的可编程性将融合蛋白靶向至目的基因从而对该基因的转录进行激活或干扰(图2)[62,81-88].不同于其他DNA靶向蛋白,CRISPR激活系统的dCas9-effector融合蛋白利用gRNA与靶序列的碱基互补配对进行精确定位并最终实现目的基因调控,同时gRNA的易编程性和dCas9蛋白较小的体量使该系统在基因组范围上进行调控成为了可能. ...
Too many transcription factors: positive and negative interactions
1
1990
... 转录因子通过结合或识别基因上游特定区域,进而调节RNA聚合酶的招募,最终使靶基因表达上调或下调[79-80].其不仅可以与启动子区域结合,多个转录因子间也可结合形成转录因子复合体进而对靶基因进行更复杂、精细的调控,促进或抑制DNA上遗传信息向RNA转录的过程.在基因组范围上对靶基因进行调控是探究基因功能、重塑代谢调控网络的重要方法[62].自20世纪末以来,研究人员就已经将自然存在的或工程化后的DNA靶向蛋白如四环素阻遏蛋白[81-82]、Gal4蛋白[83]、锌指蛋白[84]以及TALE蛋白[85-87]等与转录因子融合进行靶基因调控.但由于依赖蛋白质靶向 DNA的特性,靶向不同的DNA序列时需要工程化设计不同的蛋白质氨基酸序列,使得这些蛋白普遍可编程性较差且分子庞大,因此开发用于基因组范围的转录调控蛋白文库相当耗时且价格昂贵[9].CRISPR基因调控系统包括CRISPR激活系统(CRISPR activation system,CRISPRa)和CRISPR干扰系统(CRISPR interference system,CRISPRi),该系统将转录激活或抑制因子与dCas9蛋白融合后,借助CRISPR系统精准的靶向性和灵活的可编程性将融合蛋白靶向至目的基因从而对该基因的转录进行激活或干扰(图2)[62,81-88].不同于其他DNA靶向蛋白,CRISPR激活系统的dCas9-effector融合蛋白利用gRNA与靶序列的碱基互补配对进行精确定位并最终实现目的基因调控,同时gRNA的易编程性和dCas9蛋白较小的体量使该系统在基因组范围上进行调控成为了可能. ...
Tetracycline-reversible silencing of eukaryotic promoters
2
1995
... 转录因子通过结合或识别基因上游特定区域,进而调节RNA聚合酶的招募,最终使靶基因表达上调或下调[79-80].其不仅可以与启动子区域结合,多个转录因子间也可结合形成转录因子复合体进而对靶基因进行更复杂、精细的调控,促进或抑制DNA上遗传信息向RNA转录的过程.在基因组范围上对靶基因进行调控是探究基因功能、重塑代谢调控网络的重要方法[62].自20世纪末以来,研究人员就已经将自然存在的或工程化后的DNA靶向蛋白如四环素阻遏蛋白[81-82]、Gal4蛋白[83]、锌指蛋白[84]以及TALE蛋白[85-87]等与转录因子融合进行靶基因调控.但由于依赖蛋白质靶向 DNA的特性,靶向不同的DNA序列时需要工程化设计不同的蛋白质氨基酸序列,使得这些蛋白普遍可编程性较差且分子庞大,因此开发用于基因组范围的转录调控蛋白文库相当耗时且价格昂贵[9].CRISPR基因调控系统包括CRISPR激活系统(CRISPR activation system,CRISPRa)和CRISPR干扰系统(CRISPR interference system,CRISPRi),该系统将转录激活或抑制因子与dCas9蛋白融合后,借助CRISPR系统精准的靶向性和灵活的可编程性将融合蛋白靶向至目的基因从而对该基因的转录进行激活或干扰(图2)[62,81-88].不同于其他DNA靶向蛋白,CRISPR激活系统的dCas9-effector融合蛋白利用gRNA与靶序列的碱基互补配对进行精确定位并最终实现目的基因调控,同时gRNA的易编程性和dCas9蛋白较小的体量使该系统在基因组范围上进行调控成为了可能. ...
... ,81-88].不同于其他DNA靶向蛋白,CRISPR激活系统的dCas9-effector融合蛋白利用gRNA与靶序列的碱基互补配对进行精确定位并最终实现目的基因调控,同时gRNA的易编程性和dCas9蛋白较小的体量使该系统在基因组范围上进行调控成为了可能. ...
Tight control of gene expression in mammalian cells by tetracycline-responsive promoters
1
1992
... 转录因子通过结合或识别基因上游特定区域,进而调节RNA聚合酶的招募,最终使靶基因表达上调或下调[79-80].其不仅可以与启动子区域结合,多个转录因子间也可结合形成转录因子复合体进而对靶基因进行更复杂、精细的调控,促进或抑制DNA上遗传信息向RNA转录的过程.在基因组范围上对靶基因进行调控是探究基因功能、重塑代谢调控网络的重要方法[62].自20世纪末以来,研究人员就已经将自然存在的或工程化后的DNA靶向蛋白如四环素阻遏蛋白[81-82]、Gal4蛋白[83]、锌指蛋白[84]以及TALE蛋白[85-87]等与转录因子融合进行靶基因调控.但由于依赖蛋白质靶向 DNA的特性,靶向不同的DNA序列时需要工程化设计不同的蛋白质氨基酸序列,使得这些蛋白普遍可编程性较差且分子庞大,因此开发用于基因组范围的转录调控蛋白文库相当耗时且价格昂贵[9].CRISPR基因调控系统包括CRISPR激活系统(CRISPR activation system,CRISPRa)和CRISPR干扰系统(CRISPR interference system,CRISPRi),该系统将转录激活或抑制因子与dCas9蛋白融合后,借助CRISPR系统精准的靶向性和灵活的可编程性将融合蛋白靶向至目的基因从而对该基因的转录进行激活或干扰(图2)[62,81-88].不同于其他DNA靶向蛋白,CRISPR激活系统的dCas9-effector融合蛋白利用gRNA与靶序列的碱基互补配对进行精确定位并最终实现目的基因调控,同时gRNA的易编程性和dCas9蛋白较小的体量使该系统在基因组范围上进行调控成为了可能. ...
GAL4-VP16 is an unusually potent transcriptional activator
1
1988
... 转录因子通过结合或识别基因上游特定区域,进而调节RNA聚合酶的招募,最终使靶基因表达上调或下调[79-80].其不仅可以与启动子区域结合,多个转录因子间也可结合形成转录因子复合体进而对靶基因进行更复杂、精细的调控,促进或抑制DNA上遗传信息向RNA转录的过程.在基因组范围上对靶基因进行调控是探究基因功能、重塑代谢调控网络的重要方法[62].自20世纪末以来,研究人员就已经将自然存在的或工程化后的DNA靶向蛋白如四环素阻遏蛋白[81-82]、Gal4蛋白[83]、锌指蛋白[84]以及TALE蛋白[85-87]等与转录因子融合进行靶基因调控.但由于依赖蛋白质靶向 DNA的特性,靶向不同的DNA序列时需要工程化设计不同的蛋白质氨基酸序列,使得这些蛋白普遍可编程性较差且分子庞大,因此开发用于基因组范围的转录调控蛋白文库相当耗时且价格昂贵[9].CRISPR基因调控系统包括CRISPR激活系统(CRISPR activation system,CRISPRa)和CRISPR干扰系统(CRISPR interference system,CRISPRi),该系统将转录激活或抑制因子与dCas9蛋白融合后,借助CRISPR系统精准的靶向性和灵活的可编程性将融合蛋白靶向至目的基因从而对该基因的转录进行激活或干扰(图2)[62,81-88].不同于其他DNA靶向蛋白,CRISPR激活系统的dCas9-effector融合蛋白利用gRNA与靶序列的碱基互补配对进行精确定位并最终实现目的基因调控,同时gRNA的易编程性和dCas9蛋白较小的体量使该系统在基因组范围上进行调控成为了可能. ...
Synthetic zinc finger transcription factor action at an endogenous chromosomal site: activation of the human erythropoietin gene
1
2000
... 转录因子通过结合或识别基因上游特定区域,进而调节RNA聚合酶的招募,最终使靶基因表达上调或下调[79-80].其不仅可以与启动子区域结合,多个转录因子间也可结合形成转录因子复合体进而对靶基因进行更复杂、精细的调控,促进或抑制DNA上遗传信息向RNA转录的过程.在基因组范围上对靶基因进行调控是探究基因功能、重塑代谢调控网络的重要方法[62].自20世纪末以来,研究人员就已经将自然存在的或工程化后的DNA靶向蛋白如四环素阻遏蛋白[81-82]、Gal4蛋白[83]、锌指蛋白[84]以及TALE蛋白[85-87]等与转录因子融合进行靶基因调控.但由于依赖蛋白质靶向 DNA的特性,靶向不同的DNA序列时需要工程化设计不同的蛋白质氨基酸序列,使得这些蛋白普遍可编程性较差且分子庞大,因此开发用于基因组范围的转录调控蛋白文库相当耗时且价格昂贵[9].CRISPR基因调控系统包括CRISPR激活系统(CRISPR activation system,CRISPRa)和CRISPR干扰系统(CRISPR interference system,CRISPRi),该系统将转录激活或抑制因子与dCas9蛋白融合后,借助CRISPR系统精准的靶向性和灵活的可编程性将融合蛋白靶向至目的基因从而对该基因的转录进行激活或干扰(图2)[62,81-88].不同于其他DNA靶向蛋白,CRISPR激活系统的dCas9-effector融合蛋白利用gRNA与靶序列的碱基互补配对进行精确定位并最终实现目的基因调控,同时gRNA的易编程性和dCas9蛋白较小的体量使该系统在基因组范围上进行调控成为了可能. ...
Comprehensive interrogation of natural TALE DNA-binding modules and transcriptional repressor domains
1
2012
... 转录因子通过结合或识别基因上游特定区域,进而调节RNA聚合酶的招募,最终使靶基因表达上调或下调[79-80].其不仅可以与启动子区域结合,多个转录因子间也可结合形成转录因子复合体进而对靶基因进行更复杂、精细的调控,促进或抑制DNA上遗传信息向RNA转录的过程.在基因组范围上对靶基因进行调控是探究基因功能、重塑代谢调控网络的重要方法[62].自20世纪末以来,研究人员就已经将自然存在的或工程化后的DNA靶向蛋白如四环素阻遏蛋白[81-82]、Gal4蛋白[83]、锌指蛋白[84]以及TALE蛋白[85-87]等与转录因子融合进行靶基因调控.但由于依赖蛋白质靶向 DNA的特性,靶向不同的DNA序列时需要工程化设计不同的蛋白质氨基酸序列,使得这些蛋白普遍可编程性较差且分子庞大,因此开发用于基因组范围的转录调控蛋白文库相当耗时且价格昂贵[9].CRISPR基因调控系统包括CRISPR激活系统(CRISPR activation system,CRISPRa)和CRISPR干扰系统(CRISPR interference system,CRISPRi),该系统将转录激活或抑制因子与dCas9蛋白融合后,借助CRISPR系统精准的靶向性和灵活的可编程性将融合蛋白靶向至目的基因从而对该基因的转录进行激活或干扰(图2)[62,81-88].不同于其他DNA靶向蛋白,CRISPR激活系统的dCas9-effector融合蛋白利用gRNA与靶序列的碱基互补配对进行精确定位并最终实现目的基因调控,同时gRNA的易编程性和dCas9蛋白较小的体量使该系统在基因组范围上进行调控成为了可能. ...
Robust, synergistic regulation of human gene expression using TALE activators
0
2013
Synergistic and tunable human gene activation by combinations of synthetic transcription factors
1
2013
... 转录因子通过结合或识别基因上游特定区域,进而调节RNA聚合酶的招募,最终使靶基因表达上调或下调[79-80].其不仅可以与启动子区域结合,多个转录因子间也可结合形成转录因子复合体进而对靶基因进行更复杂、精细的调控,促进或抑制DNA上遗传信息向RNA转录的过程.在基因组范围上对靶基因进行调控是探究基因功能、重塑代谢调控网络的重要方法[62].自20世纪末以来,研究人员就已经将自然存在的或工程化后的DNA靶向蛋白如四环素阻遏蛋白[81-82]、Gal4蛋白[83]、锌指蛋白[84]以及TALE蛋白[85-87]等与转录因子融合进行靶基因调控.但由于依赖蛋白质靶向 DNA的特性,靶向不同的DNA序列时需要工程化设计不同的蛋白质氨基酸序列,使得这些蛋白普遍可编程性较差且分子庞大,因此开发用于基因组范围的转录调控蛋白文库相当耗时且价格昂贵[9].CRISPR基因调控系统包括CRISPR激活系统(CRISPR activation system,CRISPRa)和CRISPR干扰系统(CRISPR interference system,CRISPRi),该系统将转录激活或抑制因子与dCas9蛋白融合后,借助CRISPR系统精准的靶向性和灵活的可编程性将融合蛋白靶向至目的基因从而对该基因的转录进行激活或干扰(图2)[62,81-88].不同于其他DNA靶向蛋白,CRISPR激活系统的dCas9-effector融合蛋白利用gRNA与靶序列的碱基互补配对进行精确定位并最终实现目的基因调控,同时gRNA的易编程性和dCas9蛋白较小的体量使该系统在基因组范围上进行调控成为了可能. ...
Genome-scale CRISPR-mediated control of gene repression and activation
1
2014
... 转录因子通过结合或识别基因上游特定区域,进而调节RNA聚合酶的招募,最终使靶基因表达上调或下调[79-80].其不仅可以与启动子区域结合,多个转录因子间也可结合形成转录因子复合体进而对靶基因进行更复杂、精细的调控,促进或抑制DNA上遗传信息向RNA转录的过程.在基因组范围上对靶基因进行调控是探究基因功能、重塑代谢调控网络的重要方法[62].自20世纪末以来,研究人员就已经将自然存在的或工程化后的DNA靶向蛋白如四环素阻遏蛋白[81-82]、Gal4蛋白[83]、锌指蛋白[84]以及TALE蛋白[85-87]等与转录因子融合进行靶基因调控.但由于依赖蛋白质靶向 DNA的特性,靶向不同的DNA序列时需要工程化设计不同的蛋白质氨基酸序列,使得这些蛋白普遍可编程性较差且分子庞大,因此开发用于基因组范围的转录调控蛋白文库相当耗时且价格昂贵[9].CRISPR基因调控系统包括CRISPR激活系统(CRISPR activation system,CRISPRa)和CRISPR干扰系统(CRISPR interference system,CRISPRi),该系统将转录激活或抑制因子与dCas9蛋白融合后,借助CRISPR系统精准的靶向性和灵活的可编程性将融合蛋白靶向至目的基因从而对该基因的转录进行激活或干扰(图2)[62,81-88].不同于其他DNA靶向蛋白,CRISPR激活系统的dCas9-effector融合蛋白利用gRNA与靶序列的碱基互补配对进行精确定位并最终实现目的基因调控,同时gRNA的易编程性和dCas9蛋白较小的体量使该系统在基因组范围上进行调控成为了可能. ...
CRISPR interference (CRISPRi) for sequence-specific control of gene expression
1
2013
... 仅将dCas9蛋白靶向至目的基因上游时,dCas9蛋白对RNA聚合酶的空间位阻效应便可在一定程度上抑制基因表达[22],而该效应抑制表达的方式仅在原核细胞中有较高的效率,在真核细胞中由于其内部复杂的调控机制使得抑制效率较低[22,62,89].将dCas9蛋白与转录抑制结构域如KRAB[62]、MXI1[62]和TUP1[63]等蛋白融合表达则可提高CRISPRi系统在真核细胞中的抑制效率.其中KRAB锌指蛋白(KRAB-ZFP)属于哺乳动物细胞中最大的转录调节因子家族[90],该蛋白能够通过诱导靶序列的染色质结构变化从而抑制基因转录[91].KRAB与dCas9融合后被靶向至启动子区、5′非翻译区以及近端或远端增强子元件区后可以有效地沉默单个基因或非编码RNA[62,92-94].2018年,Yeo等[64]表征了20多种已知的能够诱导基因沉默的转录因子的抑制效率后,将获得的最高效的6种抑制因子——MeCP2、SIN3A、HDT1、MBD2B、NIPP1和HP1A分别与通用抑制因子KRAB融合形成双重抑制因子,再与dCas9蛋白C端融合表达并表征CRISPRi系统的抑制效率,最终获得了比单独dCas9抑制效率提高了60倍的双重抑制因子融合蛋白dCas9-KRAB-MeCP2.而之后研究人员继续表征的三重抑制因子融合蛋白可能由于功能冗余或蛋白体量较大从而影响抑制因子正常工作导致抑制效率不如双重抑制因子,证明了通过无限累加融合的方式提高融合蛋白的调控效率是不可行的[64]. ...
The signaling pathways regulated by KRAB zinc-finger proteins in cancer
1
2022
... 仅将dCas9蛋白靶向至目的基因上游时,dCas9蛋白对RNA聚合酶的空间位阻效应便可在一定程度上抑制基因表达[22],而该效应抑制表达的方式仅在原核细胞中有较高的效率,在真核细胞中由于其内部复杂的调控机制使得抑制效率较低[22,62,89].将dCas9蛋白与转录抑制结构域如KRAB[62]、MXI1[62]和TUP1[63]等蛋白融合表达则可提高CRISPRi系统在真核细胞中的抑制效率.其中KRAB锌指蛋白(KRAB-ZFP)属于哺乳动物细胞中最大的转录调节因子家族[90],该蛋白能够通过诱导靶序列的染色质结构变化从而抑制基因转录[91].KRAB与dCas9融合后被靶向至启动子区、5′非翻译区以及近端或远端增强子元件区后可以有效地沉默单个基因或非编码RNA[62,92-94].2018年,Yeo等[64]表征了20多种已知的能够诱导基因沉默的转录因子的抑制效率后,将获得的最高效的6种抑制因子——MeCP2、SIN3A、HDT1、MBD2B、NIPP1和HP1A分别与通用抑制因子KRAB融合形成双重抑制因子,再与dCas9蛋白C端融合表达并表征CRISPRi系统的抑制效率,最终获得了比单独dCas9抑制效率提高了60倍的双重抑制因子融合蛋白dCas9-KRAB-MeCP2.而之后研究人员继续表征的三重抑制因子融合蛋白可能由于功能冗余或蛋白体量较大从而影响抑制因子正常工作导致抑制效率不如双重抑制因子,证明了通过无限累加融合的方式提高融合蛋白的调控效率是不可行的[64]. ...
Cas9 effector-mediated regulation of transcription and differentiation in human pluripotent stem cells
1
2014
... 仅将dCas9蛋白靶向至目的基因上游时,dCas9蛋白对RNA聚合酶的空间位阻效应便可在一定程度上抑制基因表达[22],而该效应抑制表达的方式仅在原核细胞中有较高的效率,在真核细胞中由于其内部复杂的调控机制使得抑制效率较低[22,62,89].将dCas9蛋白与转录抑制结构域如KRAB[62]、MXI1[62]和TUP1[63]等蛋白融合表达则可提高CRISPRi系统在真核细胞中的抑制效率.其中KRAB锌指蛋白(KRAB-ZFP)属于哺乳动物细胞中最大的转录调节因子家族[90],该蛋白能够通过诱导靶序列的染色质结构变化从而抑制基因转录[91].KRAB与dCas9融合后被靶向至启动子区、5′非翻译区以及近端或远端增强子元件区后可以有效地沉默单个基因或非编码RNA[62,92-94].2018年,Yeo等[64]表征了20多种已知的能够诱导基因沉默的转录因子的抑制效率后,将获得的最高效的6种抑制因子——MeCP2、SIN3A、HDT1、MBD2B、NIPP1和HP1A分别与通用抑制因子KRAB融合形成双重抑制因子,再与dCas9蛋白C端融合表达并表征CRISPRi系统的抑制效率,最终获得了比单独dCas9抑制效率提高了60倍的双重抑制因子融合蛋白dCas9-KRAB-MeCP2.而之后研究人员继续表征的三重抑制因子融合蛋白可能由于功能冗余或蛋白体量较大从而影响抑制因子正常工作导致抑制效率不如双重抑制因子,证明了通过无限累加融合的方式提高融合蛋白的调控效率是不可行的[64]. ...
Comparison of TALE designer transcription factors and the CRISPR/dCas9 in regulation of gene expression by targeting enhancers
1
2014
... 仅将dCas9蛋白靶向至目的基因上游时,dCas9蛋白对RNA聚合酶的空间位阻效应便可在一定程度上抑制基因表达[22],而该效应抑制表达的方式仅在原核细胞中有较高的效率,在真核细胞中由于其内部复杂的调控机制使得抑制效率较低[22,62,89].将dCas9蛋白与转录抑制结构域如KRAB[62]、MXI1[62]和TUP1[63]等蛋白融合表达则可提高CRISPRi系统在真核细胞中的抑制效率.其中KRAB锌指蛋白(KRAB-ZFP)属于哺乳动物细胞中最大的转录调节因子家族[90],该蛋白能够通过诱导靶序列的染色质结构变化从而抑制基因转录[91].KRAB与dCas9融合后被靶向至启动子区、5′非翻译区以及近端或远端增强子元件区后可以有效地沉默单个基因或非编码RNA[62,92-94].2018年,Yeo等[64]表征了20多种已知的能够诱导基因沉默的转录因子的抑制效率后,将获得的最高效的6种抑制因子——MeCP2、SIN3A、HDT1、MBD2B、NIPP1和HP1A分别与通用抑制因子KRAB融合形成双重抑制因子,再与dCas9蛋白C端融合表达并表征CRISPRi系统的抑制效率,最终获得了比单独dCas9抑制效率提高了60倍的双重抑制因子融合蛋白dCas9-KRAB-MeCP2.而之后研究人员继续表征的三重抑制因子融合蛋白可能由于功能冗余或蛋白体量较大从而影响抑制因子正常工作导致抑制效率不如双重抑制因子,证明了通过无限累加融合的方式提高融合蛋白的调控效率是不可行的[64]. ...
