|
||
|
Exploration of gene functions and library construction for engineering strains from a synthetic biology perspective
Synthetic Biology Journal
2025, 6 (3):
685-700.
DOI: 10.12211/2096-8280.2024-079
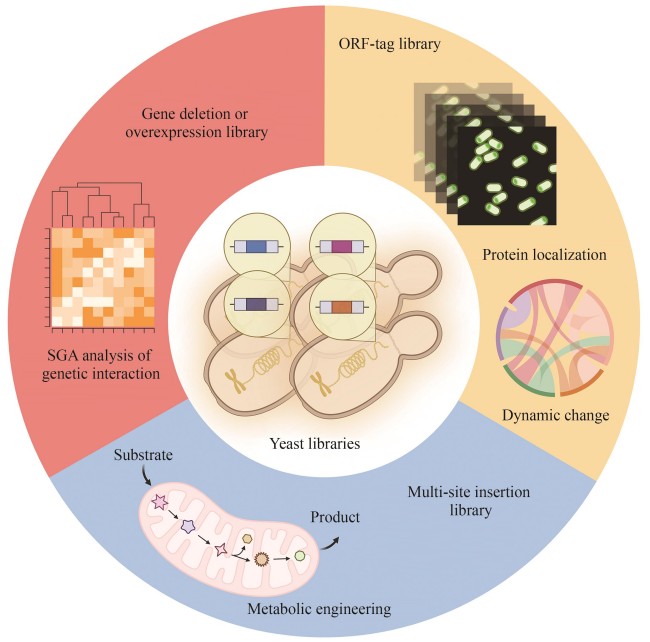
Synthetic biology, as a discipline that designs, constructs, and modifies biological systems to achieve specific functions, is widely applied in biomanufacturing, the biodegradation of environmental pollutants, and drug synthesis. Systematic exploration of gene functions and construction of libraries for engineered strains are driving forces of the development of synthetic biology. These libraries serve as foundational tools for understanding complex biological processes and engineering microorganisms for potential applications. This review focuses on the construction methods and application prospects of various yeast libraries in synthetic biology. With the rapid advancement of genome sequencing and high-throughput screening technologies, microbial libraries, such as those of Saccharomycescerevisiae and Schizosaccharomycespombe, play a pivotal role in systematic research. Yeast libraries, including gene knockout libraries, overexpression libraries, and transposon insertion libraries, provide valuable tools for optimizing gene combinations and designing metabolic pathways, thus promoting applications in metabolic engineering and synthetic biology. These libraries facilitate the development of robust industrial strains, driving improvements in biofuel production, chemical synthesis, and other biotechnological processes. In the environmental field, the screening of modified genes generates strains with pollutant degradation capabilities, contributing to ecological restoration. In drug synthesis, these libraries aid in constructing strains for the efficient production of pharmaceutical compounds, advancing the development of biopharmaceuticals. Despite these successes, there remain challenges in library construction and application, such as the high cost of library generation, difficulty in precise genome editing, and limitation in screening efficiency. In the future, advances in automation, digitization, and novel screening technologies are expected to overcome these barriers, facilitating the rapid construction and efficient screening of yeast libraries. No doubt, synthetic biology holds immense promise, with improvements in library construction and screening processes expected to accelerate the development of sustainable solutions in industrial production, environmental protection, and healthcare, thereby driving innovations in biotechnology.
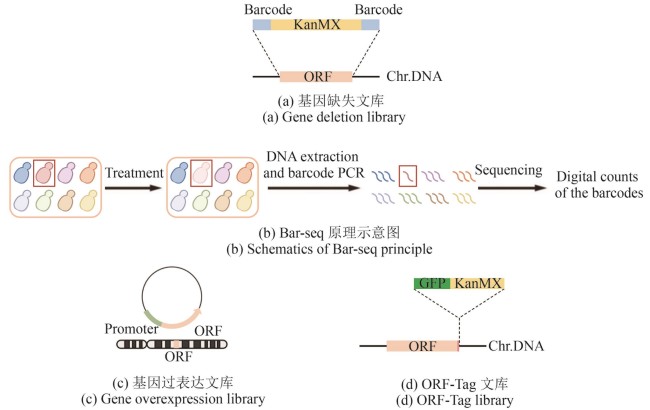
Fig. 1
Schematics for constructing classical libraries (created by biorender)
[(a) In the gene deletion library, the ORF (orange fragment) of target gene is replaced by the KanMX (kanamycin resistance selection marker, yellow fragment), flanked by unique barcodes (blue fragments). Chr.DNA: chromosomal DNA.(b) Schematic diagram of the Bar-seq principle. Under specific treatment conditions, the target strain (red) shows reduced growth (light pink), which is reflected by a lower read count of its corresponding barcode during sequencing (fewer red lines compared to others).(c) In the gene overexpression library, the ORF (orange fragment) of target gene is not only expressed from its endogenous locus but is also additionally expressed from a plasmid, driven by a constitutive promoter (light green fragment).(d) In the ORF-Tag library, the C-terminus of the target gene ORF (orange fragment), with its stop codon removed, is fused to the fluorescent protein GFP (dark green fragment) and the selection marker KanMX (yellow fragment).]
Extracts from the Article
酵母单基因缺失文库[16-18]中,每个菌株中被删除的目的基因ORF都通过同源重组法被替换成了一个卡那霉素抗性筛选标签(kanamycin modular expression,KanMX),并且两侧有一对长度为20 bp的序列各不相同的标签,被称为“分子条形码”(barcode)[图1(a)]。这些条形码能够准确、唯一地识别每个缺失突变体,这使高通量的定量平行分析成为可能,为全面、快速、经济、便捷地筛查各个生物学方面的基因功能奠定了基础[19-20]。每个基因缺失菌株携带的独特的分子条形码大大简化了在同一实验条件下对多个突变体进行平行分析的过程。例如为了筛选对生长重要的基因,通过一次实验,将所有单基因缺失菌株混合培养,并在生长过程中定期采集样本。从这些样本中提取基因组DNA,通过扩增与每个单基因缺失菌株相关联的分子条形码。即可根据该基因的丰度判断此基因对生长的重要性。对生长越重要,相应缺失菌株的分子条形码在培养过程中就减少得越快。因此,所有目的基因都可以在一次实验中被识别出来,并按照它们对适应度的相对贡献进行排序[图1(b)]。
全基因组过表达文库通过将一系列携带额外目的基因拷贝的质粒载体与强启动子相结合[图1(c)],强制性提高每个基因的表达水平,来研究基因在细胞生长、代谢通路及应激反应中的作用。质粒上的启动子通常是诱导性的,如gal1启动子[21]或nmt1启动子[22],在半乳糖碳源或去除硫胺素的条件下可以诱导有该启动子的基因强表达,这使得研究人员能够在特定条件下调控基因表达从而观察表型变化。
ORF-Tag文库通过在基因的C端或N端插入荧光标签标记目标蛋白。它有两种模式:一种是在各个基因的原生位点进行修饰[图1(d)];另一种则是在一个异位位点上额外表达融合蛋白。前者保证了目标蛋白的表达水平和模式受到的干扰最小;后者往往涉及基因过表达,一些在正常状态下不表达的基因产物也能够通过诱导表达在细胞内进行定位。但与此同时,融合蛋白的非生理表达可能导致其定位错误,对于一些剂量敏感型蛋白还可能造成细胞内分子成分之间平衡的扰动或紊乱,影响细胞正常生理活动。
Other Images/Table from this Article
|

{kind=link}