Functional annotation of native enhancers with a Cas9-histone demethylase fusion
0
2015
Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements
1
2015
... 仅将dCas9蛋白靶向至目的基因上游时,dCas9蛋白对RNA聚合酶的空间位阻效应便可在一定程度上抑制基因表达[22],而该效应抑制表达的方式仅在原核细胞中有较高的效率,在真核细胞中由于其内部复杂的调控机制使得抑制效率较低[22,62,89].将dCas9蛋白与转录抑制结构域如KRAB[62]、MXI1[62]和TUP1[63]等蛋白融合表达则可提高CRISPRi系统在真核细胞中的抑制效率.其中KRAB锌指蛋白(KRAB-ZFP)属于哺乳动物细胞中最大的转录调节因子家族[90],该蛋白能够通过诱导靶序列的染色质结构变化从而抑制基因转录[91].KRAB与dCas9融合后被靶向至启动子区、5′非翻译区以及近端或远端增强子元件区后可以有效地沉默单个基因或非编码RNA[62,92-94].2018年,Yeo等[64]表征了20多种已知的能够诱导基因沉默的转录因子的抑制效率后,将获得的最高效的6种抑制因子——MeCP2、SIN3A、HDT1、MBD2B、NIPP1和HP1A分别与通用抑制因子KRAB融合形成双重抑制因子,再与dCas9蛋白C端融合表达并表征CRISPRi系统的抑制效率,最终获得了比单独dCas9抑制效率提高了60倍的双重抑制因子融合蛋白dCas9-KRAB-MeCP2.而之后研究人员继续表征的三重抑制因子融合蛋白可能由于功能冗余或蛋白体量较大从而影响抑制因子正常工作导致抑制效率不如双重抑制因子,证明了通过无限累加融合的方式提高融合蛋白的调控效率是不可行的[64]. ...
A protein-tagging system for signal amplification in gene expression and fluorescence imaging
1
2014
... 将dCas9与各种转录激活因子融合后,则可在gRNA的靶向作用下精确激活目的基因,该系统称为CRISPR激活系统或CRISPRa.最初的CRISPRa系统中dCas9仅融合了单个反式激活因子p65(也被称为RelA,是构成NF-κB转录因子家族的五种成员之一)或VP64 (由单纯疱疹病毒蛋白VP16的四个重复结构域构成),这些早期工作共同构成了最初的可编程dCas9基因激活工具箱,但效果差强人意[23-28,39-44,62,65-66].为了不断提高CRISPRa系统的激活效率,研究人员尝试了用各种方式将更复杂或多拷贝的激活结构域应用于CRISPRa.2015年,Chavez等[67]通过蛋白质理性设计,构建了三重融合激活因子VP64-p65-Rta(VPR)并应用于CRISPRa系统,其中Rta是来源于EB病毒(epstein-barr virus)的反式激活因子.与dCas9-VP64相比,dCas9-VPR使人胚胎肾细胞293(HEK 293T cells)的内源靶基因表达上调了22~320倍,同时证明了该系统也能够在多重基因的激活表达与刺激人诱导多功能干细胞(induced pluripotent stem cells,iPSCs)的神经元分化上发挥作用.同年,张锋研究团队[68]开发的基于CRISPRa的协同激活介导系统(synergistic activation mediator system, SAM)通过修饰sgRNA使其携带了两个MS2发夹二级结构,每个发夹结构都能招募一个MS2-p65-HSF1融合蛋白,其中包括反式激活因子p65、MS2结合蛋白以及热休克蛋白1(heat shock protein 1),最终证明该修饰后的gRNA与dCas9-VP64的协同作用能够使10个靶基因的表达均上调2倍以上.2022年,山东大学候进课题组[69]构建的CRISPR介导的蛋白质标签信号放大系统首次将SPY系统(包括SpyTag和SpyCatcher)和SunTag系统分别引入dCas9介导的CRISPRa和dCpf1(一种丧失核酸酶活性的Cas9同源蛋白)介导的CRISPRi系统并进行骨架长度和转录因子优化后,成功使酿酒酵母内目的基因转录水平分别提高了34.9倍以及受到几乎完全的抑制.其中,SunTag系统是由若干个拷贝的GCN4肽以及作为标签的抗GCN4抗体的单链可变片段scFv组成[95],与dCas9融合的多拷贝GCN4肽通过招募多拷贝的转录因子与GCN4抗体标签的融合蛋白从而进行转录调控,该系统除了与CRISPR系统结合后用于碱基编辑以及基因调控,还在哺乳动物和植物细胞的DNA的甲基化和去甲基化诱导中发挥作用[96-100];而SPY系统作为一种广泛使用的蛋白质组装系统首次在酿酒酵母的转录调控中得到了应用[69].可见在dCas9与转录因子融合的基础上,CRISPRa与CRISPRi系统能够利用融合多重结构域或融合蛋白质骨架系统的方式提高调控效率.而通过利用RNA与蛋白质间的相互作用,将多个适配子融合在gRNA中使其同时含有目的基因靶向序列和转录因子招募序列的方式能够产生与dCas9融合多个转录激活或抑制结构域的策略类似的效果[101].此外,针对不同细胞乃至不同dCas9同源蛋白,CRISPRa与CRISPRi系统均需对不同转录激活或抑制结构域乃至其组合进行优化才能发挥该系统的最大效能. ...
BE-PLUS: a new base editing tool with broadened editing window and enhanced fidelity
1
2018
... 将dCas9与各种转录激活因子融合后,则可在gRNA的靶向作用下精确激活目的基因,该系统称为CRISPR激活系统或CRISPRa.最初的CRISPRa系统中dCas9仅融合了单个反式激活因子p65(也被称为RelA,是构成NF-κB转录因子家族的五种成员之一)或VP64 (由单纯疱疹病毒蛋白VP16的四个重复结构域构成),这些早期工作共同构成了最初的可编程dCas9基因激活工具箱,但效果差强人意[23-28,39-44,62,65-66].为了不断提高CRISPRa系统的激活效率,研究人员尝试了用各种方式将更复杂或多拷贝的激活结构域应用于CRISPRa.2015年,Chavez等[67]通过蛋白质理性设计,构建了三重融合激活因子VP64-p65-Rta(VPR)并应用于CRISPRa系统,其中Rta是来源于EB病毒(epstein-barr virus)的反式激活因子.与dCas9-VP64相比,dCas9-VPR使人胚胎肾细胞293(HEK 293T cells)的内源靶基因表达上调了22~320倍,同时证明了该系统也能够在多重基因的激活表达与刺激人诱导多功能干细胞(induced pluripotent stem cells,iPSCs)的神经元分化上发挥作用.同年,张锋研究团队[68]开发的基于CRISPRa的协同激活介导系统(synergistic activation mediator system, SAM)通过修饰sgRNA使其携带了两个MS2发夹二级结构,每个发夹结构都能招募一个MS2-p65-HSF1融合蛋白,其中包括反式激活因子p65、MS2结合蛋白以及热休克蛋白1(heat shock protein 1),最终证明该修饰后的gRNA与dCas9-VP64的协同作用能够使10个靶基因的表达均上调2倍以上.2022年,山东大学候进课题组[69]构建的CRISPR介导的蛋白质标签信号放大系统首次将SPY系统(包括SpyTag和SpyCatcher)和SunTag系统分别引入dCas9介导的CRISPRa和dCpf1(一种丧失核酸酶活性的Cas9同源蛋白)介导的CRISPRi系统并进行骨架长度和转录因子优化后,成功使酿酒酵母内目的基因转录水平分别提高了34.9倍以及受到几乎完全的抑制.其中,SunTag系统是由若干个拷贝的GCN4肽以及作为标签的抗GCN4抗体的单链可变片段scFv组成[95],与dCas9融合的多拷贝GCN4肽通过招募多拷贝的转录因子与GCN4抗体标签的融合蛋白从而进行转录调控,该系统除了与CRISPR系统结合后用于碱基编辑以及基因调控,还在哺乳动物和植物细胞的DNA的甲基化和去甲基化诱导中发挥作用[96-100];而SPY系统作为一种广泛使用的蛋白质组装系统首次在酿酒酵母的转录调控中得到了应用[69].可见在dCas9与转录因子融合的基础上,CRISPRa与CRISPRi系统能够利用融合多重结构域或融合蛋白质骨架系统的方式提高调控效率.而通过利用RNA与蛋白质间的相互作用,将多个适配子融合在gRNA中使其同时含有目的基因靶向序列和转录因子招募序列的方式能够产生与dCas9融合多个转录激活或抑制结构域的策略类似的效果[101].此外,针对不同细胞乃至不同dCas9同源蛋白,CRISPRa与CRISPRi系统均需对不同转录激活或抑制结构域乃至其组合进行优化才能发挥该系统的最大效能. ...
Targeted DNA demethylation of the Arabidopsis genome using the human TET1 catalytic domain
0
2018
Targeted DNA demethylation in vivo using dCas9-peptide repeat and scFv-TET1 catalytic domain fusions
0
2016
DNA epigenome editing using CRISPR-Cas suntag-directed DNMT3A
0
2017
Gene activation in human cells using CRISPR/Cpf1-p300 and CRISPR/Cpf1-SunTag systems
1
2018
... 将dCas9与各种转录激活因子融合后,则可在gRNA的靶向作用下精确激活目的基因,该系统称为CRISPR激活系统或CRISPRa.最初的CRISPRa系统中dCas9仅融合了单个反式激活因子p65(也被称为RelA,是构成NF-κB转录因子家族的五种成员之一)或VP64 (由单纯疱疹病毒蛋白VP16的四个重复结构域构成),这些早期工作共同构成了最初的可编程dCas9基因激活工具箱,但效果差强人意[23-28,39-44,62,65-66].为了不断提高CRISPRa系统的激活效率,研究人员尝试了用各种方式将更复杂或多拷贝的激活结构域应用于CRISPRa.2015年,Chavez等[67]通过蛋白质理性设计,构建了三重融合激活因子VP64-p65-Rta(VPR)并应用于CRISPRa系统,其中Rta是来源于EB病毒(epstein-barr virus)的反式激活因子.与dCas9-VP64相比,dCas9-VPR使人胚胎肾细胞293(HEK 293T cells)的内源靶基因表达上调了22~320倍,同时证明了该系统也能够在多重基因的激活表达与刺激人诱导多功能干细胞(induced pluripotent stem cells,iPSCs)的神经元分化上发挥作用.同年,张锋研究团队[68]开发的基于CRISPRa的协同激活介导系统(synergistic activation mediator system, SAM)通过修饰sgRNA使其携带了两个MS2发夹二级结构,每个发夹结构都能招募一个MS2-p65-HSF1融合蛋白,其中包括反式激活因子p65、MS2结合蛋白以及热休克蛋白1(heat shock protein 1),最终证明该修饰后的gRNA与dCas9-VP64的协同作用能够使10个靶基因的表达均上调2倍以上.2022年,山东大学候进课题组[69]构建的CRISPR介导的蛋白质标签信号放大系统首次将SPY系统(包括SpyTag和SpyCatcher)和SunTag系统分别引入dCas9介导的CRISPRa和dCpf1(一种丧失核酸酶活性的Cas9同源蛋白)介导的CRISPRi系统并进行骨架长度和转录因子优化后,成功使酿酒酵母内目的基因转录水平分别提高了34.9倍以及受到几乎完全的抑制.其中,SunTag系统是由若干个拷贝的GCN4肽以及作为标签的抗GCN4抗体的单链可变片段scFv组成[95],与dCas9融合的多拷贝GCN4肽通过招募多拷贝的转录因子与GCN4抗体标签的融合蛋白从而进行转录调控,该系统除了与CRISPR系统结合后用于碱基编辑以及基因调控,还在哺乳动物和植物细胞的DNA的甲基化和去甲基化诱导中发挥作用[96-100];而SPY系统作为一种广泛使用的蛋白质组装系统首次在酿酒酵母的转录调控中得到了应用[69].可见在dCas9与转录因子融合的基础上,CRISPRa与CRISPRi系统能够利用融合多重结构域或融合蛋白质骨架系统的方式提高调控效率.而通过利用RNA与蛋白质间的相互作用,将多个适配子融合在gRNA中使其同时含有目的基因靶向序列和转录因子招募序列的方式能够产生与dCas9融合多个转录激活或抑制结构域的策略类似的效果[101].此外,针对不同细胞乃至不同dCas9同源蛋白,CRISPRa与CRISPRi系统均需对不同转录激活或抑制结构域乃至其组合进行优化才能发挥该系统的最大效能. ...
Spatiotemporal control of CRISPR/Cas9 gene editing
1
2021
... 将dCas9与各种转录激活因子融合后,则可在gRNA的靶向作用下精确激活目的基因,该系统称为CRISPR激活系统或CRISPRa.最初的CRISPRa系统中dCas9仅融合了单个反式激活因子p65(也被称为RelA,是构成NF-κB转录因子家族的五种成员之一)或VP64 (由单纯疱疹病毒蛋白VP16的四个重复结构域构成),这些早期工作共同构成了最初的可编程dCas9基因激活工具箱,但效果差强人意[23-28,39-44,62,65-66].为了不断提高CRISPRa系统的激活效率,研究人员尝试了用各种方式将更复杂或多拷贝的激活结构域应用于CRISPRa.2015年,Chavez等[67]通过蛋白质理性设计,构建了三重融合激活因子VP64-p65-Rta(VPR)并应用于CRISPRa系统,其中Rta是来源于EB病毒(epstein-barr virus)的反式激活因子.与dCas9-VP64相比,dCas9-VPR使人胚胎肾细胞293(HEK 293T cells)的内源靶基因表达上调了22~320倍,同时证明了该系统也能够在多重基因的激活表达与刺激人诱导多功能干细胞(induced pluripotent stem cells,iPSCs)的神经元分化上发挥作用.同年,张锋研究团队[68]开发的基于CRISPRa的协同激活介导系统(synergistic activation mediator system, SAM)通过修饰sgRNA使其携带了两个MS2发夹二级结构,每个发夹结构都能招募一个MS2-p65-HSF1融合蛋白,其中包括反式激活因子p65、MS2结合蛋白以及热休克蛋白1(heat shock protein 1),最终证明该修饰后的gRNA与dCas9-VP64的协同作用能够使10个靶基因的表达均上调2倍以上.2022年,山东大学候进课题组[69]构建的CRISPR介导的蛋白质标签信号放大系统首次将SPY系统(包括SpyTag和SpyCatcher)和SunTag系统分别引入dCas9介导的CRISPRa和dCpf1(一种丧失核酸酶活性的Cas9同源蛋白)介导的CRISPRi系统并进行骨架长度和转录因子优化后,成功使酿酒酵母内目的基因转录水平分别提高了34.9倍以及受到几乎完全的抑制.其中,SunTag系统是由若干个拷贝的GCN4肽以及作为标签的抗GCN4抗体的单链可变片段scFv组成[95],与dCas9融合的多拷贝GCN4肽通过招募多拷贝的转录因子与GCN4抗体标签的融合蛋白从而进行转录调控,该系统除了与CRISPR系统结合后用于碱基编辑以及基因调控,还在哺乳动物和植物细胞的DNA的甲基化和去甲基化诱导中发挥作用[96-100];而SPY系统作为一种广泛使用的蛋白质组装系统首次在酿酒酵母的转录调控中得到了应用[69].可见在dCas9与转录因子融合的基础上,CRISPRa与CRISPRi系统能够利用融合多重结构域或融合蛋白质骨架系统的方式提高调控效率.而通过利用RNA与蛋白质间的相互作用,将多个适配子融合在gRNA中使其同时含有目的基因靶向序列和转录因子招募序列的方式能够产生与dCas9融合多个转录激活或抑制结构域的策略类似的效果[101].此外,针对不同细胞乃至不同dCas9同源蛋白,CRISPRa与CRISPRi系统均需对不同转录激活或抑制结构域乃至其组合进行优化才能发挥该系统的最大效能. ...
Epigenetic modification enhances ergot alkaloid production of Claviceps purpurea
1
2019
... 通过使用基于CRISPR/dCas9系统的一系列融合蛋白工具可以实现诸多靶向的表观遗传修饰,如DNA甲基化和去甲基化、组蛋白乙酰基化/去乙酰化和甲基化/去甲基化等,从而调控基因的表达并在许多细胞过程、癌症治疗及细胞工厂中发挥关键作用(图2)[30].其中DNA甲基化如5′胞嘧啶甲基化(5mC)是一种稳定的抑制性化学修饰调控.2016年,研究人员将dCas9、KRAB转录抑制因子以及DNA甲基化酶(DNA methyltransferase)的催化结构域融合蛋白基因与7个gRNA在K562细胞中共表达后获得了β2-微球蛋白启动子-增强子78%的稳定沉默[70].2021年,Nunez等[71]开发的CRISPRoff甲基化修饰工具能够在细胞分裂及分化过程中稳定维持靶基因的甲基化并抑制其表达水平.DNA去甲基化是将甲基胞嘧啶双加氧酶1(methylcytosine dioxygenase 1,TET1)的肽链C端催化结构域与dCas9融合后,再靶向至目的基因启动子或增强子区,从而实现目的基因的表达上调.2016年,Choudhury等[72]将dCas9- TET1靶向至乳腺癌基因1(breast cancer 1,BRCA1)启动子后成功地上调其表达水平.BRCA1作为抑制恶性肿瘤发生的关键基因之一,其成功的靶向上调为研究癌变新疗法带来新的思路,证明了通过去甲基化靶向激活人类肿瘤抑制基因的疗法是可行的.同样的,将dCas9与人类组蛋白乙酰基转移酶p300的催化核心融合后靶向至启动子和增强子区从而催化组蛋白H3 第27位赖氨酸残基的乙酰基化也会造成较强的基因上调[73].2019年,Chen等[102]通过提高麦角生物碱合成基因簇(ergot alkaloid synthesis gene cluster)的组蛋白乙酰化水平,使麦角菌Cp-1菌株(Claviceps pururea Cp-1 strain)中的麦角生物碱含量提高了一倍.通过融合dCas9与适当的表观遗传效应蛋白,CRISPR表观遗传工程工具则可利用CRISPR系统稳健地靶向性实现各种所需的表观遗传修饰,而不同工具间的适当组合,理论上讲可以为表观遗传学工具箱带来更多新型和功能复杂的应用策略,如利用与CRISPRa和CRISPRi优化策略类似的方式优化CRISPR表观遗传工程工具箱的特异性、稳定性和性能[103]. ...
Engineering of effector domains for targeted DNA methylation with reduced off-target effects
1
2020
... 通过使用基于CRISPR/dCas9系统的一系列融合蛋白工具可以实现诸多靶向的表观遗传修饰,如DNA甲基化和去甲基化、组蛋白乙酰基化/去乙酰化和甲基化/去甲基化等,从而调控基因的表达并在许多细胞过程、癌症治疗及细胞工厂中发挥关键作用(图2)[30].其中DNA甲基化如5′胞嘧啶甲基化(5mC)是一种稳定的抑制性化学修饰调控.2016年,研究人员将dCas9、KRAB转录抑制因子以及DNA甲基化酶(DNA methyltransferase)的催化结构域融合蛋白基因与7个gRNA在K562细胞中共表达后获得了β2-微球蛋白启动子-增强子78%的稳定沉默[70].2021年,Nunez等[71]开发的CRISPRoff甲基化修饰工具能够在细胞分裂及分化过程中稳定维持靶基因的甲基化并抑制其表达水平.DNA去甲基化是将甲基胞嘧啶双加氧酶1(methylcytosine dioxygenase 1,TET1)的肽链C端催化结构域与dCas9融合后,再靶向至目的基因启动子或增强子区,从而实现目的基因的表达上调.2016年,Choudhury等[72]将dCas9- TET1靶向至乳腺癌基因1(breast cancer 1,BRCA1)启动子后成功地上调其表达水平.BRCA1作为抑制恶性肿瘤发生的关键基因之一,其成功的靶向上调为研究癌变新疗法带来新的思路,证明了通过去甲基化靶向激活人类肿瘤抑制基因的疗法是可行的.同样的,将dCas9与人类组蛋白乙酰基转移酶p300的催化核心融合后靶向至启动子和增强子区从而催化组蛋白H3 第27位赖氨酸残基的乙酰基化也会造成较强的基因上调[73].2019年,Chen等[102]通过提高麦角生物碱合成基因簇(ergot alkaloid synthesis gene cluster)的组蛋白乙酰化水平,使麦角菌Cp-1菌株(Claviceps pururea Cp-1 strain)中的麦角生物碱含量提高了一倍.通过融合dCas9与适当的表观遗传效应蛋白,CRISPR表观遗传工程工具则可利用CRISPR系统稳健地靶向性实现各种所需的表观遗传修饰,而不同工具间的适当组合,理论上讲可以为表观遗传学工具箱带来更多新型和功能复杂的应用策略,如利用与CRISPRa和CRISPRi优化策略类似的方式优化CRISPR表观遗传工程工具箱的特异性、稳定性和性能[103]. ...
acute and chronic diseases and injuries in 188 countries, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013
1
301
... 单点突变是人类遗传病中最常见的基因变异,且该变异通常会造成严重的后果.如镰刀形红细胞贫血症患者由于其血红蛋白基因中一对碱基对发生突变使得血红蛋白β亚基的N端第6个氨基酸发生改变,最终使血红蛋白结构发生改变并失去氧气输送功能[104].因此,利用CRISPR/Ca9系统的靶向性进行高效的单核苷酸转换是建立遗传病模型、开发基因纠正疗法的重要手段,同时促进了基础生物学研究和合成生物学的发展.通过共递送一段含有待编辑碱基的同源DNA,CRIPSR/Cas9系统可在HDR活性高的细胞中以较高的效率将目的碱基替换为任意碱基[16],而在HDR活性较低的细胞如大多数细菌和有丝分裂后期细胞中,该策略则无法正常发挥作用.此外,诱导HDR时Cas9对靶序列产生的双链切割带来的脱靶诱变风险以及HDR进行DNA修复时对细胞活力产生的不利影响均会使CRIPSR/Cas9系统在遗传病的基因疗法等应用场景上受到限制[105-106]. ...
CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response
1
2018
... 单点突变是人类遗传病中最常见的基因变异,且该变异通常会造成严重的后果.如镰刀形红细胞贫血症患者由于其血红蛋白基因中一对碱基对发生突变使得血红蛋白β亚基的N端第6个氨基酸发生改变,最终使血红蛋白结构发生改变并失去氧气输送功能[104].因此,利用CRISPR/Ca9系统的靶向性进行高效的单核苷酸转换是建立遗传病模型、开发基因纠正疗法的重要手段,同时促进了基础生物学研究和合成生物学的发展.通过共递送一段含有待编辑碱基的同源DNA,CRIPSR/Cas9系统可在HDR活性高的细胞中以较高的效率将目的碱基替换为任意碱基[16],而在HDR活性较低的细胞如大多数细菌和有丝分裂后期细胞中,该策略则无法正常发挥作用.此外,诱导HDR时Cas9对靶序列产生的双链切割带来的脱靶诱变风险以及HDR进行DNA修复时对细胞活力产生的不利影响均会使CRIPSR/Cas9系统在遗传病的基因疗法等应用场景上受到限制[105-106]. ...
p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells
1
2018
... 单点突变是人类遗传病中最常见的基因变异,且该变异通常会造成严重的后果.如镰刀形红细胞贫血症患者由于其血红蛋白基因中一对碱基对发生突变使得血红蛋白β亚基的N端第6个氨基酸发生改变,最终使血红蛋白结构发生改变并失去氧气输送功能[104].因此,利用CRISPR/Ca9系统的靶向性进行高效的单核苷酸转换是建立遗传病模型、开发基因纠正疗法的重要手段,同时促进了基础生物学研究和合成生物学的发展.通过共递送一段含有待编辑碱基的同源DNA,CRIPSR/Cas9系统可在HDR活性高的细胞中以较高的效率将目的碱基替换为任意碱基[16],而在HDR活性较低的细胞如大多数细菌和有丝分裂后期细胞中,该策略则无法正常发挥作用.此外,诱导HDR时Cas9对靶序列产生的双链切割带来的脱靶诱变风险以及HDR进行DNA修复时对细胞活力产生的不利影响均会使CRIPSR/Cas9系统在遗传病的基因疗法等应用场景上受到限制[105-106]. ...
单碱基编辑工具—腺嘌呤碱基编辑器ABE的研究进展
1
2019
... 为了改善CRIPSR/Cas9系统的碱基编辑工具箱,研究人员通过将nCas9与胞嘧啶脱氨酶(cytosine deaminase)或腺嘌呤脱氨酶(adenine deaminase)融合的方式在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换(图2),其中相应的融合蛋白分别被称为胞嘧啶碱基编辑器(cytosine base editors,CBEs)[30,74]和腺嘌呤碱基编辑器(adenine base editors,ABEs)[75].CBEs中的胞嘧啶脱氨酶能使被nCas9释放的单链DNA(single strand DNA,ssDNA)上的胞嘧啶C转变为尿嘧啶U,随后该位置产生的U:G错配碱基对被修复为T:A碱基对;相应的,ABEs中的腺嘌呤脱氨酶使腺嘌呤A水解脱氨为次黄嘌呤I,随后DNA聚合酶能将次黄嘌呤I识别为鸟嘌呤G进行复制,互补链上原来与腺嘌呤A配对的胸腺嘧啶T则会相应变成胞嘧啶C,至此完成碱基置换全过程[107].由于不存在天然的ssDNA腺嘌呤脱氨酶[75],Kim等[108]通过定向进化改造得到一种大肠杆菌(Escherichia coli)tRNA来源的腺嘌呤脱氨酶ecTadA(E.coli TadA)突变体TadA*.由于在天然环境(大肠杆菌)中TadA是以同型二聚体的方式发挥作用,所以最初的TadA*-nCas9融合蛋白在哺乳动物细胞中的编辑效率并不高,直到研究人员将野生型TadA单体、进化的突变体TadA*及nCas9整合到一起(TadA- TadA*- nCas9),才使ABE在哺乳动物中的编辑效率大大提高.2018年,David R. Liu团队[109]工程化改造并优化了ABE7.10后获得的ABEmax最高可获得前者7.9倍的编辑效率,是目前为止在哺乳动物中编辑效率最高的ABEs.为了方便体内编辑,该团队[110]在2022年7月发表的紧凑型ABEs递送系统通过将小型腺嘌呤编辑器SaABE8e及其PAM变体SaKKH-ABE8e的基因长度最小化以及AAV组件基因长度最小化后使得ABEs能够进行单AAV递送,且在肝脏(66%)、心脏(33%)和肌肉细胞(22%)中的编辑效率均高于双AAV递送系统.同年,吉林大学李占军团队和赖学良团队[111]合作开发的紧凑型的Cas9(Cje2Cas9和Cje3Cas9)能够与sgRNA一起包装到单个AAV中,并在成年小鼠肝脏中以12%的效率成功编辑Pcsk9基因,最终有效降低了小鼠体内的胆固醇水平.高效的单AAV递送系统的开发不仅确立了其相对于双AAV递送系统的优势,还促进了CRISPR/Cas9系统作为体内碱基编辑工具时的研究和治疗应用,具有巨大的临床治疗价值.为了改善编辑效率,Zhang等[76]通过将单链DNA结合结构域(single-stranded DNA-binding domain,ssDBD)融合到胞嘧啶脱氨酶与nCas9之间从而延长ssDNA的暴露时间,可在不增加插入缺失突变(indels)、细胞毒性或脱靶效应等负面效应概率的条件下极大地提高CBEs的编辑效率.ssDBD的融合还使CBEs的编辑窗口范围得到了提高,从而拓宽了其靶向作用范围及用于疾病建模和基因治疗的适用范围.David R. Liu团队[30]在CBE的基础上融合了尿嘧啶糖基化酶抑制剂(uracil glycosylase inhibitor,UGI),作为一种体量较小的蛋白(9.5kDa),UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率.该团队还发现通过招募多拷贝的脱氨酶或更改其空间位置以优化编辑窗口能有效提高编辑效率和特异性.此外,与nCas9融合的脱氨酶若工程化改造后具有序列偏好性,则可通过降低旁观者编辑效应从而提高编辑精度.尽管ABEs和CBEs碱基编辑器的性能不断提升,A:T到G:C和C:G到T:A碱基对的单向固定替换仍限制着BEs作为单碱基改变工具使用时的灵活性.2020年,Zhao等[77]通过组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶(uracil-DNA glycosylase,Ung)实现了大肠杆菌中C到A 87.2%的编辑效率和哺乳动物细胞C到G 5.3%~53.0%的编辑效率.先导编辑作为一种多功能编辑手段,虽然同时具备可编程性和高精确度,但对于单碱基编辑的效率仍与BEs有较大差距[28].2021年,Song等[78]构建的hyPE2在PE2的基础上融合加入了一个ssDBD结构域Rad51,使得PE2的编辑效率最高提升了2.6倍,同时该团队还通过深度学习开发了能够预测不同的hyPE2与PE2相比编辑效率提升倍数的计算模型PEselector,该工具的网址为http://deepcrispr.info/PEselector.2022年,Velimirovic等[112]通过高通量肽链自编辑测序(peptide self-editing sequencing assay,PepSEq)测出12000个85个氨基酸的肽段对PE效率的影响,从而通过肽链融合提高PE的效率.综上所述,基于CRIPSR/Cas9系统的碱基编辑器编辑效率、编辑精度的不断提高以及编辑窗口乃至碱基固定替换模式的扩展相对于大量已知且不断增加的遗传病相关点突变数量仍捉襟见肘,各种脱氨酶赋予CRIPSR/Cas9系统的碱基编辑能力在其他结构域如UGI和ssDBD加持下不仅能够发挥CRIPSR系统高效率、高灵活、易编程的优势,也能够充分发挥脱氨酶的效能,但仍需进一步优化它们以有效地转换碱基和在无旁观者编辑效应的条件下进行目的碱基替换. ...
Advances in research on single base editing tool—adenine base editor ABE
1
2019
... 为了改善CRIPSR/Cas9系统的碱基编辑工具箱,研究人员通过将nCas9与胞嘧啶脱氨酶(cytosine deaminase)或腺嘌呤脱氨酶(adenine deaminase)融合的方式在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换(图2),其中相应的融合蛋白分别被称为胞嘧啶碱基编辑器(cytosine base editors,CBEs)[30,74]和腺嘌呤碱基编辑器(adenine base editors,ABEs)[75].CBEs中的胞嘧啶脱氨酶能使被nCas9释放的单链DNA(single strand DNA,ssDNA)上的胞嘧啶C转变为尿嘧啶U,随后该位置产生的U:G错配碱基对被修复为T:A碱基对;相应的,ABEs中的腺嘌呤脱氨酶使腺嘌呤A水解脱氨为次黄嘌呤I,随后DNA聚合酶能将次黄嘌呤I识别为鸟嘌呤G进行复制,互补链上原来与腺嘌呤A配对的胸腺嘧啶T则会相应变成胞嘧啶C,至此完成碱基置换全过程[107].由于不存在天然的ssDNA腺嘌呤脱氨酶[75],Kim等[108]通过定向进化改造得到一种大肠杆菌(Escherichia coli)tRNA来源的腺嘌呤脱氨酶ecTadA(E.coli TadA)突变体TadA*.由于在天然环境(大肠杆菌)中TadA是以同型二聚体的方式发挥作用,所以最初的TadA*-nCas9融合蛋白在哺乳动物细胞中的编辑效率并不高,直到研究人员将野生型TadA单体、进化的突变体TadA*及nCas9整合到一起(TadA- TadA*- nCas9),才使ABE在哺乳动物中的编辑效率大大提高.2018年,David R. Liu团队[109]工程化改造并优化了ABE7.10后获得的ABEmax最高可获得前者7.9倍的编辑效率,是目前为止在哺乳动物中编辑效率最高的ABEs.为了方便体内编辑,该团队[110]在2022年7月发表的紧凑型ABEs递送系统通过将小型腺嘌呤编辑器SaABE8e及其PAM变体SaKKH-ABE8e的基因长度最小化以及AAV组件基因长度最小化后使得ABEs能够进行单AAV递送,且在肝脏(66%)、心脏(33%)和肌肉细胞(22%)中的编辑效率均高于双AAV递送系统.同年,吉林大学李占军团队和赖学良团队[111]合作开发的紧凑型的Cas9(Cje2Cas9和Cje3Cas9)能够与sgRNA一起包装到单个AAV中,并在成年小鼠肝脏中以12%的效率成功编辑Pcsk9基因,最终有效降低了小鼠体内的胆固醇水平.高效的单AAV递送系统的开发不仅确立了其相对于双AAV递送系统的优势,还促进了CRISPR/Cas9系统作为体内碱基编辑工具时的研究和治疗应用,具有巨大的临床治疗价值.为了改善编辑效率,Zhang等[76]通过将单链DNA结合结构域(single-stranded DNA-binding domain,ssDBD)融合到胞嘧啶脱氨酶与nCas9之间从而延长ssDNA的暴露时间,可在不增加插入缺失突变(indels)、细胞毒性或脱靶效应等负面效应概率的条件下极大地提高CBEs的编辑效率.ssDBD的融合还使CBEs的编辑窗口范围得到了提高,从而拓宽了其靶向作用范围及用于疾病建模和基因治疗的适用范围.David R. Liu团队[30]在CBE的基础上融合了尿嘧啶糖基化酶抑制剂(uracil glycosylase inhibitor,UGI),作为一种体量较小的蛋白(9.5kDa),UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率.该团队还发现通过招募多拷贝的脱氨酶或更改其空间位置以优化编辑窗口能有效提高编辑效率和特异性.此外,与nCas9融合的脱氨酶若工程化改造后具有序列偏好性,则可通过降低旁观者编辑效应从而提高编辑精度.尽管ABEs和CBEs碱基编辑器的性能不断提升,A:T到G:C和C:G到T:A碱基对的单向固定替换仍限制着BEs作为单碱基改变工具使用时的灵活性.2020年,Zhao等[77]通过组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶(uracil-DNA glycosylase,Ung)实现了大肠杆菌中C到A 87.2%的编辑效率和哺乳动物细胞C到G 5.3%~53.0%的编辑效率.先导编辑作为一种多功能编辑手段,虽然同时具备可编程性和高精确度,但对于单碱基编辑的效率仍与BEs有较大差距[28].2021年,Song等[78]构建的hyPE2在PE2的基础上融合加入了一个ssDBD结构域Rad51,使得PE2的编辑效率最高提升了2.6倍,同时该团队还通过深度学习开发了能够预测不同的hyPE2与PE2相比编辑效率提升倍数的计算模型PEselector,该工具的网址为http://deepcrispr.info/PEselector.2022年,Velimirovic等[112]通过高通量肽链自编辑测序(peptide self-editing sequencing assay,PepSEq)测出12000个85个氨基酸的肽段对PE效率的影响,从而通过肽链融合提高PE的效率.综上所述,基于CRIPSR/Cas9系统的碱基编辑器编辑效率、编辑精度的不断提高以及编辑窗口乃至碱基固定替换模式的扩展相对于大量已知且不断增加的遗传病相关点突变数量仍捉襟见肘,各种脱氨酶赋予CRIPSR/Cas9系统的碱基编辑能力在其他结构域如UGI和ssDBD加持下不仅能够发挥CRIPSR系统高效率、高灵活、易编程的优势,也能够充分发挥脱氨酶的效能,但仍需进一步优化它们以有效地转换碱基和在无旁观者编辑效应的条件下进行目的碱基替换. ...
Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions
1
2017
... 为了改善CRIPSR/Cas9系统的碱基编辑工具箱,研究人员通过将nCas9与胞嘧啶脱氨酶(cytosine deaminase)或腺嘌呤脱氨酶(adenine deaminase)融合的方式在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换(图2),其中相应的融合蛋白分别被称为胞嘧啶碱基编辑器(cytosine base editors,CBEs)[30,74]和腺嘌呤碱基编辑器(adenine base editors,ABEs)[75].CBEs中的胞嘧啶脱氨酶能使被nCas9释放的单链DNA(single strand DNA,ssDNA)上的胞嘧啶C转变为尿嘧啶U,随后该位置产生的U:G错配碱基对被修复为T:A碱基对;相应的,ABEs中的腺嘌呤脱氨酶使腺嘌呤A水解脱氨为次黄嘌呤I,随后DNA聚合酶能将次黄嘌呤I识别为鸟嘌呤G进行复制,互补链上原来与腺嘌呤A配对的胸腺嘧啶T则会相应变成胞嘧啶C,至此完成碱基置换全过程[107].由于不存在天然的ssDNA腺嘌呤脱氨酶[75],Kim等[108]通过定向进化改造得到一种大肠杆菌(Escherichia coli)tRNA来源的腺嘌呤脱氨酶ecTadA(E.coli TadA)突变体TadA*.由于在天然环境(大肠杆菌)中TadA是以同型二聚体的方式发挥作用,所以最初的TadA*-nCas9融合蛋白在哺乳动物细胞中的编辑效率并不高,直到研究人员将野生型TadA单体、进化的突变体TadA*及nCas9整合到一起(TadA- TadA*- nCas9),才使ABE在哺乳动物中的编辑效率大大提高.2018年,David R. Liu团队[109]工程化改造并优化了ABE7.10后获得的ABEmax最高可获得前者7.9倍的编辑效率,是目前为止在哺乳动物中编辑效率最高的ABEs.为了方便体内编辑,该团队[110]在2022年7月发表的紧凑型ABEs递送系统通过将小型腺嘌呤编辑器SaABE8e及其PAM变体SaKKH-ABE8e的基因长度最小化以及AAV组件基因长度最小化后使得ABEs能够进行单AAV递送,且在肝脏(66%)、心脏(33%)和肌肉细胞(22%)中的编辑效率均高于双AAV递送系统.同年,吉林大学李占军团队和赖学良团队[111]合作开发的紧凑型的Cas9(Cje2Cas9和Cje3Cas9)能够与sgRNA一起包装到单个AAV中,并在成年小鼠肝脏中以12%的效率成功编辑Pcsk9基因,最终有效降低了小鼠体内的胆固醇水平.高效的单AAV递送系统的开发不仅确立了其相对于双AAV递送系统的优势,还促进了CRISPR/Cas9系统作为体内碱基编辑工具时的研究和治疗应用,具有巨大的临床治疗价值.为了改善编辑效率,Zhang等[76]通过将单链DNA结合结构域(single-stranded DNA-binding domain,ssDBD)融合到胞嘧啶脱氨酶与nCas9之间从而延长ssDNA的暴露时间,可在不增加插入缺失突变(indels)、细胞毒性或脱靶效应等负面效应概率的条件下极大地提高CBEs的编辑效率.ssDBD的融合还使CBEs的编辑窗口范围得到了提高,从而拓宽了其靶向作用范围及用于疾病建模和基因治疗的适用范围.David R. Liu团队[30]在CBE的基础上融合了尿嘧啶糖基化酶抑制剂(uracil glycosylase inhibitor,UGI),作为一种体量较小的蛋白(9.5kDa),UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率.该团队还发现通过招募多拷贝的脱氨酶或更改其空间位置以优化编辑窗口能有效提高编辑效率和特异性.此外,与nCas9融合的脱氨酶若工程化改造后具有序列偏好性,则可通过降低旁观者编辑效应从而提高编辑精度.尽管ABEs和CBEs碱基编辑器的性能不断提升,A:T到G:C和C:G到T:A碱基对的单向固定替换仍限制着BEs作为单碱基改变工具使用时的灵活性.2020年,Zhao等[77]通过组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶(uracil-DNA glycosylase,Ung)实现了大肠杆菌中C到A 87.2%的编辑效率和哺乳动物细胞C到G 5.3%~53.0%的编辑效率.先导编辑作为一种多功能编辑手段,虽然同时具备可编程性和高精确度,但对于单碱基编辑的效率仍与BEs有较大差距[28].2021年,Song等[78]构建的hyPE2在PE2的基础上融合加入了一个ssDBD结构域Rad51,使得PE2的编辑效率最高提升了2.6倍,同时该团队还通过深度学习开发了能够预测不同的hyPE2与PE2相比编辑效率提升倍数的计算模型PEselector,该工具的网址为http://deepcrispr.info/PEselector.2022年,Velimirovic等[112]通过高通量肽链自编辑测序(peptide self-editing sequencing assay,PepSEq)测出12000个85个氨基酸的肽段对PE效率的影响,从而通过肽链融合提高PE的效率.综上所述,基于CRIPSR/Cas9系统的碱基编辑器编辑效率、编辑精度的不断提高以及编辑窗口乃至碱基固定替换模式的扩展相对于大量已知且不断增加的遗传病相关点突变数量仍捉襟见肘,各种脱氨酶赋予CRIPSR/Cas9系统的碱基编辑能力在其他结构域如UGI和ssDBD加持下不仅能够发挥CRIPSR系统高效率、高灵活、易编程的优势,也能够充分发挥脱氨酶的效能,但仍需进一步优化它们以有效地转换碱基和在无旁观者编辑效应的条件下进行目的碱基替换. ...
Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction
1
2018
... 为了改善CRIPSR/Cas9系统的碱基编辑工具箱,研究人员通过将nCas9与胞嘧啶脱氨酶(cytosine deaminase)或腺嘌呤脱氨酶(adenine deaminase)融合的方式在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换(图2),其中相应的融合蛋白分别被称为胞嘧啶碱基编辑器(cytosine base editors,CBEs)[30,74]和腺嘌呤碱基编辑器(adenine base editors,ABEs)[75].CBEs中的胞嘧啶脱氨酶能使被nCas9释放的单链DNA(single strand DNA,ssDNA)上的胞嘧啶C转变为尿嘧啶U,随后该位置产生的U:G错配碱基对被修复为T:A碱基对;相应的,ABEs中的腺嘌呤脱氨酶使腺嘌呤A水解脱氨为次黄嘌呤I,随后DNA聚合酶能将次黄嘌呤I识别为鸟嘌呤G进行复制,互补链上原来与腺嘌呤A配对的胸腺嘧啶T则会相应变成胞嘧啶C,至此完成碱基置换全过程[107].由于不存在天然的ssDNA腺嘌呤脱氨酶[75],Kim等[108]通过定向进化改造得到一种大肠杆菌(Escherichia coli)tRNA来源的腺嘌呤脱氨酶ecTadA(E.coli TadA)突变体TadA*.由于在天然环境(大肠杆菌)中TadA是以同型二聚体的方式发挥作用,所以最初的TadA*-nCas9融合蛋白在哺乳动物细胞中的编辑效率并不高,直到研究人员将野生型TadA单体、进化的突变体TadA*及nCas9整合到一起(TadA- TadA*- nCas9),才使ABE在哺乳动物中的编辑效率大大提高.2018年,David R. Liu团队[109]工程化改造并优化了ABE7.10后获得的ABEmax最高可获得前者7.9倍的编辑效率,是目前为止在哺乳动物中编辑效率最高的ABEs.为了方便体内编辑,该团队[110]在2022年7月发表的紧凑型ABEs递送系统通过将小型腺嘌呤编辑器SaABE8e及其PAM变体SaKKH-ABE8e的基因长度最小化以及AAV组件基因长度最小化后使得ABEs能够进行单AAV递送,且在肝脏(66%)、心脏(33%)和肌肉细胞(22%)中的编辑效率均高于双AAV递送系统.同年,吉林大学李占军团队和赖学良团队[111]合作开发的紧凑型的Cas9(Cje2Cas9和Cje3Cas9)能够与sgRNA一起包装到单个AAV中,并在成年小鼠肝脏中以12%的效率成功编辑Pcsk9基因,最终有效降低了小鼠体内的胆固醇水平.高效的单AAV递送系统的开发不仅确立了其相对于双AAV递送系统的优势,还促进了CRISPR/Cas9系统作为体内碱基编辑工具时的研究和治疗应用,具有巨大的临床治疗价值.为了改善编辑效率,Zhang等[76]通过将单链DNA结合结构域(single-stranded DNA-binding domain,ssDBD)融合到胞嘧啶脱氨酶与nCas9之间从而延长ssDNA的暴露时间,可在不增加插入缺失突变(indels)、细胞毒性或脱靶效应等负面效应概率的条件下极大地提高CBEs的编辑效率.ssDBD的融合还使CBEs的编辑窗口范围得到了提高,从而拓宽了其靶向作用范围及用于疾病建模和基因治疗的适用范围.David R. Liu团队[30]在CBE的基础上融合了尿嘧啶糖基化酶抑制剂(uracil glycosylase inhibitor,UGI),作为一种体量较小的蛋白(9.5kDa),UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率.该团队还发现通过招募多拷贝的脱氨酶或更改其空间位置以优化编辑窗口能有效提高编辑效率和特异性.此外,与nCas9融合的脱氨酶若工程化改造后具有序列偏好性,则可通过降低旁观者编辑效应从而提高编辑精度.尽管ABEs和CBEs碱基编辑器的性能不断提升,A:T到G:C和C:G到T:A碱基对的单向固定替换仍限制着BEs作为单碱基改变工具使用时的灵活性.2020年,Zhao等[77]通过组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶(uracil-DNA glycosylase,Ung)实现了大肠杆菌中C到A 87.2%的编辑效率和哺乳动物细胞C到G 5.3%~53.0%的编辑效率.先导编辑作为一种多功能编辑手段,虽然同时具备可编程性和高精确度,但对于单碱基编辑的效率仍与BEs有较大差距[28].2021年,Song等[78]构建的hyPE2在PE2的基础上融合加入了一个ssDBD结构域Rad51,使得PE2的编辑效率最高提升了2.6倍,同时该团队还通过深度学习开发了能够预测不同的hyPE2与PE2相比编辑效率提升倍数的计算模型PEselector,该工具的网址为http://deepcrispr.info/PEselector.2022年,Velimirovic等[112]通过高通量肽链自编辑测序(peptide self-editing sequencing assay,PepSEq)测出12000个85个氨基酸的肽段对PE效率的影响,从而通过肽链融合提高PE的效率.综上所述,基于CRIPSR/Cas9系统的碱基编辑器编辑效率、编辑精度的不断提高以及编辑窗口乃至碱基固定替换模式的扩展相对于大量已知且不断增加的遗传病相关点突变数量仍捉襟见肘,各种脱氨酶赋予CRIPSR/Cas9系统的碱基编辑能力在其他结构域如UGI和ssDBD加持下不仅能够发挥CRIPSR系统高效率、高灵活、易编程的优势,也能够充分发挥脱氨酶的效能,但仍需进一步优化它们以有效地转换碱基和在无旁观者编辑效应的条件下进行目的碱基替换. ...
Efficient in vivo base editing via single adeno-associated viruses with size-optimized genomes encoding compact adenine base editors
1
2022
... 为了改善CRIPSR/Cas9系统的碱基编辑工具箱,研究人员通过将nCas9与胞嘧啶脱氨酶(cytosine deaminase)或腺嘌呤脱氨酶(adenine deaminase)融合的方式在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换(图2),其中相应的融合蛋白分别被称为胞嘧啶碱基编辑器(cytosine base editors,CBEs)[30,74]和腺嘌呤碱基编辑器(adenine base editors,ABEs)[75].CBEs中的胞嘧啶脱氨酶能使被nCas9释放的单链DNA(single strand DNA,ssDNA)上的胞嘧啶C转变为尿嘧啶U,随后该位置产生的U:G错配碱基对被修复为T:A碱基对;相应的,ABEs中的腺嘌呤脱氨酶使腺嘌呤A水解脱氨为次黄嘌呤I,随后DNA聚合酶能将次黄嘌呤I识别为鸟嘌呤G进行复制,互补链上原来与腺嘌呤A配对的胸腺嘧啶T则会相应变成胞嘧啶C,至此完成碱基置换全过程[107].由于不存在天然的ssDNA腺嘌呤脱氨酶[75],Kim等[108]通过定向进化改造得到一种大肠杆菌(Escherichia coli)tRNA来源的腺嘌呤脱氨酶ecTadA(E.coli TadA)突变体TadA*.由于在天然环境(大肠杆菌)中TadA是以同型二聚体的方式发挥作用,所以最初的TadA*-nCas9融合蛋白在哺乳动物细胞中的编辑效率并不高,直到研究人员将野生型TadA单体、进化的突变体TadA*及nCas9整合到一起(TadA- TadA*- nCas9),才使ABE在哺乳动物中的编辑效率大大提高.2018年,David R. Liu团队[109]工程化改造并优化了ABE7.10后获得的ABEmax最高可获得前者7.9倍的编辑效率,是目前为止在哺乳动物中编辑效率最高的ABEs.为了方便体内编辑,该团队[110]在2022年7月发表的紧凑型ABEs递送系统通过将小型腺嘌呤编辑器SaABE8e及其PAM变体SaKKH-ABE8e的基因长度最小化以及AAV组件基因长度最小化后使得ABEs能够进行单AAV递送,且在肝脏(66%)、心脏(33%)和肌肉细胞(22%)中的编辑效率均高于双AAV递送系统.同年,吉林大学李占军团队和赖学良团队[111]合作开发的紧凑型的Cas9(Cje2Cas9和Cje3Cas9)能够与sgRNA一起包装到单个AAV中,并在成年小鼠肝脏中以12%的效率成功编辑Pcsk9基因,最终有效降低了小鼠体内的胆固醇水平.高效的单AAV递送系统的开发不仅确立了其相对于双AAV递送系统的优势,还促进了CRISPR/Cas9系统作为体内碱基编辑工具时的研究和治疗应用,具有巨大的临床治疗价值.为了改善编辑效率,Zhang等[76]通过将单链DNA结合结构域(single-stranded DNA-binding domain,ssDBD)融合到胞嘧啶脱氨酶与nCas9之间从而延长ssDNA的暴露时间,可在不增加插入缺失突变(indels)、细胞毒性或脱靶效应等负面效应概率的条件下极大地提高CBEs的编辑效率.ssDBD的融合还使CBEs的编辑窗口范围得到了提高,从而拓宽了其靶向作用范围及用于疾病建模和基因治疗的适用范围.David R. Liu团队[30]在CBE的基础上融合了尿嘧啶糖基化酶抑制剂(uracil glycosylase inhibitor,UGI),作为一种体量较小的蛋白(9.5kDa),UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率.该团队还发现通过招募多拷贝的脱氨酶或更改其空间位置以优化编辑窗口能有效提高编辑效率和特异性.此外,与nCas9融合的脱氨酶若工程化改造后具有序列偏好性,则可通过降低旁观者编辑效应从而提高编辑精度.尽管ABEs和CBEs碱基编辑器的性能不断提升,A:T到G:C和C:G到T:A碱基对的单向固定替换仍限制着BEs作为单碱基改变工具使用时的灵活性.2020年,Zhao等[77]通过组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶(uracil-DNA glycosylase,Ung)实现了大肠杆菌中C到A 87.2%的编辑效率和哺乳动物细胞C到G 5.3%~53.0%的编辑效率.先导编辑作为一种多功能编辑手段,虽然同时具备可编程性和高精确度,但对于单碱基编辑的效率仍与BEs有较大差距[28].2021年,Song等[78]构建的hyPE2在PE2的基础上融合加入了一个ssDBD结构域Rad51,使得PE2的编辑效率最高提升了2.6倍,同时该团队还通过深度学习开发了能够预测不同的hyPE2与PE2相比编辑效率提升倍数的计算模型PEselector,该工具的网址为http://deepcrispr.info/PEselector.2022年,Velimirovic等[112]通过高通量肽链自编辑测序(peptide self-editing sequencing assay,PepSEq)测出12000个85个氨基酸的肽段对PE效率的影响,从而通过肽链融合提高PE的效率.综上所述,基于CRIPSR/Cas9系统的碱基编辑器编辑效率、编辑精度的不断提高以及编辑窗口乃至碱基固定替换模式的扩展相对于大量已知且不断增加的遗传病相关点突变数量仍捉襟见肘,各种脱氨酶赋予CRIPSR/Cas9系统的碱基编辑能力在其他结构域如UGI和ssDBD加持下不仅能够发挥CRIPSR系统高效率、高灵活、易编程的优势,也能够充分发挥脱氨酶的效能,但仍需进一步优化它们以有效地转换碱基和在无旁观者编辑效应的条件下进行目的碱基替换. ...
Compact Cje3Cas9 for efficient in vivo genome editing and adenine base editing
1
2022
... 为了改善CRIPSR/Cas9系统的碱基编辑工具箱,研究人员通过将nCas9与胞嘧啶脱氨酶(cytosine deaminase)或腺嘌呤脱氨酶(adenine deaminase)融合的方式在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换(图2),其中相应的融合蛋白分别被称为胞嘧啶碱基编辑器(cytosine base editors,CBEs)[30,74]和腺嘌呤碱基编辑器(adenine base editors,ABEs)[75].CBEs中的胞嘧啶脱氨酶能使被nCas9释放的单链DNA(single strand DNA,ssDNA)上的胞嘧啶C转变为尿嘧啶U,随后该位置产生的U:G错配碱基对被修复为T:A碱基对;相应的,ABEs中的腺嘌呤脱氨酶使腺嘌呤A水解脱氨为次黄嘌呤I,随后DNA聚合酶能将次黄嘌呤I识别为鸟嘌呤G进行复制,互补链上原来与腺嘌呤A配对的胸腺嘧啶T则会相应变成胞嘧啶C,至此完成碱基置换全过程[107].由于不存在天然的ssDNA腺嘌呤脱氨酶[75],Kim等[108]通过定向进化改造得到一种大肠杆菌(Escherichia coli)tRNA来源的腺嘌呤脱氨酶ecTadA(E.coli TadA)突变体TadA*.由于在天然环境(大肠杆菌)中TadA是以同型二聚体的方式发挥作用,所以最初的TadA*-nCas9融合蛋白在哺乳动物细胞中的编辑效率并不高,直到研究人员将野生型TadA单体、进化的突变体TadA*及nCas9整合到一起(TadA- TadA*- nCas9),才使ABE在哺乳动物中的编辑效率大大提高.2018年,David R. Liu团队[109]工程化改造并优化了ABE7.10后获得的ABEmax最高可获得前者7.9倍的编辑效率,是目前为止在哺乳动物中编辑效率最高的ABEs.为了方便体内编辑,该团队[110]在2022年7月发表的紧凑型ABEs递送系统通过将小型腺嘌呤编辑器SaABE8e及其PAM变体SaKKH-ABE8e的基因长度最小化以及AAV组件基因长度最小化后使得ABEs能够进行单AAV递送,且在肝脏(66%)、心脏(33%)和肌肉细胞(22%)中的编辑效率均高于双AAV递送系统.同年,吉林大学李占军团队和赖学良团队[111]合作开发的紧凑型的Cas9(Cje2Cas9和Cje3Cas9)能够与sgRNA一起包装到单个AAV中,并在成年小鼠肝脏中以12%的效率成功编辑Pcsk9基因,最终有效降低了小鼠体内的胆固醇水平.高效的单AAV递送系统的开发不仅确立了其相对于双AAV递送系统的优势,还促进了CRISPR/Cas9系统作为体内碱基编辑工具时的研究和治疗应用,具有巨大的临床治疗价值.为了改善编辑效率,Zhang等[76]通过将单链DNA结合结构域(single-stranded DNA-binding domain,ssDBD)融合到胞嘧啶脱氨酶与nCas9之间从而延长ssDNA的暴露时间,可在不增加插入缺失突变(indels)、细胞毒性或脱靶效应等负面效应概率的条件下极大地提高CBEs的编辑效率.ssDBD的融合还使CBEs的编辑窗口范围得到了提高,从而拓宽了其靶向作用范围及用于疾病建模和基因治疗的适用范围.David R. Liu团队[30]在CBE的基础上融合了尿嘧啶糖基化酶抑制剂(uracil glycosylase inhibitor,UGI),作为一种体量较小的蛋白(9.5kDa),UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率.该团队还发现通过招募多拷贝的脱氨酶或更改其空间位置以优化编辑窗口能有效提高编辑效率和特异性.此外,与nCas9融合的脱氨酶若工程化改造后具有序列偏好性,则可通过降低旁观者编辑效应从而提高编辑精度.尽管ABEs和CBEs碱基编辑器的性能不断提升,A:T到G:C和C:G到T:A碱基对的单向固定替换仍限制着BEs作为单碱基改变工具使用时的灵活性.2020年,Zhao等[77]通过组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶(uracil-DNA glycosylase,Ung)实现了大肠杆菌中C到A 87.2%的编辑效率和哺乳动物细胞C到G 5.3%~53.0%的编辑效率.先导编辑作为一种多功能编辑手段,虽然同时具备可编程性和高精确度,但对于单碱基编辑的效率仍与BEs有较大差距[28].2021年,Song等[78]构建的hyPE2在PE2的基础上融合加入了一个ssDBD结构域Rad51,使得PE2的编辑效率最高提升了2.6倍,同时该团队还通过深度学习开发了能够预测不同的hyPE2与PE2相比编辑效率提升倍数的计算模型PEselector,该工具的网址为http://deepcrispr.info/PEselector.2022年,Velimirovic等[112]通过高通量肽链自编辑测序(peptide self-editing sequencing assay,PepSEq)测出12000个85个氨基酸的肽段对PE效率的影响,从而通过肽链融合提高PE的效率.综上所述,基于CRIPSR/Cas9系统的碱基编辑器编辑效率、编辑精度的不断提高以及编辑窗口乃至碱基固定替换模式的扩展相对于大量已知且不断增加的遗传病相关点突变数量仍捉襟见肘,各种脱氨酶赋予CRIPSR/Cas9系统的碱基编辑能力在其他结构域如UGI和ssDBD加持下不仅能够发挥CRIPSR系统高效率、高灵活、易编程的优势,也能够充分发挥脱氨酶的效能,但仍需进一步优化它们以有效地转换碱基和在无旁观者编辑效应的条件下进行目的碱基替换. ...
Peptide fusion improves prime editing efficiency
1
2022
... 为了改善CRIPSR/Cas9系统的碱基编辑工具箱,研究人员通过将nCas9与胞嘧啶脱氨酶(cytosine deaminase)或腺嘌呤脱氨酶(adenine deaminase)融合的方式在不造成DNA双链断裂的条件下使目的碱基产生C:G与T:A或A:T与G:C的碱基对替换(图2),其中相应的融合蛋白分别被称为胞嘧啶碱基编辑器(cytosine base editors,CBEs)[30,74]和腺嘌呤碱基编辑器(adenine base editors,ABEs)[75].CBEs中的胞嘧啶脱氨酶能使被nCas9释放的单链DNA(single strand DNA,ssDNA)上的胞嘧啶C转变为尿嘧啶U,随后该位置产生的U:G错配碱基对被修复为T:A碱基对;相应的,ABEs中的腺嘌呤脱氨酶使腺嘌呤A水解脱氨为次黄嘌呤I,随后DNA聚合酶能将次黄嘌呤I识别为鸟嘌呤G进行复制,互补链上原来与腺嘌呤A配对的胸腺嘧啶T则会相应变成胞嘧啶C,至此完成碱基置换全过程[107].由于不存在天然的ssDNA腺嘌呤脱氨酶[75],Kim等[108]通过定向进化改造得到一种大肠杆菌(Escherichia coli)tRNA来源的腺嘌呤脱氨酶ecTadA(E.coli TadA)突变体TadA*.由于在天然环境(大肠杆菌)中TadA是以同型二聚体的方式发挥作用,所以最初的TadA*-nCas9融合蛋白在哺乳动物细胞中的编辑效率并不高,直到研究人员将野生型TadA单体、进化的突变体TadA*及nCas9整合到一起(TadA- TadA*- nCas9),才使ABE在哺乳动物中的编辑效率大大提高.2018年,David R. Liu团队[109]工程化改造并优化了ABE7.10后获得的ABEmax最高可获得前者7.9倍的编辑效率,是目前为止在哺乳动物中编辑效率最高的ABEs.为了方便体内编辑,该团队[110]在2022年7月发表的紧凑型ABEs递送系统通过将小型腺嘌呤编辑器SaABE8e及其PAM变体SaKKH-ABE8e的基因长度最小化以及AAV组件基因长度最小化后使得ABEs能够进行单AAV递送,且在肝脏(66%)、心脏(33%)和肌肉细胞(22%)中的编辑效率均高于双AAV递送系统.同年,吉林大学李占军团队和赖学良团队[111]合作开发的紧凑型的Cas9(Cje2Cas9和Cje3Cas9)能够与sgRNA一起包装到单个AAV中,并在成年小鼠肝脏中以12%的效率成功编辑Pcsk9基因,最终有效降低了小鼠体内的胆固醇水平.高效的单AAV递送系统的开发不仅确立了其相对于双AAV递送系统的优势,还促进了CRISPR/Cas9系统作为体内碱基编辑工具时的研究和治疗应用,具有巨大的临床治疗价值.为了改善编辑效率,Zhang等[76]通过将单链DNA结合结构域(single-stranded DNA-binding domain,ssDBD)融合到胞嘧啶脱氨酶与nCas9之间从而延长ssDNA的暴露时间,可在不增加插入缺失突变(indels)、细胞毒性或脱靶效应等负面效应概率的条件下极大地提高CBEs的编辑效率.ssDBD的融合还使CBEs的编辑窗口范围得到了提高,从而拓宽了其靶向作用范围及用于疾病建模和基因治疗的适用范围.David R. Liu团队[30]在CBE的基础上融合了尿嘧啶糖基化酶抑制剂(uracil glycosylase inhibitor,UGI),作为一种体量较小的蛋白(9.5kDa),UGI能够防止胞嘧啶C脱氨后的尿嘧啶U被切除,从而显著提高了CBEs的编辑效率.该团队还发现通过招募多拷贝的脱氨酶或更改其空间位置以优化编辑窗口能有效提高编辑效率和特异性.此外,与nCas9融合的脱氨酶若工程化改造后具有序列偏好性,则可通过降低旁观者编辑效应从而提高编辑精度.尽管ABEs和CBEs碱基编辑器的性能不断提升,A:T到G:C和C:G到T:A碱基对的单向固定替换仍限制着BEs作为单碱基改变工具使用时的灵活性.2020年,Zhao等[77]通过组合nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA糖基化酶(uracil-DNA glycosylase,Ung)实现了大肠杆菌中C到A 87.2%的编辑效率和哺乳动物细胞C到G 5.3%~53.0%的编辑效率.先导编辑作为一种多功能编辑手段,虽然同时具备可编程性和高精确度,但对于单碱基编辑的效率仍与BEs有较大差距[28].2021年,Song等[78]构建的hyPE2在PE2的基础上融合加入了一个ssDBD结构域Rad51,使得PE2的编辑效率最高提升了2.6倍,同时该团队还通过深度学习开发了能够预测不同的hyPE2与PE2相比编辑效率提升倍数的计算模型PEselector,该工具的网址为http://deepcrispr.info/PEselector.2022年,Velimirovic等[112]通过高通量肽链自编辑测序(peptide self-editing sequencing assay,PepSEq)测出12000个85个氨基酸的肽段对PE效率的影响,从而通过肽链融合提高PE的效率.综上所述,基于CRIPSR/Cas9系统的碱基编辑器编辑效率、编辑精度的不断提高以及编辑窗口乃至碱基固定替换模式的扩展相对于大量已知且不断增加的遗传病相关点突变数量仍捉襟见肘,各种脱氨酶赋予CRIPSR/Cas9系统的碱基编辑能力在其他结构域如UGI和ssDBD加持下不仅能够发挥CRIPSR系统高效率、高灵活、易编程的优势,也能够充分发挥脱氨酶的效能,但仍需进一步优化它们以有效地转换碱基和在无旁观者编辑效应的条件下进行目的碱基替换. ...
Multiplexed CRISPR/Cas9 genome editing and gene regulation using Csy4 in Saccharomyces cerevisiae
2
2018
... 目前,多gRNA的表达策略主要有三种(图3):①每个不同gRNA表达盒通过Pol Ⅲ启动子单独启动表达,表达盒之间串联在一起[4];②多个不同gRNA在一个表达盒内由Pol Ⅱ或Pol Ⅲ启动表达为一条转录本,然后再通过mRNA上gRNA之间的tRNA序列切割加工机制、核酶自切割能力或Csy4核酸酶切割释放为成熟的gRNA[11];③一条转录本内的多个gRNA也可以在CRISPR系统自然的自我加工活性的基础上被切割释放为成熟gRNA.2013年,Cong等[4]依据CRISPR基因位点的间隔区排列的自然结构,首次将Pol Ⅲ驱动的表达盒串联实现两个靶基因间隔区的多表达并成功编辑.2018年,Ferreira等[113]首次在酿酒酵母(Saccharomyces cerevisiae)中利用铜绿假单胞菌(Pseudomonas aeruginosa)来源的核酸内切酶Csy4处理单转录本中的多个gRNA,并最终获得高达96%四基因同时敲除效率,并借助CRISPRi系统对3个靶基因进行了稳定的扰动,证明了使用Csy4可以快速有效地进行多重基因组编辑和基因调控.基于此,McCarty等[114]开发的酵母模块化克隆多gRNA快速组装策略通过两步PCR反应及一步Golden Gate反应可在两天内实现单阵列12个gRNA的组装,每个gRNA两侧都有Csy4识别位点从而将转录本加工为成熟的gRNA.同年,刘子鹤团队[115]基于tRNA转录本切割机制构建了一种gRNA-tRNA阵列多gRNA表达系统(GTR-CRISPR系统)并成功在酿酒酵母中以80%的效率同时敲除8个基因.通过使用更快速的Lightning GTR-CRISPR系统,该团队成功在10天中敲除了8个目的基因,简化了酿酒酵母的脂质代谢网络,最终获得了游离脂肪酸30倍的提升效果.该系统的开发在极大地提高了CRISPR/Cas9系统用于酿酒酵母基因组多重编辑效率的同时也揭示该系统在多重应用领域蕴藏的巨大潜力. ...
... Research on the applications of the multiplexed CRISPR/Cas9 system
Table 3| 宿主细胞 | 多重CRISPR策略 | 靶向目标数量 | 应用/概念验证 | 参考文献 |
|---|
| 哺乳动物细胞 | 2个间隔区通过天然CRISPR序列自我加工 | 2个靶标(1个基因) | 对1个靶基因敲除,效率为1.6% | [4] |
| 酿酒酵母 | Casy4核酸酶加工 | 3个基因 | 同时对3个不同启动子进行激活,使得3个报告基因荧光强度提升了2倍 | [113] |
| 酿酒酵母 | 基于Csy4核酸酶加工的多gRNA快速组装 | 12个靶标(3个基因) | 通过12个gRNA靶向使得3个报告基因荧光强度分别被抑制了92%、81%和95% | [114] |
| 酿酒酵母 | 基于tRNA转录本切割机制的多gRNA表达系统 | 8个基因 | 通过两轮8个基因的敲除获得了30倍的游离脂肪酸产量 | [115] |
| 酿酒酵母 | 多个gRNA表达盒通过pol Ⅲ启动子启动 | 3个基因 | 将适配子与gRNA融合进行多基因调控,获得了不同的紫罗兰素生物合成产物 | [118] |
| 酿酒酵母 | 基于Csy4核酸酶加工的多gRNA快速组装策略及多Cas9正交组合调控 | 3个基因 | 通过组合调控使两个报告基因表达分别被抑制和激活5倍,同时以95%的效率敲除第三个基因 | [119] |
| 酿酒酵母 | 通过质粒表达多个gRNA表达盒以及截短gRNA用于调控 | 3个基因 | 通过对三个基因分别进行编辑、激活和抑制获得了α-檀香烯2.66倍的产量 | [43] |
| 马克思克鲁维酵母(Kluyveromyces marxianus) | 基于tRNA转录本切割机制的多gRNA表达系统 | 6个靶标(4个基因) | 通过对4个基因进行调控,使乙酸乙酯的产量提升了3.8倍 | [120] |
| 大肠杆菌 | 多个gRNA表达盒通过质粒表达 | 3个基因 | 通过对3个基因进行组合调控得到了2.3倍苹果酸产量菌株 | [121] |
| 大肠杆菌 | 多个gRNA表达盒通过质粒表达 | 3个基因 | 对苹果酸合成途径基因进行组合调控后获得了2.3倍苹果酸产量菌株 | [121] |
| 枯草芽孢杆菌(Bacillus subtilis) | 多个gRNA表达盒整合至基因组 | 3个基因 | 通过多基因动态组合调控提高了N-乙酰葡糖胺产量 | [122] |
| 天蓝色链霉菌(Streptomyces coelicolor) | 多个gRNA表达盒与dCas9在质粒上表达 | 4个基因 | 通过对4个靶基因同时进行抑制,使其mRNA表达量降为对照的2%~32% | [123] |
2015年,Zalatan等[118]通过多sgRNA表达盒策略及sgRNA工程将适配体与识别区结合,将配体与转录因子融合表达后同时对3个目标基因进行激活或抑制调控,最终在酿酒酵母中获得了不同的紫罗兰素生物合成途径产物.Lobs等[120]在对一株马克思克鲁维酵母(Kluyveromyces marxianus)中乙酸酯的主要代谢途径和前体物质进行鉴定后,利用CRISPRi系统对ACO2b、SDH2、RIP1和MSS51四个基因进行多重调控,最终获得了乙酸乙酯3.8倍的提升.2017年,Lian等[119]在表征了多种同源Cas9蛋白的基因敲除、转录激活和转录抑制效率后,优化并组合三种同源蛋白最终构建了一套完全正交的三功能CRISPR代谢调控系统.由于正交的Cas9同源蛋白所识别的PAM序列不同,发挥不同功能的同源Cas9之间互不干扰,使得两个被调控报告基因的表达强度均被激活和抑制了5倍,并使第三个报告基因的敲除效率达到了95%.该系统通过一步组合调控使得改造的酿酒酵母中胡萝卜素产量提升了3倍,同时使表面葡聚糖酶展示量提高了2.5倍.原核生物具有相对较低的代谢网络复杂性,同时其无核膜包被的拟核能够实现转录与翻译同时进行,这使得原核生物(如大肠杆菌)成为多基因操作及表达调控的模式生物[122-123].2018年,Gao等[121]通过多重CRISPRi系统对苹果酸生物合成途径中的基因进行组合调控后得到了2.3倍的苹果酸产量菌株. ...
Rapid assembly of gRNA arrays via modular cloning in yeast
2
2019
... 目前,多gRNA的表达策略主要有三种(图3):①每个不同gRNA表达盒通过Pol Ⅲ启动子单独启动表达,表达盒之间串联在一起[4];②多个不同gRNA在一个表达盒内由Pol Ⅱ或Pol Ⅲ启动表达为一条转录本,然后再通过mRNA上gRNA之间的tRNA序列切割加工机制、核酶自切割能力或Csy4核酸酶切割释放为成熟的gRNA[11];③一条转录本内的多个gRNA也可以在CRISPR系统自然的自我加工活性的基础上被切割释放为成熟gRNA.2013年,Cong等[4]依据CRISPR基因位点的间隔区排列的自然结构,首次将Pol Ⅲ驱动的表达盒串联实现两个靶基因间隔区的多表达并成功编辑.2018年,Ferreira等[113]首次在酿酒酵母(Saccharomyces cerevisiae)中利用铜绿假单胞菌(Pseudomonas aeruginosa)来源的核酸内切酶Csy4处理单转录本中的多个gRNA,并最终获得高达96%四基因同时敲除效率,并借助CRISPRi系统对3个靶基因进行了稳定的扰动,证明了使用Csy4可以快速有效地进行多重基因组编辑和基因调控.基于此,McCarty等[114]开发的酵母模块化克隆多gRNA快速组装策略通过两步PCR反应及一步Golden Gate反应可在两天内实现单阵列12个gRNA的组装,每个gRNA两侧都有Csy4识别位点从而将转录本加工为成熟的gRNA.同年,刘子鹤团队[115]基于tRNA转录本切割机制构建了一种gRNA-tRNA阵列多gRNA表达系统(GTR-CRISPR系统)并成功在酿酒酵母中以80%的效率同时敲除8个基因.通过使用更快速的Lightning GTR-CRISPR系统,该团队成功在10天中敲除了8个目的基因,简化了酿酒酵母的脂质代谢网络,最终获得了游离脂肪酸30倍的提升效果.该系统的开发在极大地提高了CRISPR/Cas9系统用于酿酒酵母基因组多重编辑效率的同时也揭示该系统在多重应用领域蕴藏的巨大潜力. ...
... Research on the applications of the multiplexed CRISPR/Cas9 system
Table 3| 宿主细胞 | 多重CRISPR策略 | 靶向目标数量 | 应用/概念验证 | 参考文献 |
|---|
| 哺乳动物细胞 | 2个间隔区通过天然CRISPR序列自我加工 | 2个靶标(1个基因) | 对1个靶基因敲除,效率为1.6% | [4] |
| 酿酒酵母 | Casy4核酸酶加工 | 3个基因 | 同时对3个不同启动子进行激活,使得3个报告基因荧光强度提升了2倍 | [113] |
| 酿酒酵母 | 基于Csy4核酸酶加工的多gRNA快速组装 | 12个靶标(3个基因) | 通过12个gRNA靶向使得3个报告基因荧光强度分别被抑制了92%、81%和95% | [114] |
| 酿酒酵母 | 基于tRNA转录本切割机制的多gRNA表达系统 | 8个基因 | 通过两轮8个基因的敲除获得了30倍的游离脂肪酸产量 | [115] |
| 酿酒酵母 | 多个gRNA表达盒通过pol Ⅲ启动子启动 | 3个基因 | 将适配子与gRNA融合进行多基因调控,获得了不同的紫罗兰素生物合成产物 | [118] |
| 酿酒酵母 | 基于Csy4核酸酶加工的多gRNA快速组装策略及多Cas9正交组合调控 | 3个基因 | 通过组合调控使两个报告基因表达分别被抑制和激活5倍,同时以95%的效率敲除第三个基因 | [119] |
| 酿酒酵母 | 通过质粒表达多个gRNA表达盒以及截短gRNA用于调控 | 3个基因 | 通过对三个基因分别进行编辑、激活和抑制获得了α-檀香烯2.66倍的产量 | [43] |
| 马克思克鲁维酵母(Kluyveromyces marxianus) | 基于tRNA转录本切割机制的多gRNA表达系统 | 6个靶标(4个基因) | 通过对4个基因进行调控,使乙酸乙酯的产量提升了3.8倍 | [120] |
| 大肠杆菌 | 多个gRNA表达盒通过质粒表达 | 3个基因 | 通过对3个基因进行组合调控得到了2.3倍苹果酸产量菌株 | [121] |
| 大肠杆菌 | 多个gRNA表达盒通过质粒表达 | 3个基因 | 对苹果酸合成途径基因进行组合调控后获得了2.3倍苹果酸产量菌株 | [121] |
| 枯草芽孢杆菌(Bacillus subtilis) | 多个gRNA表达盒整合至基因组 | 3个基因 | 通过多基因动态组合调控提高了N-乙酰葡糖胺产量 | [122] |
| 天蓝色链霉菌(Streptomyces coelicolor) | 多个gRNA表达盒与dCas9在质粒上表达 | 4个基因 | 通过对4个靶基因同时进行抑制,使其mRNA表达量降为对照的2%~32% | [123] |
2015年,Zalatan等[118]通过多sgRNA表达盒策略及sgRNA工程将适配体与识别区结合,将配体与转录因子融合表达后同时对3个目标基因进行激活或抑制调控,最终在酿酒酵母中获得了不同的紫罗兰素生物合成途径产物.Lobs等[120]在对一株马克思克鲁维酵母(Kluyveromyces marxianus)中乙酸酯的主要代谢途径和前体物质进行鉴定后,利用CRISPRi系统对ACO2b、SDH2、RIP1和MSS51四个基因进行多重调控,最终获得了乙酸乙酯3.8倍的提升.2017年,Lian等[119]在表征了多种同源Cas9蛋白的基因敲除、转录激活和转录抑制效率后,优化并组合三种同源蛋白最终构建了一套完全正交的三功能CRISPR代谢调控系统.由于正交的Cas9同源蛋白所识别的PAM序列不同,发挥不同功能的同源Cas9之间互不干扰,使得两个被调控报告基因的表达强度均被激活和抑制了5倍,并使第三个报告基因的敲除效率达到了95%.该系统通过一步组合调控使得改造的酿酒酵母中胡萝卜素产量提升了3倍,同时使表面葡聚糖酶展示量提高了2.5倍.原核生物具有相对较低的代谢网络复杂性,同时其无核膜包被的拟核能够实现转录与翻译同时进行,这使得原核生物(如大肠杆菌)成为多基因操作及表达调控的模式生物[122-123].2018年,Gao等[121]通过多重CRISPRi系统对苹果酸生物合成途径中的基因进行组合调控后得到了2.3倍的苹果酸产量菌株. ...
A gRNA-tRNA array for CRISPR-Cas9 based rapid multiplexed genome editing in Saccharomyces cerevisiae
2
2019
... 目前,多gRNA的表达策略主要有三种(图3):①每个不同gRNA表达盒通过Pol Ⅲ启动子单独启动表达,表达盒之间串联在一起[4];②多个不同gRNA在一个表达盒内由Pol Ⅱ或Pol Ⅲ启动表达为一条转录本,然后再通过mRNA上gRNA之间的tRNA序列切割加工机制、核酶自切割能力或Csy4核酸酶切割释放为成熟的gRNA[11];③一条转录本内的多个gRNA也可以在CRISPR系统自然的自我加工活性的基础上被切割释放为成熟gRNA.2013年,Cong等[4]依据CRISPR基因位点的间隔区排列的自然结构,首次将Pol Ⅲ驱动的表达盒串联实现两个靶基因间隔区的多表达并成功编辑.2018年,Ferreira等[113]首次在酿酒酵母(Saccharomyces cerevisiae)中利用铜绿假单胞菌(Pseudomonas aeruginosa)来源的核酸内切酶Csy4处理单转录本中的多个gRNA,并最终获得高达96%四基因同时敲除效率,并借助CRISPRi系统对3个靶基因进行了稳定的扰动,证明了使用Csy4可以快速有效地进行多重基因组编辑和基因调控.基于此,McCarty等[114]开发的酵母模块化克隆多gRNA快速组装策略通过两步PCR反应及一步Golden Gate反应可在两天内实现单阵列12个gRNA的组装,每个gRNA两侧都有Csy4识别位点从而将转录本加工为成熟的gRNA.同年,刘子鹤团队[115]基于tRNA转录本切割机制构建了一种gRNA-tRNA阵列多gRNA表达系统(GTR-CRISPR系统)并成功在酿酒酵母中以80%的效率同时敲除8个基因.通过使用更快速的Lightning GTR-CRISPR系统,该团队成功在10天中敲除了8个目的基因,简化了酿酒酵母的脂质代谢网络,最终获得了游离脂肪酸30倍的提升效果.该系统的开发在极大地提高了CRISPR/Cas9系统用于酿酒酵母基因组多重编辑效率的同时也揭示该系统在多重应用领域蕴藏的巨大潜力. ...
... Research on the applications of the multiplexed CRISPR/Cas9 system
Table 3| 宿主细胞 | 多重CRISPR策略 | 靶向目标数量 | 应用/概念验证 | 参考文献 |
|---|
| 哺乳动物细胞 | 2个间隔区通过天然CRISPR序列自我加工 | 2个靶标(1个基因) | 对1个靶基因敲除,效率为1.6% | [4] |
| 酿酒酵母 | Casy4核酸酶加工 | 3个基因 | 同时对3个不同启动子进行激活,使得3个报告基因荧光强度提升了2倍 | [113] |
| 酿酒酵母 | 基于Csy4核酸酶加工的多gRNA快速组装 | 12个靶标(3个基因) | 通过12个gRNA靶向使得3个报告基因荧光强度分别被抑制了92%、81%和95% | [114] |
| 酿酒酵母 | 基于tRNA转录本切割机制的多gRNA表达系统 | 8个基因 | 通过两轮8个基因的敲除获得了30倍的游离脂肪酸产量 | [115] |
| 酿酒酵母 | 多个gRNA表达盒通过pol Ⅲ启动子启动 | 3个基因 | 将适配子与gRNA融合进行多基因调控,获得了不同的紫罗兰素生物合成产物 | [118] |
| 酿酒酵母 | 基于Csy4核酸酶加工的多gRNA快速组装策略及多Cas9正交组合调控 | 3个基因 | 通过组合调控使两个报告基因表达分别被抑制和激活5倍,同时以95%的效率敲除第三个基因 | [119] |
| 酿酒酵母 | 通过质粒表达多个gRNA表达盒以及截短gRNA用于调控 | 3个基因 | 通过对三个基因分别进行编辑、激活和抑制获得了α-檀香烯2.66倍的产量 | [43] |
| 马克思克鲁维酵母(Kluyveromyces marxianus) | 基于tRNA转录本切割机制的多gRNA表达系统 | 6个靶标(4个基因) | 通过对4个基因进行调控,使乙酸乙酯的产量提升了3.8倍 | [120] |
| 大肠杆菌 | 多个gRNA表达盒通过质粒表达 | 3个基因 | 通过对3个基因进行组合调控得到了2.3倍苹果酸产量菌株 | [121] |
| 大肠杆菌 | 多个gRNA表达盒通过质粒表达 | 3个基因 | 对苹果酸合成途径基因进行组合调控后获得了2.3倍苹果酸产量菌株 | [121] |
| 枯草芽孢杆菌(Bacillus subtilis) | 多个gRNA表达盒整合至基因组 | 3个基因 | 通过多基因动态组合调控提高了N-乙酰葡糖胺产量 | [122] |
| 天蓝色链霉菌(Streptomyces coelicolor) | 多个gRNA表达盒与dCas9在质粒上表达 | 4个基因 | 通过对4个靶基因同时进行抑制,使其mRNA表达量降为对照的2%~32% | [123] |
2015年,Zalatan等[118]通过多sgRNA表达盒策略及sgRNA工程将适配体与识别区结合,将配体与转录因子融合表达后同时对3个目标基因进行激活或抑制调控,最终在酿酒酵母中获得了不同的紫罗兰素生物合成途径产物.Lobs等[120]在对一株马克思克鲁维酵母(Kluyveromyces marxianus)中乙酸酯的主要代谢途径和前体物质进行鉴定后,利用CRISPRi系统对ACO2b、SDH2、RIP1和MSS51四个基因进行多重调控,最终获得了乙酸乙酯3.8倍的提升.2017年,Lian等[119]在表征了多种同源Cas9蛋白的基因敲除、转录激活和转录抑制效率后,优化并组合三种同源蛋白最终构建了一套完全正交的三功能CRISPR代谢调控系统.由于正交的Cas9同源蛋白所识别的PAM序列不同,发挥不同功能的同源Cas9之间互不干扰,使得两个被调控报告基因的表达强度均被激活和抑制了5倍,并使第三个报告基因的敲除效率达到了95%.该系统通过一步组合调控使得改造的酿酒酵母中胡萝卜素产量提升了3倍,同时使表面葡聚糖酶展示量提高了2.5倍.原核生物具有相对较低的代谢网络复杂性,同时其无核膜包被的拟核能够实现转录与翻译同时进行,这使得原核生物(如大肠杆菌)成为多基因操作及表达调控的模式生物[122-123].2018年,Gao等[121]通过多重CRISPRi系统对苹果酸生物合成途径中的基因进行组合调控后得到了2.3倍的苹果酸产量菌株. ...
Multiplexed CRISPR technologies for gene editing and transcriptional regulation
1
2020
... 基于gRNA的多表达、多个同源Cas9或dCas9蛋白同时表达则可实现CRISPR系统的多路复用正交编辑或多功能组合代谢工程[36,116].自2013年Jiang等[117]证明了肺炎链球菌原生CRISPR/Cas9系统具备多重应用潜力后,研究人员基于gRNA多表达策略在大肠杆菌、酿酒酵母等模式生物体中开展了多项CRISPR系统的多路复用应用研究,从而在合成生物学及生物制造领域实现对底盘细胞的改造,最终提升目标产品产量或改变细胞特性(表3). ...
RNA-guided editing of bacterial genomes using CRISPR-Cas systems
1
2013
... 基于gRNA的多表达、多个同源Cas9或dCas9蛋白同时表达则可实现CRISPR系统的多路复用正交编辑或多功能组合代谢工程[36,116].自2013年Jiang等[117]证明了肺炎链球菌原生CRISPR/Cas9系统具备多重应用潜力后,研究人员基于gRNA多表达策略在大肠杆菌、酿酒酵母等模式生物体中开展了多项CRISPR系统的多路复用应用研究,从而在合成生物学及生物制造领域实现对底盘细胞的改造,最终提升目标产品产量或改变细胞特性(表3). ...
Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds
2
2015
... Research on the applications of the multiplexed CRISPR/Cas9 system
Table 3| 宿主细胞 | 多重CRISPR策略 | 靶向目标数量 | 应用/概念验证 | 参考文献 |
|---|
| 哺乳动物细胞 | 2个间隔区通过天然CRISPR序列自我加工 | 2个靶标(1个基因) | 对1个靶基因敲除,效率为1.6% | [4] |
| 酿酒酵母 | Casy4核酸酶加工 | 3个基因 | 同时对3个不同启动子进行激活,使得3个报告基因荧光强度提升了2倍 | [113] |
| 酿酒酵母 | 基于Csy4核酸酶加工的多gRNA快速组装 | 12个靶标(3个基因) | 通过12个gRNA靶向使得3个报告基因荧光强度分别被抑制了92%、81%和95% | [114] |
| 酿酒酵母 | 基于tRNA转录本切割机制的多gRNA表达系统 | 8个基因 | 通过两轮8个基因的敲除获得了30倍的游离脂肪酸产量 | [115] |
| 酿酒酵母 | 多个gRNA表达盒通过pol Ⅲ启动子启动 | 3个基因 | 将适配子与gRNA融合进行多基因调控,获得了不同的紫罗兰素生物合成产物 | [118] |
| 酿酒酵母 | 基于Csy4核酸酶加工的多gRNA快速组装策略及多Cas9正交组合调控 | 3个基因 | 通过组合调控使两个报告基因表达分别被抑制和激活5倍,同时以95%的效率敲除第三个基因 | [119] |
| 酿酒酵母 | 通过质粒表达多个gRNA表达盒以及截短gRNA用于调控 | 3个基因 | 通过对三个基因分别进行编辑、激活和抑制获得了α-檀香烯2.66倍的产量 | [43] |
| 马克思克鲁维酵母(Kluyveromyces marxianus) | 基于tRNA转录本切割机制的多gRNA表达系统 | 6个靶标(4个基因) | 通过对4个基因进行调控,使乙酸乙酯的产量提升了3.8倍 | [120] |
| 大肠杆菌 | 多个gRNA表达盒通过质粒表达 | 3个基因 | 通过对3个基因进行组合调控得到了2.3倍苹果酸产量菌株 | [121] |
| 大肠杆菌 | 多个gRNA表达盒通过质粒表达 | 3个基因 | 对苹果酸合成途径基因进行组合调控后获得了2.3倍苹果酸产量菌株 | [121] |
| 枯草芽孢杆菌(Bacillus subtilis) | 多个gRNA表达盒整合至基因组 | 3个基因 | 通过多基因动态组合调控提高了N-乙酰葡糖胺产量 | [122] |
| 天蓝色链霉菌(Streptomyces coelicolor) | 多个gRNA表达盒与dCas9在质粒上表达 | 4个基因 | 通过对4个靶基因同时进行抑制,使其mRNA表达量降为对照的2%~32% | [123] |
2015年,Zalatan等[118]通过多sgRNA表达盒策略及sgRNA工程将适配体与识别区结合,将配体与转录因子融合表达后同时对3个目标基因进行激活或抑制调控,最终在酿酒酵母中获得了不同的紫罗兰素生物合成途径产物.Lobs等[120]在对一株马克思克鲁维酵母(Kluyveromyces marxianus)中乙酸酯的主要代谢途径和前体物质进行鉴定后,利用CRISPRi系统对ACO2b、SDH2、RIP1和MSS51四个基因进行多重调控,最终获得了乙酸乙酯3.8倍的提升.2017年,Lian等[119]在表征了多种同源Cas9蛋白的基因敲除、转录激活和转录抑制效率后,优化并组合三种同源蛋白最终构建了一套完全正交的三功能CRISPR代谢调控系统.由于正交的Cas9同源蛋白所识别的PAM序列不同,发挥不同功能的同源Cas9之间互不干扰,使得两个被调控报告基因的表达强度均被激活和抑制了5倍,并使第三个报告基因的敲除效率达到了95%.该系统通过一步组合调控使得改造的酿酒酵母中胡萝卜素产量提升了3倍,同时使表面葡聚糖酶展示量提高了2.5倍.原核生物具有相对较低的代谢网络复杂性,同时其无核膜包被的拟核能够实现转录与翻译同时进行,这使得原核生物(如大肠杆菌)成为多基因操作及表达调控的模式生物[122-123].2018年,Gao等[121]通过多重CRISPRi系统对苹果酸生物合成途径中的基因进行组合调控后得到了2.3倍的苹果酸产量菌株. ...
... 2015年,Zalatan等[118]通过多sgRNA表达盒策略及sgRNA工程将适配体与识别区结合,将配体与转录因子融合表达后同时对3个目标基因进行激活或抑制调控,最终在酿酒酵母中获得了不同的紫罗兰素生物合成途径产物.Lobs等[120]在对一株马克思克鲁维酵母(Kluyveromyces marxianus)中乙酸酯的主要代谢途径和前体物质进行鉴定后,利用CRISPRi系统对ACO2b、SDH2、RIP1和MSS51四个基因进行多重调控,最终获得了乙酸乙酯3.8倍的提升.2017年,Lian等[119]在表征了多种同源Cas9蛋白的基因敲除、转录激活和转录抑制效率后,优化并组合三种同源蛋白最终构建了一套完全正交的三功能CRISPR代谢调控系统.由于正交的Cas9同源蛋白所识别的PAM序列不同,发挥不同功能的同源Cas9之间互不干扰,使得两个被调控报告基因的表达强度均被激活和抑制了5倍,并使第三个报告基因的敲除效率达到了95%.该系统通过一步组合调控使得改造的酿酒酵母中胡萝卜素产量提升了3倍,同时使表面葡聚糖酶展示量提高了2.5倍.原核生物具有相对较低的代谢网络复杂性,同时其无核膜包被的拟核能够实现转录与翻译同时进行,这使得原核生物(如大肠杆菌)成为多基因操作及表达调控的模式生物[122-123].2018年,Gao等[121]通过多重CRISPRi系统对苹果酸生物合成途径中的基因进行组合调控后得到了2.3倍的苹果酸产量菌株. ...
Combinatorial metabolic engineering using an orthogonal tri-functional CRISPR system
2
2017
... Research on the applications of the multiplexed CRISPR/Cas9 system
Table 3| 宿主细胞 | 多重CRISPR策略 | 靶向目标数量 | 应用/概念验证 | 参考文献 |
|---|
| 哺乳动物细胞 | 2个间隔区通过天然CRISPR序列自我加工 | 2个靶标(1个基因) | 对1个靶基因敲除,效率为1.6% | [4] |
| 酿酒酵母 | Casy4核酸酶加工 | 3个基因 | 同时对3个不同启动子进行激活,使得3个报告基因荧光强度提升了2倍 | [113] |
| 酿酒酵母 | 基于Csy4核酸酶加工的多gRNA快速组装 | 12个靶标(3个基因) | 通过12个gRNA靶向使得3个报告基因荧光强度分别被抑制了92%、81%和95% | [114] |
| 酿酒酵母 | 基于tRNA转录本切割机制的多gRNA表达系统 | 8个基因 | 通过两轮8个基因的敲除获得了30倍的游离脂肪酸产量 | [115] |
| 酿酒酵母 | 多个gRNA表达盒通过pol Ⅲ启动子启动 | 3个基因 | 将适配子与gRNA融合进行多基因调控,获得了不同的紫罗兰素生物合成产物 | [118] |
| 酿酒酵母 | 基于Csy4核酸酶加工的多gRNA快速组装策略及多Cas9正交组合调控 | 3个基因 | 通过组合调控使两个报告基因表达分别被抑制和激活5倍,同时以95%的效率敲除第三个基因 | [119] |
| 酿酒酵母 | 通过质粒表达多个gRNA表达盒以及截短gRNA用于调控 | 3个基因 | 通过对三个基因分别进行编辑、激活和抑制获得了α-檀香烯2.66倍的产量 | [43] |
| 马克思克鲁维酵母(Kluyveromyces marxianus) | 基于tRNA转录本切割机制的多gRNA表达系统 | 6个靶标(4个基因) | 通过对4个基因进行调控,使乙酸乙酯的产量提升了3.8倍 | [120] |
| 大肠杆菌 | 多个gRNA表达盒通过质粒表达 | 3个基因 | 通过对3个基因进行组合调控得到了2.3倍苹果酸产量菌株 | [121] |
| 大肠杆菌 | 多个gRNA表达盒通过质粒表达 | 3个基因 | 对苹果酸合成途径基因进行组合调控后获得了2.3倍苹果酸产量菌株 | [121] |
| 枯草芽孢杆菌(Bacillus subtilis) | 多个gRNA表达盒整合至基因组 | 3个基因 | 通过多基因动态组合调控提高了N-乙酰葡糖胺产量 | [122] |
| 天蓝色链霉菌(Streptomyces coelicolor) | 多个gRNA表达盒与dCas9在质粒上表达 | 4个基因 | 通过对4个靶基因同时进行抑制,使其mRNA表达量降为对照的2%~32% | [123] |
2015年,Zalatan等[118]通过多sgRNA表达盒策略及sgRNA工程将适配体与识别区结合,将配体与转录因子融合表达后同时对3个目标基因进行激活或抑制调控,最终在酿酒酵母中获得了不同的紫罗兰素生物合成途径产物.Lobs等[120]在对一株马克思克鲁维酵母(Kluyveromyces marxianus)中乙酸酯的主要代谢途径和前体物质进行鉴定后,利用CRISPRi系统对ACO2b、SDH2、RIP1和MSS51四个基因进行多重调控,最终获得了乙酸乙酯3.8倍的提升.2017年,Lian等[119]在表征了多种同源Cas9蛋白的基因敲除、转录激活和转录抑制效率后,优化并组合三种同源蛋白最终构建了一套完全正交的三功能CRISPR代谢调控系统.由于正交的Cas9同源蛋白所识别的PAM序列不同,发挥不同功能的同源Cas9之间互不干扰,使得两个被调控报告基因的表达强度均被激活和抑制了5倍,并使第三个报告基因的敲除效率达到了95%.该系统通过一步组合调控使得改造的酿酒酵母中胡萝卜素产量提升了3倍,同时使表面葡聚糖酶展示量提高了2.5倍.原核生物具有相对较低的代谢网络复杂性,同时其无核膜包被的拟核能够实现转录与翻译同时进行,这使得原核生物(如大肠杆菌)成为多基因操作及表达调控的模式生物[122-123].2018年,Gao等[121]通过多重CRISPRi系统对苹果酸生物合成途径中的基因进行组合调控后得到了2.3倍的苹果酸产量菌株. ...
... 2015年,Zalatan等[118]通过多sgRNA表达盒策略及sgRNA工程将适配体与识别区结合,将配体与转录因子融合表达后同时对3个目标基因进行激活或抑制调控,最终在酿酒酵母中获得了不同的紫罗兰素生物合成途径产物.Lobs等[120]在对一株马克思克鲁维酵母(Kluyveromyces marxianus)中乙酸酯的主要代谢途径和前体物质进行鉴定后,利用CRISPRi系统对ACO2b、SDH2、RIP1和MSS51四个基因进行多重调控,最终获得了乙酸乙酯3.8倍的提升.2017年,Lian等[119]在表征了多种同源Cas9蛋白的基因敲除、转录激活和转录抑制效率后,优化并组合三种同源蛋白最终构建了一套完全正交的三功能CRISPR代谢调控系统.由于正交的Cas9同源蛋白所识别的PAM序列不同,发挥不同功能的同源Cas9之间互不干扰,使得两个被调控报告基因的表达强度均被激活和抑制了5倍,并使第三个报告基因的敲除效率达到了95%.该系统通过一步组合调控使得改造的酿酒酵母中胡萝卜素产量提升了3倍,同时使表面葡聚糖酶展示量提高了2.5倍.原核生物具有相对较低的代谢网络复杂性,同时其无核膜包被的拟核能够实现转录与翻译同时进行,这使得原核生物(如大肠杆菌)成为多基因操作及表达调控的模式生物[122-123].2018年,Gao等[121]通过多重CRISPRi系统对苹果酸生物合成途径中的基因进行组合调控后得到了2.3倍的苹果酸产量菌株. ...
Highly multiplexed CRISPRi repression of respiratory functions enhances mitochondrial localized ethyl acetate biosynthesis in Kluyveromyces marxianus
2
2018
... Research on the applications of the multiplexed CRISPR/Cas9 system
Table 3| 宿主细胞 | 多重CRISPR策略 | 靶向目标数量 | 应用/概念验证 | 参考文献 |
|---|
| 哺乳动物细胞 | 2个间隔区通过天然CRISPR序列自我加工 | 2个靶标(1个基因) | 对1个靶基因敲除,效率为1.6% | [4] |
| 酿酒酵母 | Casy4核酸酶加工 | 3个基因 | 同时对3个不同启动子进行激活,使得3个报告基因荧光强度提升了2倍 | [113] |
| 酿酒酵母 | 基于Csy4核酸酶加工的多gRNA快速组装 | 12个靶标(3个基因) | 通过12个gRNA靶向使得3个报告基因荧光强度分别被抑制了92%、81%和95% | [114] |
| 酿酒酵母 | 基于tRNA转录本切割机制的多gRNA表达系统 | 8个基因 | 通过两轮8个基因的敲除获得了30倍的游离脂肪酸产量 | [115] |
| 酿酒酵母 | 多个gRNA表达盒通过pol Ⅲ启动子启动 | 3个基因 | 将适配子与gRNA融合进行多基因调控,获得了不同的紫罗兰素生物合成产物 | [118] |
| 酿酒酵母 | 基于Csy4核酸酶加工的多gRNA快速组装策略及多Cas9正交组合调控 | 3个基因 | 通过组合调控使两个报告基因表达分别被抑制和激活5倍,同时以95%的效率敲除第三个基因 | [119] |
| 酿酒酵母 | 通过质粒表达多个gRNA表达盒以及截短gRNA用于调控 | 3个基因 | 通过对三个基因分别进行编辑、激活和抑制获得了α-檀香烯2.66倍的产量 | [43] |
| 马克思克鲁维酵母(Kluyveromyces marxianus) | 基于tRNA转录本切割机制的多gRNA表达系统 | 6个靶标(4个基因) | 通过对4个基因进行调控,使乙酸乙酯的产量提升了3.8倍 | [120] |
| 大肠杆菌 | 多个gRNA表达盒通过质粒表达 | 3个基因 | 通过对3个基因进行组合调控得到了2.3倍苹果酸产量菌株 | [121] |
| 大肠杆菌 | 多个gRNA表达盒通过质粒表达 | 3个基因 | 对苹果酸合成途径基因进行组合调控后获得了2.3倍苹果酸产量菌株 | [121] |
| 枯草芽孢杆菌(Bacillus subtilis) | 多个gRNA表达盒整合至基因组 | 3个基因 | 通过多基因动态组合调控提高了N-乙酰葡糖胺产量 | [122] |
| 天蓝色链霉菌(Streptomyces coelicolor) | 多个gRNA表达盒与dCas9在质粒上表达 | 4个基因 | 通过对4个靶基因同时进行抑制,使其mRNA表达量降为对照的2%~32% | [123] |
2015年,Zalatan等[118]通过多sgRNA表达盒策略及sgRNA工程将适配体与识别区结合,将配体与转录因子融合表达后同时对3个目标基因进行激活或抑制调控,最终在酿酒酵母中获得了不同的紫罗兰素生物合成途径产物.Lobs等[120]在对一株马克思克鲁维酵母(Kluyveromyces marxianus)中乙酸酯的主要代谢途径和前体物质进行鉴定后,利用CRISPRi系统对ACO2b、SDH2、RIP1和MSS51四个基因进行多重调控,最终获得了乙酸乙酯3.8倍的提升.2017年,Lian等[119]在表征了多种同源Cas9蛋白的基因敲除、转录激活和转录抑制效率后,优化并组合三种同源蛋白最终构建了一套完全正交的三功能CRISPR代谢调控系统.由于正交的Cas9同源蛋白所识别的PAM序列不同,发挥不同功能的同源Cas9之间互不干扰,使得两个被调控报告基因的表达强度均被激活和抑制了5倍,并使第三个报告基因的敲除效率达到了95%.该系统通过一步组合调控使得改造的酿酒酵母中胡萝卜素产量提升了3倍,同时使表面葡聚糖酶展示量提高了2.5倍.原核生物具有相对较低的代谢网络复杂性,同时其无核膜包被的拟核能够实现转录与翻译同时进行,这使得原核生物(如大肠杆菌)成为多基因操作及表达调控的模式生物[122-123].2018年,Gao等[121]通过多重CRISPRi系统对苹果酸生物合成途径中的基因进行组合调控后得到了2.3倍的苹果酸产量菌株. ...
... 2015年,Zalatan等[118]通过多sgRNA表达盒策略及sgRNA工程将适配体与识别区结合,将配体与转录因子融合表达后同时对3个目标基因进行激活或抑制调控,最终在酿酒酵母中获得了不同的紫罗兰素生物合成途径产物.Lobs等[120]在对一株马克思克鲁维酵母(Kluyveromyces marxianus)中乙酸酯的主要代谢途径和前体物质进行鉴定后,利用CRISPRi系统对ACO2b、SDH2、RIP1和MSS51四个基因进行多重调控,最终获得了乙酸乙酯3.8倍的提升.2017年,Lian等[119]在表征了多种同源Cas9蛋白的基因敲除、转录激活和转录抑制效率后,优化并组合三种同源蛋白最终构建了一套完全正交的三功能CRISPR代谢调控系统.由于正交的Cas9同源蛋白所识别的PAM序列不同,发挥不同功能的同源Cas9之间互不干扰,使得两个被调控报告基因的表达强度均被激活和抑制了5倍,并使第三个报告基因的敲除效率达到了95%.该系统通过一步组合调控使得改造的酿酒酵母中胡萝卜素产量提升了3倍,同时使表面葡聚糖酶展示量提高了2.5倍.原核生物具有相对较低的代谢网络复杂性,同时其无核膜包被的拟核能够实现转录与翻译同时进行,这使得原核生物(如大肠杆菌)成为多基因操作及表达调控的模式生物[122-123].2018年,Gao等[121]通过多重CRISPRi系统对苹果酸生物合成途径中的基因进行组合调控后得到了2.3倍的苹果酸产量菌株. ...
Engineering Escherichia coli for malate production by integrating modular pathway characterization with CRISPRi-guided multiplexed metabolic tuning
3
2018
... Research on the applications of the multiplexed CRISPR/Cas9 system
Table 3| 宿主细胞 | 多重CRISPR策略 | 靶向目标数量 | 应用/概念验证 | 参考文献 |
|---|
| 哺乳动物细胞 | 2个间隔区通过天然CRISPR序列自我加工 | 2个靶标(1个基因) | 对1个靶基因敲除,效率为1.6% | [4] |
| 酿酒酵母 | Casy4核酸酶加工 | 3个基因 | 同时对3个不同启动子进行激活,使得3个报告基因荧光强度提升了2倍 | [113] |
| 酿酒酵母 | 基于Csy4核酸酶加工的多gRNA快速组装 | 12个靶标(3个基因) | 通过12个gRNA靶向使得3个报告基因荧光强度分别被抑制了92%、81%和95% | [114] |
| 酿酒酵母 | 基于tRNA转录本切割机制的多gRNA表达系统 | 8个基因 | 通过两轮8个基因的敲除获得了30倍的游离脂肪酸产量 | [115] |
| 酿酒酵母 | 多个gRNA表达盒通过pol Ⅲ启动子启动 | 3个基因 | 将适配子与gRNA融合进行多基因调控,获得了不同的紫罗兰素生物合成产物 | [118] |
| 酿酒酵母 | 基于Csy4核酸酶加工的多gRNA快速组装策略及多Cas9正交组合调控 | 3个基因 | 通过组合调控使两个报告基因表达分别被抑制和激活5倍,同时以95%的效率敲除第三个基因 | [119] |
| 酿酒酵母 | 通过质粒表达多个gRNA表达盒以及截短gRNA用于调控 | 3个基因 | 通过对三个基因分别进行编辑、激活和抑制获得了α-檀香烯2.66倍的产量 | [43] |
| 马克思克鲁维酵母(Kluyveromyces marxianus) | 基于tRNA转录本切割机制的多gRNA表达系统 | 6个靶标(4个基因) | 通过对4个基因进行调控,使乙酸乙酯的产量提升了3.8倍 | [120] |
| 大肠杆菌 | 多个gRNA表达盒通过质粒表达 | 3个基因 | 通过对3个基因进行组合调控得到了2.3倍苹果酸产量菌株 | [121] |
| 大肠杆菌 | 多个gRNA表达盒通过质粒表达 | 3个基因 | 对苹果酸合成途径基因进行组合调控后获得了2.3倍苹果酸产量菌株 | [121] |
| 枯草芽孢杆菌(Bacillus subtilis) | 多个gRNA表达盒整合至基因组 | 3个基因 | 通过多基因动态组合调控提高了N-乙酰葡糖胺产量 | [122] |
| 天蓝色链霉菌(Streptomyces coelicolor) | 多个gRNA表达盒与dCas9在质粒上表达 | 4个基因 | 通过对4个靶基因同时进行抑制,使其mRNA表达量降为对照的2%~32% | [123] |
2015年,Zalatan等[118]通过多sgRNA表达盒策略及sgRNA工程将适配体与识别区结合,将配体与转录因子融合表达后同时对3个目标基因进行激活或抑制调控,最终在酿酒酵母中获得了不同的紫罗兰素生物合成途径产物.Lobs等[120]在对一株马克思克鲁维酵母(Kluyveromyces marxianus)中乙酸酯的主要代谢途径和前体物质进行鉴定后,利用CRISPRi系统对ACO2b、SDH2、RIP1和MSS51四个基因进行多重调控,最终获得了乙酸乙酯3.8倍的提升.2017年,Lian等[119]在表征了多种同源Cas9蛋白的基因敲除、转录激活和转录抑制效率后,优化并组合三种同源蛋白最终构建了一套完全正交的三功能CRISPR代谢调控系统.由于正交的Cas9同源蛋白所识别的PAM序列不同,发挥不同功能的同源Cas9之间互不干扰,使得两个被调控报告基因的表达强度均被激活和抑制了5倍,并使第三个报告基因的敲除效率达到了95%.该系统通过一步组合调控使得改造的酿酒酵母中胡萝卜素产量提升了3倍,同时使表面葡聚糖酶展示量提高了2.5倍.原核生物具有相对较低的代谢网络复杂性,同时其无核膜包被的拟核能够实现转录与翻译同时进行,这使得原核生物(如大肠杆菌)成为多基因操作及表达调控的模式生物[122-123].2018年,Gao等[121]通过多重CRISPRi系统对苹果酸生物合成途径中的基因进行组合调控后得到了2.3倍的苹果酸产量菌株. ...
... [
121]
| 枯草芽孢杆菌(Bacillus subtilis) | 多个gRNA表达盒整合至基因组 | 3个基因 | 通过多基因动态组合调控提高了N-乙酰葡糖胺产量 | [122] |
| 天蓝色链霉菌(Streptomyces coelicolor) | 多个gRNA表达盒与dCas9在质粒上表达 | 4个基因 | 通过对4个靶基因同时进行抑制,使其mRNA表达量降为对照的2%~32% | [123] |
2015年,Zalatan等[118]通过多sgRNA表达盒策略及sgRNA工程将适配体与识别区结合,将配体与转录因子融合表达后同时对3个目标基因进行激活或抑制调控,最终在酿酒酵母中获得了不同的紫罗兰素生物合成途径产物.Lobs等[120]在对一株马克思克鲁维酵母(Kluyveromyces marxianus)中乙酸酯的主要代谢途径和前体物质进行鉴定后,利用CRISPRi系统对ACO2b、SDH2、RIP1和MSS51四个基因进行多重调控,最终获得了乙酸乙酯3.8倍的提升.2017年,Lian等[119]在表征了多种同源Cas9蛋白的基因敲除、转录激活和转录抑制效率后,优化并组合三种同源蛋白最终构建了一套完全正交的三功能CRISPR代谢调控系统.由于正交的Cas9同源蛋白所识别的PAM序列不同,发挥不同功能的同源Cas9之间互不干扰,使得两个被调控报告基因的表达强度均被激活和抑制了5倍,并使第三个报告基因的敲除效率达到了95%.该系统通过一步组合调控使得改造的酿酒酵母中胡萝卜素产量提升了3倍,同时使表面葡聚糖酶展示量提高了2.5倍.原核生物具有相对较低的代谢网络复杂性,同时其无核膜包被的拟核能够实现转录与翻译同时进行,这使得原核生物(如大肠杆菌)成为多基因操作及表达调控的模式生物[122-123].2018年,Gao等[121]通过多重CRISPRi系统对苹果酸生物合成途径中的基因进行组合调控后得到了2.3倍的苹果酸产量菌株. ...
... 2015年,Zalatan等[118]通过多sgRNA表达盒策略及sgRNA工程将适配体与识别区结合,将配体与转录因子融合表达后同时对3个目标基因进行激活或抑制调控,最终在酿酒酵母中获得了不同的紫罗兰素生物合成途径产物.Lobs等[120]在对一株马克思克鲁维酵母(Kluyveromyces marxianus)中乙酸酯的主要代谢途径和前体物质进行鉴定后,利用CRISPRi系统对ACO2b、SDH2、RIP1和MSS51四个基因进行多重调控,最终获得了乙酸乙酯3.8倍的提升.2017年,Lian等[119]在表征了多种同源Cas9蛋白的基因敲除、转录激活和转录抑制效率后,优化并组合三种同源蛋白最终构建了一套完全正交的三功能CRISPR代谢调控系统.由于正交的Cas9同源蛋白所识别的PAM序列不同,发挥不同功能的同源Cas9之间互不干扰,使得两个被调控报告基因的表达强度均被激活和抑制了5倍,并使第三个报告基因的敲除效率达到了95%.该系统通过一步组合调控使得改造的酿酒酵母中胡萝卜素产量提升了3倍,同时使表面葡聚糖酶展示量提高了2.5倍.原核生物具有相对较低的代谢网络复杂性,同时其无核膜包被的拟核能够实现转录与翻译同时进行,这使得原核生物(如大肠杆菌)成为多基因操作及表达调控的模式生物[122-123].2018年,Gao等[121]通过多重CRISPRi系统对苹果酸生物合成途径中的基因进行组合调控后得到了2.3倍的苹果酸产量菌株. ...
CRISPRi allows optimal temporal control of N-acetylglucosamine bioproduction by a dynamic coordination of glucose and xylose metabolism in Bacillus subtilis
2
2018
... Research on the applications of the multiplexed CRISPR/Cas9 system
Table 3| 宿主细胞 | 多重CRISPR策略 | 靶向目标数量 | 应用/概念验证 | 参考文献 |
|---|
| 哺乳动物细胞 | 2个间隔区通过天然CRISPR序列自我加工 | 2个靶标(1个基因) | 对1个靶基因敲除,效率为1.6% | [4] |
| 酿酒酵母 | Casy4核酸酶加工 | 3个基因 | 同时对3个不同启动子进行激活,使得3个报告基因荧光强度提升了2倍 | [113] |
| 酿酒酵母 | 基于Csy4核酸酶加工的多gRNA快速组装 | 12个靶标(3个基因) | 通过12个gRNA靶向使得3个报告基因荧光强度分别被抑制了92%、81%和95% | [114] |
| 酿酒酵母 | 基于tRNA转录本切割机制的多gRNA表达系统 | 8个基因 | 通过两轮8个基因的敲除获得了30倍的游离脂肪酸产量 | [115] |
| 酿酒酵母 | 多个gRNA表达盒通过pol Ⅲ启动子启动 | 3个基因 | 将适配子与gRNA融合进行多基因调控,获得了不同的紫罗兰素生物合成产物 | [118] |
| 酿酒酵母 | 基于Csy4核酸酶加工的多gRNA快速组装策略及多Cas9正交组合调控 | 3个基因 | 通过组合调控使两个报告基因表达分别被抑制和激活5倍,同时以95%的效率敲除第三个基因 | [119] |
| 酿酒酵母 | 通过质粒表达多个gRNA表达盒以及截短gRNA用于调控 | 3个基因 | 通过对三个基因分别进行编辑、激活和抑制获得了α-檀香烯2.66倍的产量 | [43] |
| 马克思克鲁维酵母(Kluyveromyces marxianus) | 基于tRNA转录本切割机制的多gRNA表达系统 | 6个靶标(4个基因) | 通过对4个基因进行调控,使乙酸乙酯的产量提升了3.8倍 | [120] |
| 大肠杆菌 | 多个gRNA表达盒通过质粒表达 | 3个基因 | 通过对3个基因进行组合调控得到了2.3倍苹果酸产量菌株 | [121] |
| 大肠杆菌 | 多个gRNA表达盒通过质粒表达 | 3个基因 | 对苹果酸合成途径基因进行组合调控后获得了2.3倍苹果酸产量菌株 | [121] |
| 枯草芽孢杆菌(Bacillus subtilis) | 多个gRNA表达盒整合至基因组 | 3个基因 | 通过多基因动态组合调控提高了N-乙酰葡糖胺产量 | [122] |
| 天蓝色链霉菌(Streptomyces coelicolor) | 多个gRNA表达盒与dCas9在质粒上表达 | 4个基因 | 通过对4个靶基因同时进行抑制,使其mRNA表达量降为对照的2%~32% | [123] |
2015年,Zalatan等[118]通过多sgRNA表达盒策略及sgRNA工程将适配体与识别区结合,将配体与转录因子融合表达后同时对3个目标基因进行激活或抑制调控,最终在酿酒酵母中获得了不同的紫罗兰素生物合成途径产物.Lobs等[120]在对一株马克思克鲁维酵母(Kluyveromyces marxianus)中乙酸酯的主要代谢途径和前体物质进行鉴定后,利用CRISPRi系统对ACO2b、SDH2、RIP1和MSS51四个基因进行多重调控,最终获得了乙酸乙酯3.8倍的提升.2017年,Lian等[119]在表征了多种同源Cas9蛋白的基因敲除、转录激活和转录抑制效率后,优化并组合三种同源蛋白最终构建了一套完全正交的三功能CRISPR代谢调控系统.由于正交的Cas9同源蛋白所识别的PAM序列不同,发挥不同功能的同源Cas9之间互不干扰,使得两个被调控报告基因的表达强度均被激活和抑制了5倍,并使第三个报告基因的敲除效率达到了95%.该系统通过一步组合调控使得改造的酿酒酵母中胡萝卜素产量提升了3倍,同时使表面葡聚糖酶展示量提高了2.5倍.原核生物具有相对较低的代谢网络复杂性,同时其无核膜包被的拟核能够实现转录与翻译同时进行,这使得原核生物(如大肠杆菌)成为多基因操作及表达调控的模式生物[122-123].2018年,Gao等[121]通过多重CRISPRi系统对苹果酸生物合成途径中的基因进行组合调控后得到了2.3倍的苹果酸产量菌株. ...
... 2015年,Zalatan等[118]通过多sgRNA表达盒策略及sgRNA工程将适配体与识别区结合,将配体与转录因子融合表达后同时对3个目标基因进行激活或抑制调控,最终在酿酒酵母中获得了不同的紫罗兰素生物合成途径产物.Lobs等[120]在对一株马克思克鲁维酵母(Kluyveromyces marxianus)中乙酸酯的主要代谢途径和前体物质进行鉴定后,利用CRISPRi系统对ACO2b、SDH2、RIP1和MSS51四个基因进行多重调控,最终获得了乙酸乙酯3.8倍的提升.2017年,Lian等[119]在表征了多种同源Cas9蛋白的基因敲除、转录激活和转录抑制效率后,优化并组合三种同源蛋白最终构建了一套完全正交的三功能CRISPR代谢调控系统.由于正交的Cas9同源蛋白所识别的PAM序列不同,发挥不同功能的同源Cas9之间互不干扰,使得两个被调控报告基因的表达强度均被激活和抑制了5倍,并使第三个报告基因的敲除效率达到了95%.该系统通过一步组合调控使得改造的酿酒酵母中胡萝卜素产量提升了3倍,同时使表面葡聚糖酶展示量提高了2.5倍.原核生物具有相对较低的代谢网络复杂性,同时其无核膜包被的拟核能够实现转录与翻译同时进行,这使得原核生物(如大肠杆菌)成为多基因操作及表达调控的模式生物[122-123].2018年,Gao等[121]通过多重CRISPRi系统对苹果酸生物合成途径中的基因进行组合调控后得到了2.3倍的苹果酸产量菌株. ...
CRISPR/dCas9-mediated multiplex gene repression in streptomyces
2
2018
... Research on the applications of the multiplexed CRISPR/Cas9 system
Table 3| 宿主细胞 | 多重CRISPR策略 | 靶向目标数量 | 应用/概念验证 | 参考文献 |
|---|
| 哺乳动物细胞 | 2个间隔区通过天然CRISPR序列自我加工 | 2个靶标(1个基因) | 对1个靶基因敲除,效率为1.6% | [4] |
| 酿酒酵母 | Casy4核酸酶加工 | 3个基因 | 同时对3个不同启动子进行激活,使得3个报告基因荧光强度提升了2倍 | [113] |
| 酿酒酵母 | 基于Csy4核酸酶加工的多gRNA快速组装 | 12个靶标(3个基因) | 通过12个gRNA靶向使得3个报告基因荧光强度分别被抑制了92%、81%和95% | [114] |
| 酿酒酵母 | 基于tRNA转录本切割机制的多gRNA表达系统 | 8个基因 | 通过两轮8个基因的敲除获得了30倍的游离脂肪酸产量 | [115] |
| 酿酒酵母 | 多个gRNA表达盒通过pol Ⅲ启动子启动 | 3个基因 | 将适配子与gRNA融合进行多基因调控,获得了不同的紫罗兰素生物合成产物 | [118] |
| 酿酒酵母 | 基于Csy4核酸酶加工的多gRNA快速组装策略及多Cas9正交组合调控 | 3个基因 | 通过组合调控使两个报告基因表达分别被抑制和激活5倍,同时以95%的效率敲除第三个基因 | [119] |
| 酿酒酵母 | 通过质粒表达多个gRNA表达盒以及截短gRNA用于调控 | 3个基因 | 通过对三个基因分别进行编辑、激活和抑制获得了α-檀香烯2.66倍的产量 | [43] |
| 马克思克鲁维酵母(Kluyveromyces marxianus) | 基于tRNA转录本切割机制的多gRNA表达系统 | 6个靶标(4个基因) | 通过对4个基因进行调控,使乙酸乙酯的产量提升了3.8倍 | [120] |
| 大肠杆菌 | 多个gRNA表达盒通过质粒表达 | 3个基因 | 通过对3个基因进行组合调控得到了2.3倍苹果酸产量菌株 | [121] |
| 大肠杆菌 | 多个gRNA表达盒通过质粒表达 | 3个基因 | 对苹果酸合成途径基因进行组合调控后获得了2.3倍苹果酸产量菌株 | [121] |
| 枯草芽孢杆菌(Bacillus subtilis) | 多个gRNA表达盒整合至基因组 | 3个基因 | 通过多基因动态组合调控提高了N-乙酰葡糖胺产量 | [122] |
| 天蓝色链霉菌(Streptomyces coelicolor) | 多个gRNA表达盒与dCas9在质粒上表达 | 4个基因 | 通过对4个靶基因同时进行抑制,使其mRNA表达量降为对照的2%~32% | [123] |
2015年,Zalatan等[118]通过多sgRNA表达盒策略及sgRNA工程将适配体与识别区结合,将配体与转录因子融合表达后同时对3个目标基因进行激活或抑制调控,最终在酿酒酵母中获得了不同的紫罗兰素生物合成途径产物.Lobs等[120]在对一株马克思克鲁维酵母(Kluyveromyces marxianus)中乙酸酯的主要代谢途径和前体物质进行鉴定后,利用CRISPRi系统对ACO2b、SDH2、RIP1和MSS51四个基因进行多重调控,最终获得了乙酸乙酯3.8倍的提升.2017年,Lian等[119]在表征了多种同源Cas9蛋白的基因敲除、转录激活和转录抑制效率后,优化并组合三种同源蛋白最终构建了一套完全正交的三功能CRISPR代谢调控系统.由于正交的Cas9同源蛋白所识别的PAM序列不同,发挥不同功能的同源Cas9之间互不干扰,使得两个被调控报告基因的表达强度均被激活和抑制了5倍,并使第三个报告基因的敲除效率达到了95%.该系统通过一步组合调控使得改造的酿酒酵母中胡萝卜素产量提升了3倍,同时使表面葡聚糖酶展示量提高了2.5倍.原核生物具有相对较低的代谢网络复杂性,同时其无核膜包被的拟核能够实现转录与翻译同时进行,这使得原核生物(如大肠杆菌)成为多基因操作及表达调控的模式生物[122-123].2018年,Gao等[121]通过多重CRISPRi系统对苹果酸生物合成途径中的基因进行组合调控后得到了2.3倍的苹果酸产量菌株. ...
... 2015年,Zalatan等[118]通过多sgRNA表达盒策略及sgRNA工程将适配体与识别区结合,将配体与转录因子融合表达后同时对3个目标基因进行激活或抑制调控,最终在酿酒酵母中获得了不同的紫罗兰素生物合成途径产物.Lobs等[120]在对一株马克思克鲁维酵母(Kluyveromyces marxianus)中乙酸酯的主要代谢途径和前体物质进行鉴定后,利用CRISPRi系统对ACO2b、SDH2、RIP1和MSS51四个基因进行多重调控,最终获得了乙酸乙酯3.8倍的提升.2017年,Lian等[119]在表征了多种同源Cas9蛋白的基因敲除、转录激活和转录抑制效率后,优化并组合三种同源蛋白最终构建了一套完全正交的三功能CRISPR代谢调控系统.由于正交的Cas9同源蛋白所识别的PAM序列不同,发挥不同功能的同源Cas9之间互不干扰,使得两个被调控报告基因的表达强度均被激活和抑制了5倍,并使第三个报告基因的敲除效率达到了95%.该系统通过一步组合调控使得改造的酿酒酵母中胡萝卜素产量提升了3倍,同时使表面葡聚糖酶展示量提高了2.5倍.原核生物具有相对较低的代谢网络复杂性,同时其无核膜包被的拟核能够实现转录与翻译同时进行,这使得原核生物(如大肠杆菌)成为多基因操作及表达调控的模式生物[122-123].2018年,Gao等[121]通过多重CRISPRi系统对苹果酸生物合成途径中的基因进行组合调控后得到了2.3倍的苹果酸产量菌株. ...
Shortening the sgRNA-DNA interface enables SpCas9 and eSpCas9(1.1) to nick the target DNA strand
1
2020
... 此外,通过修饰gRNA如使其融合适配子或改变gRNA识别区长度,就可能实现CRISPR系统的多重编辑或调控.2020年,Fan等[124]通过研究证实了不同长度的gRNA-DNA互补区会对HNH和RuvC结构域切割靶向链和非靶向链的能力产生影响,并基于此开发了一种使两个靶基因分别产生DSB和C-T碱基转换的双功能编辑系统.同年,浙江大学徐志南组[43]通过gRNA工程构建了一种能同时进行基因激活、抑制和编辑的三功能CRISPR系统(CRISPR-ARE).该系统在仅使用Cas9-VPR一种CRISPR融合蛋白的条件下,利用截短的14bp gRNA实现靶基因的激活和抑制,并利用正常的20bp gRNA实现目标基因敲除.尽管目前的gRNA多表达策略已逐渐成熟,对于gRNA在细胞内的多路复用机制及其对Cas9竞争作用的了解仍有待加强.多gRNA策略(包括Csy4、核酶和tRNA等)的处理效率将在确定理论上的gRNA数量上限方面发挥关键作用,在未来的研究中,创建和处理长阵列的策略将会得到优化和加强. ...
A chemical-inducible CRISPR-Cas9 system for rapid control of genome editing
1
2016
... 尽管研究人员针对CRISPR系统的PAM序列依赖性、编辑脱靶率等问题给出了一些解决方案如发掘Cas9同源蛋白、通过工程化改造Cas9或gRNA从而拓宽PAM序列识别范围和提高中靶率等,这些问题依然困扰着CRISPR系统在更多生物中进行快速、高效、精准的基因编辑和调控,尤其是在最终将CRISPR系统作为基因疗法使用时,其风险控制、脱靶编辑等问题一直都被科研人员重视.为了减少脱靶效应、提高基因编辑的精确性和可控性,Liu等[125]将一种Cas9突变体iCas9与雌激素受体的激素结合结构域(hormone-binding domain of the estrogen receptor,ERT2)融合后,可使iCas9的核酸酶活性获得4-羟基三苯甲胺(4-hydroxytamoxifen,4-HT)诱导性,从而快速和可逆地控制该系统在基因组编辑功能上的作用.2017年,Shin 等[126]发现了抗CRISPR 蛋白AcrIIA4对CRISPR/ Cas9系统的抑制作用,使人为控制CRISPR系统作为疗法使用时的风险成为可能.2021年,Jain等[127]将AcrIIA4与二氢叶酸还原酶(dihydrofolate reductase,DHRF)融合后使其仅在甲氧苄啶(trimethoprim,TMP)存在时才能抑制Cas9和DNA的结合能力,从而实现对Cas9编辑和dCas9基因组扰动的化学调控,提高了CRISPR/Cas9系统的特异性和生物安全性.2022年,北京大学胡家志课题组[128]揭示了CRISPR/Cas9系统在基因编辑过程中造成染色体易位的分子机制,随后将优化过的人源TREX2蛋白与Cas9相偶联生成Cas9TX,其能抑制基因编辑过程中的染色体易位、大片段缺失等染色体结构异常的产生,使CRISPR/Cas9的基因编辑安全性大大提高.这些研究最终将服务于基于CRISPR/Cas9工具箱的单基因突变遗传病的基因疗法,并为全人类带来福祉.目前,碱基编辑已成功在其他哺乳动物中用于疾病治疗,并已被证明具有治疗人类单基因突变遗传病的潜力.2020年,Yeh等[129]通过双AAV递送CBEMax系统成功治愈小鼠Tmc1基因隐性突变导致的耳聋,使其能够重新对中等程度的声音做出反应.Wang等[130]通过对β-地中海贫血和镰刀状贫血患者造血干细胞中的BCL11A增强子位点进行编辑或修复发生突变的HBB基因,以及进行自体造血干细胞移植使其在体内分化产生正常的红细胞,从而治疗由β-globin珠蛋白突变引发的遗传病.CRISPR作为近年来发展起来的新兴技术,在为生命科学带来一系列革命性应用技术的同时,还存在一些问题和困难,随着越来越多热忱的研究者的加入,CRIPSR/Cas9系统的功能将会进一步完善,这些阻碍与挑战也将被不断去除和攻破. ...
Disabling Cas9 by an anti-CRISPR DNA mimic
1
2017
... 尽管研究人员针对CRISPR系统的PAM序列依赖性、编辑脱靶率等问题给出了一些解决方案如发掘Cas9同源蛋白、通过工程化改造Cas9或gRNA从而拓宽PAM序列识别范围和提高中靶率等,这些问题依然困扰着CRISPR系统在更多生物中进行快速、高效、精准的基因编辑和调控,尤其是在最终将CRISPR系统作为基因疗法使用时,其风险控制、脱靶编辑等问题一直都被科研人员重视.为了减少脱靶效应、提高基因编辑的精确性和可控性,Liu等[125]将一种Cas9突变体iCas9与雌激素受体的激素结合结构域(hormone-binding domain of the estrogen receptor,ERT2)融合后,可使iCas9的核酸酶活性获得4-羟基三苯甲胺(4-hydroxytamoxifen,4-HT)诱导性,从而快速和可逆地控制该系统在基因组编辑功能上的作用.2017年,Shin 等[126]发现了抗CRISPR 蛋白AcrIIA4对CRISPR/ Cas9系统的抑制作用,使人为控制CRISPR系统作为疗法使用时的风险成为可能.2021年,Jain等[127]将AcrIIA4与二氢叶酸还原酶(dihydrofolate reductase,DHRF)融合后使其仅在甲氧苄啶(trimethoprim,TMP)存在时才能抑制Cas9和DNA的结合能力,从而实现对Cas9编辑和dCas9基因组扰动的化学调控,提高了CRISPR/Cas9系统的特异性和生物安全性.2022年,北京大学胡家志课题组[128]揭示了CRISPR/Cas9系统在基因编辑过程中造成染色体易位的分子机制,随后将优化过的人源TREX2蛋白与Cas9相偶联生成Cas9TX,其能抑制基因编辑过程中的染色体易位、大片段缺失等染色体结构异常的产生,使CRISPR/Cas9的基因编辑安全性大大提高.这些研究最终将服务于基于CRISPR/Cas9工具箱的单基因突变遗传病的基因疗法,并为全人类带来福祉.目前,碱基编辑已成功在其他哺乳动物中用于疾病治疗,并已被证明具有治疗人类单基因突变遗传病的潜力.2020年,Yeh等[129]通过双AAV递送CBEMax系统成功治愈小鼠Tmc1基因隐性突变导致的耳聋,使其能够重新对中等程度的声音做出反应.Wang等[130]通过对β-地中海贫血和镰刀状贫血患者造血干细胞中的BCL11A增强子位点进行编辑或修复发生突变的HBB基因,以及进行自体造血干细胞移植使其在体内分化产生正常的红细胞,从而治疗由β-globin珠蛋白突变引发的遗传病.CRISPR作为近年来发展起来的新兴技术,在为生命科学带来一系列革命性应用技术的同时,还存在一些问题和困难,随着越来越多热忱的研究者的加入,CRIPSR/Cas9系统的功能将会进一步完善,这些阻碍与挑战也将被不断去除和攻破. ...
Precise regulation of Cas9-mediated genome engineering by anti-CRISPR-based inducible CRISPR controllers
1
2021
... 尽管研究人员针对CRISPR系统的PAM序列依赖性、编辑脱靶率等问题给出了一些解决方案如发掘Cas9同源蛋白、通过工程化改造Cas9或gRNA从而拓宽PAM序列识别范围和提高中靶率等,这些问题依然困扰着CRISPR系统在更多生物中进行快速、高效、精准的基因编辑和调控,尤其是在最终将CRISPR系统作为基因疗法使用时,其风险控制、脱靶编辑等问题一直都被科研人员重视.为了减少脱靶效应、提高基因编辑的精确性和可控性,Liu等[125]将一种Cas9突变体iCas9与雌激素受体的激素结合结构域(hormone-binding domain of the estrogen receptor,ERT2)融合后,可使iCas9的核酸酶活性获得4-羟基三苯甲胺(4-hydroxytamoxifen,4-HT)诱导性,从而快速和可逆地控制该系统在基因组编辑功能上的作用.2017年,Shin 等[126]发现了抗CRISPR 蛋白AcrIIA4对CRISPR/ Cas9系统的抑制作用,使人为控制CRISPR系统作为疗法使用时的风险成为可能.2021年,Jain等[127]将AcrIIA4与二氢叶酸还原酶(dihydrofolate reductase,DHRF)融合后使其仅在甲氧苄啶(trimethoprim,TMP)存在时才能抑制Cas9和DNA的结合能力,从而实现对Cas9编辑和dCas9基因组扰动的化学调控,提高了CRISPR/Cas9系统的特异性和生物安全性.2022年,北京大学胡家志课题组[128]揭示了CRISPR/Cas9系统在基因编辑过程中造成染色体易位的分子机制,随后将优化过的人源TREX2蛋白与Cas9相偶联生成Cas9TX,其能抑制基因编辑过程中的染色体易位、大片段缺失等染色体结构异常的产生,使CRISPR/Cas9的基因编辑安全性大大提高.这些研究最终将服务于基于CRISPR/Cas9工具箱的单基因突变遗传病的基因疗法,并为全人类带来福祉.目前,碱基编辑已成功在其他哺乳动物中用于疾病治疗,并已被证明具有治疗人类单基因突变遗传病的潜力.2020年,Yeh等[129]通过双AAV递送CBEMax系统成功治愈小鼠Tmc1基因隐性突变导致的耳聋,使其能够重新对中等程度的声音做出反应.Wang等[130]通过对β-地中海贫血和镰刀状贫血患者造血干细胞中的BCL11A增强子位点进行编辑或修复发生突变的HBB基因,以及进行自体造血干细胞移植使其在体内分化产生正常的红细胞,从而治疗由β-globin珠蛋白突变引发的遗传病.CRISPR作为近年来发展起来的新兴技术,在为生命科学带来一系列革命性应用技术的同时,还存在一些问题和困难,随着越来越多热忱的研究者的加入,CRIPSR/Cas9系统的功能将会进一步完善,这些阻碍与挑战也将被不断去除和攻破. ...
Cas9 exo-endonuclease eliminates chromosomal translocations during genome editing
1
2022
... 尽管研究人员针对CRISPR系统的PAM序列依赖性、编辑脱靶率等问题给出了一些解决方案如发掘Cas9同源蛋白、通过工程化改造Cas9或gRNA从而拓宽PAM序列识别范围和提高中靶率等,这些问题依然困扰着CRISPR系统在更多生物中进行快速、高效、精准的基因编辑和调控,尤其是在最终将CRISPR系统作为基因疗法使用时,其风险控制、脱靶编辑等问题一直都被科研人员重视.为了减少脱靶效应、提高基因编辑的精确性和可控性,Liu等[125]将一种Cas9突变体iCas9与雌激素受体的激素结合结构域(hormone-binding domain of the estrogen receptor,ERT2)融合后,可使iCas9的核酸酶活性获得4-羟基三苯甲胺(4-hydroxytamoxifen,4-HT)诱导性,从而快速和可逆地控制该系统在基因组编辑功能上的作用.2017年,Shin 等[126]发现了抗CRISPR 蛋白AcrIIA4对CRISPR/ Cas9系统的抑制作用,使人为控制CRISPR系统作为疗法使用时的风险成为可能.2021年,Jain等[127]将AcrIIA4与二氢叶酸还原酶(dihydrofolate reductase,DHRF)融合后使其仅在甲氧苄啶(trimethoprim,TMP)存在时才能抑制Cas9和DNA的结合能力,从而实现对Cas9编辑和dCas9基因组扰动的化学调控,提高了CRISPR/Cas9系统的特异性和生物安全性.2022年,北京大学胡家志课题组[128]揭示了CRISPR/Cas9系统在基因编辑过程中造成染色体易位的分子机制,随后将优化过的人源TREX2蛋白与Cas9相偶联生成Cas9TX,其能抑制基因编辑过程中的染色体易位、大片段缺失等染色体结构异常的产生,使CRISPR/Cas9的基因编辑安全性大大提高.这些研究最终将服务于基于CRISPR/Cas9工具箱的单基因突变遗传病的基因疗法,并为全人类带来福祉.目前,碱基编辑已成功在其他哺乳动物中用于疾病治疗,并已被证明具有治疗人类单基因突变遗传病的潜力.2020年,Yeh等[129]通过双AAV递送CBEMax系统成功治愈小鼠Tmc1基因隐性突变导致的耳聋,使其能够重新对中等程度的声音做出反应.Wang等[130]通过对β-地中海贫血和镰刀状贫血患者造血干细胞中的BCL11A增强子位点进行编辑或修复发生突变的HBB基因,以及进行自体造血干细胞移植使其在体内分化产生正常的红细胞,从而治疗由β-globin珠蛋白突变引发的遗传病.CRISPR作为近年来发展起来的新兴技术,在为生命科学带来一系列革命性应用技术的同时,还存在一些问题和困难,随着越来越多热忱的研究者的加入,CRIPSR/Cas9系统的功能将会进一步完善,这些阻碍与挑战也将被不断去除和攻破. ...
In vivo base editing restores sensory transduction and transiently improves auditory function in a mouse model of recessive deafness
1
2020
... 尽管研究人员针对CRISPR系统的PAM序列依赖性、编辑脱靶率等问题给出了一些解决方案如发掘Cas9同源蛋白、通过工程化改造Cas9或gRNA从而拓宽PAM序列识别范围和提高中靶率等,这些问题依然困扰着CRISPR系统在更多生物中进行快速、高效、精准的基因编辑和调控,尤其是在最终将CRISPR系统作为基因疗法使用时,其风险控制、脱靶编辑等问题一直都被科研人员重视.为了减少脱靶效应、提高基因编辑的精确性和可控性,Liu等[125]将一种Cas9突变体iCas9与雌激素受体的激素结合结构域(hormone-binding domain of the estrogen receptor,ERT2)融合后,可使iCas9的核酸酶活性获得4-羟基三苯甲胺(4-hydroxytamoxifen,4-HT)诱导性,从而快速和可逆地控制该系统在基因组编辑功能上的作用.2017年,Shin 等[126]发现了抗CRISPR 蛋白AcrIIA4对CRISPR/ Cas9系统的抑制作用,使人为控制CRISPR系统作为疗法使用时的风险成为可能.2021年,Jain等[127]将AcrIIA4与二氢叶酸还原酶(dihydrofolate reductase,DHRF)融合后使其仅在甲氧苄啶(trimethoprim,TMP)存在时才能抑制Cas9和DNA的结合能力,从而实现对Cas9编辑和dCas9基因组扰动的化学调控,提高了CRISPR/Cas9系统的特异性和生物安全性.2022年,北京大学胡家志课题组[128]揭示了CRISPR/Cas9系统在基因编辑过程中造成染色体易位的分子机制,随后将优化过的人源TREX2蛋白与Cas9相偶联生成Cas9TX,其能抑制基因编辑过程中的染色体易位、大片段缺失等染色体结构异常的产生,使CRISPR/Cas9的基因编辑安全性大大提高.这些研究最终将服务于基于CRISPR/Cas9工具箱的单基因突变遗传病的基因疗法,并为全人类带来福祉.目前,碱基编辑已成功在其他哺乳动物中用于疾病治疗,并已被证明具有治疗人类单基因突变遗传病的潜力.2020年,Yeh等[129]通过双AAV递送CBEMax系统成功治愈小鼠Tmc1基因隐性突变导致的耳聋,使其能够重新对中等程度的声音做出反应.Wang等[130]通过对β-地中海贫血和镰刀状贫血患者造血干细胞中的BCL11A增强子位点进行编辑或修复发生突变的HBB基因,以及进行自体造血干细胞移植使其在体内分化产生正常的红细胞,从而治疗由β-globin珠蛋白突变引发的遗传病.CRISPR作为近年来发展起来的新兴技术,在为生命科学带来一系列革命性应用技术的同时,还存在一些问题和困难,随着越来越多热忱的研究者的加入,CRIPSR/Cas9系统的功能将会进一步完善,这些阻碍与挑战也将被不断去除和攻破. ...
Reactivation of γ-globin expression through Cas9 or base editor to treat β-hemoglobinopathies
1
2020
... 尽管研究人员针对CRISPR系统的PAM序列依赖性、编辑脱靶率等问题给出了一些解决方案如发掘Cas9同源蛋白、通过工程化改造Cas9或gRNA从而拓宽PAM序列识别范围和提高中靶率等,这些问题依然困扰着CRISPR系统在更多生物中进行快速、高效、精准的基因编辑和调控,尤其是在最终将CRISPR系统作为基因疗法使用时,其风险控制、脱靶编辑等问题一直都被科研人员重视.为了减少脱靶效应、提高基因编辑的精确性和可控性,Liu等[125]将一种Cas9突变体iCas9与雌激素受体的激素结合结构域(hormone-binding domain of the estrogen receptor,ERT2)融合后,可使iCas9的核酸酶活性获得4-羟基三苯甲胺(4-hydroxytamoxifen,4-HT)诱导性,从而快速和可逆地控制该系统在基因组编辑功能上的作用.2017年,Shin 等[126]发现了抗CRISPR 蛋白AcrIIA4对CRISPR/ Cas9系统的抑制作用,使人为控制CRISPR系统作为疗法使用时的风险成为可能.2021年,Jain等[127]将AcrIIA4与二氢叶酸还原酶(dihydrofolate reductase,DHRF)融合后使其仅在甲氧苄啶(trimethoprim,TMP)存在时才能抑制Cas9和DNA的结合能力,从而实现对Cas9编辑和dCas9基因组扰动的化学调控,提高了CRISPR/Cas9系统的特异性和生物安全性.2022年,北京大学胡家志课题组[128]揭示了CRISPR/Cas9系统在基因编辑过程中造成染色体易位的分子机制,随后将优化过的人源TREX2蛋白与Cas9相偶联生成Cas9TX,其能抑制基因编辑过程中的染色体易位、大片段缺失等染色体结构异常的产生,使CRISPR/Cas9的基因编辑安全性大大提高.这些研究最终将服务于基于CRISPR/Cas9工具箱的单基因突变遗传病的基因疗法,并为全人类带来福祉.目前,碱基编辑已成功在其他哺乳动物中用于疾病治疗,并已被证明具有治疗人类单基因突变遗传病的潜力.2020年,Yeh等[129]通过双AAV递送CBEMax系统成功治愈小鼠Tmc1基因隐性突变导致的耳聋,使其能够重新对中等程度的声音做出反应.Wang等[130]通过对β-地中海贫血和镰刀状贫血患者造血干细胞中的BCL11A增强子位点进行编辑或修复发生突变的HBB基因,以及进行自体造血干细胞移植使其在体内分化产生正常的红细胞,从而治疗由β-globin珠蛋白突变引发的遗传病.CRISPR作为近年来发展起来的新兴技术,在为生命科学带来一系列革命性应用技术的同时,还存在一些问题和困难,随着越来越多热忱的研究者的加入,CRIPSR/Cas9系统的功能将会进一步完善,这些阻碍与挑战也将被不断去除和攻破. ...



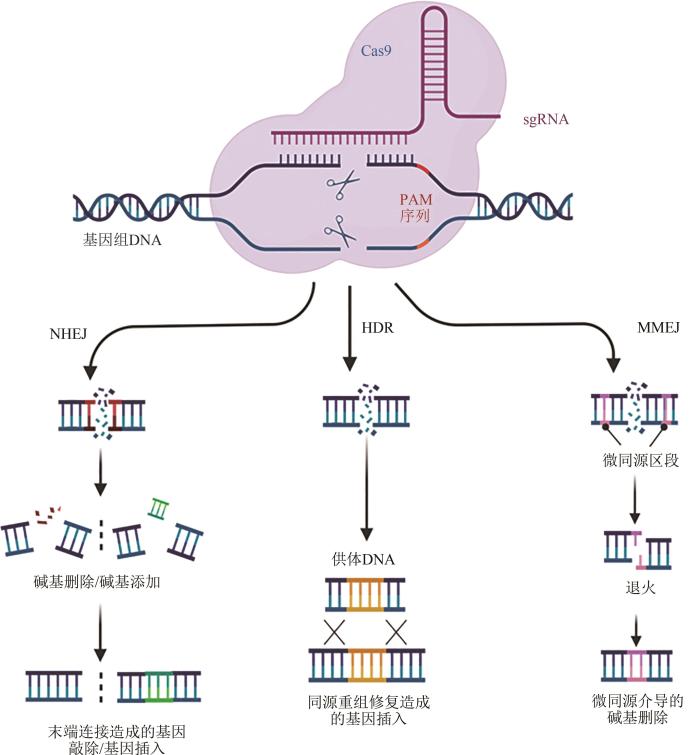
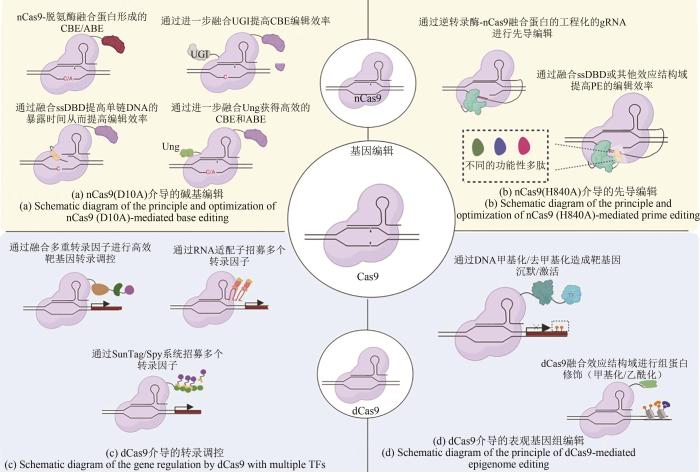
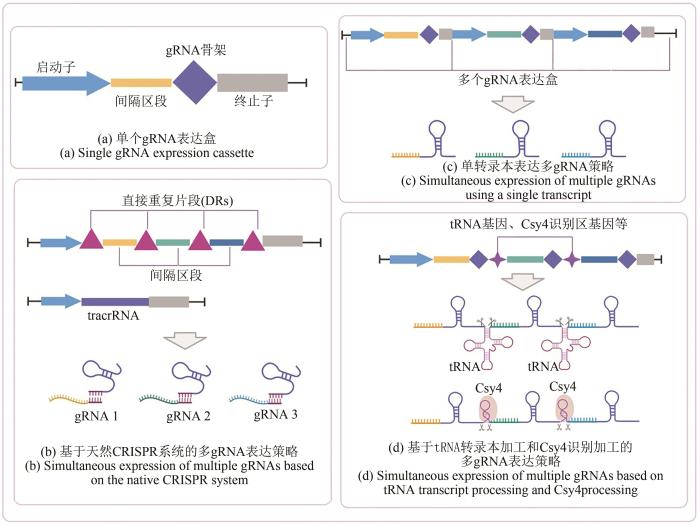


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}