Synthetic Biology Journal ›› 2023, Vol. 4 ›› Issue (3): 507-523.DOI: 10.12211/2096-8280.2022-079
• Invited Review • Previous Articles Next Articles
Prediction of protein complex structure: methods and progress
HUANG He1, WU Tong1, WANG Wenda1, LI Jiashan1, SUN Daiwen1, YE Qiwei2, GONG Xinqi1,2
- 1.Institute for Mathematical Sciences,Renmin University of China,Beijing 100872,China
2.Beijing Academy of Artificial Intelligence,Beijing 100084,China
-
Received:2022-12-31Revised:2023-03-20Online:2023-07-05Published:2023-06-30 -
Contact:GONG Xinqi
蛋白质复合物结构预测:方法与进展
黄鹤1, 吴桐1, 王闻达1, 李佳珊1, 孙黛雯1, 叶启威2, 龚新奇1,2
- 1.中国人民大学数学科学研究院,北京 100872
2.北京智源人工智能研究院,北京 100084
-
通讯作者:龚新奇 -
作者简介:黄鹤 (1995—),男,博士研究生。研究方向为蛋白质复合物结构预测算法等。 E-mail:hehuang@ruc.edu.cn龚新奇 (1978—),男,教授,博士生导师。龚新奇 课题组(数学智能应用实验室)在生物信息学方向构建数学模型、开发计算方法和并应用于研究多聚体超大蛋白质相互作用复合物的结构、网络和动力学等;在机器学习方向利用深度学习和大数据方法设计新的算法框架解决生物大分子和医疗图像的计算。 E-mail:xinqigong@ruc.edu.cn
CLC Number:
Cite this article
HUANG He, WU Tong, WANG Wenda, LI Jiashan, SUN Daiwen, YE Qiwei, GONG Xinqi. Prediction of protein complex structure: methods and progress[J]. Synthetic Biology Journal, 2023, 4(3): 507-523.
黄鹤, 吴桐, 王闻达, 李佳珊, 孙黛雯, 叶启威, 龚新奇. 蛋白质复合物结构预测:方法与进展[J]. 合成生物学, 2023, 4(3): 507-523.
share this article
Add to citation manager EndNote|Ris|BibTeX
URL: https://synbioj.cip.com.cn/EN/10.12211/2096-8280.2022-079

Fig. 1 AI-based methods for predicting protein structure
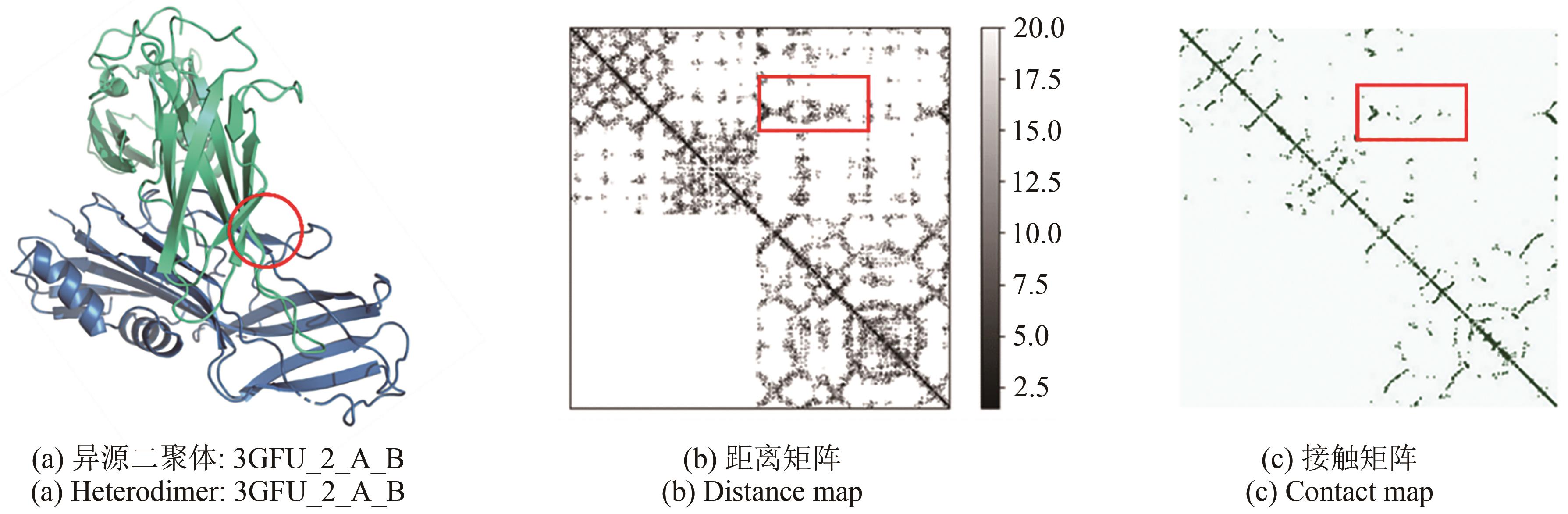
Fig. 2 Interactions between residues with the inter-chains of proteins
| 方法 | 输入特征 | 网络架构 | 任务 | ||||
|---|---|---|---|---|---|---|---|
| 共进化 | 单体距离图 | 单体结构 | 蛋白语言模型 | 残差网络 | 同源 | 异源 | |
| ComplexContact[ | √ | √ | √ | ||||
| Glinter[ | √ | √ | √ | √ + 图学习 | √ | √ | |
| DeepHomo[ | √ | √ | √ | √ | |||
| DeepHomo2[ | √ | √ | √ | √ | √ | ||
| DRcon[ | √ | √ | √ + 空洞卷积 | √ | |||
| DeepInteract[ | √ | 几何深度学习 | √ | ||||
| CDPred[ | √ | √ | √ | √ + 注意力机制 | √ | √ | |
| PDII[ | √ | 图像修复 | √ | √ | |||
| PGT[ | √ | √ | √ + 图注意力+三角更新 | √ | |||
Table 1 Overview of methods for predicting interactions between the inter-chains of proteins[48,61-67,69]
| 方法 | 输入特征 | 网络架构 | 任务 | ||||
|---|---|---|---|---|---|---|---|
| 共进化 | 单体距离图 | 单体结构 | 蛋白语言模型 | 残差网络 | 同源 | 异源 | |
| ComplexContact[ | √ | √ | √ | ||||
| Glinter[ | √ | √ | √ | √ + 图学习 | √ | √ | |
| DeepHomo[ | √ | √ | √ | √ | |||
| DeepHomo2[ | √ | √ | √ | √ | √ | ||
| DRcon[ | √ | √ | √ + 空洞卷积 | √ | |||
| DeepInteract[ | √ | 几何深度学习 | √ | ||||
| CDPred[ | √ | √ | √ | √ + 注意力机制 | √ | √ | |
| PDII[ | √ | 图像修复 | √ | √ | |||
| PGT[ | √ | √ | √ + 图注意力+三角更新 | √ | |||
| 35 | RIVES A, MEIER J, SERCU T, et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences[J]. Proceedings of the National Academy of Sciences of the United States of America, 2021, 118(15): e2016239118 |
| 36 | RAO R, LIU J, VERKUIL R, et al. MSA Transformer[EB/OL]. 2021[2022-12-10]. . |
| 37 | FANG X, WANG F, LIU L, et al. HelixFold-single: MSA-free protein structure prediction by using protein language model as an alternative[EB/OL]. arXiv, 2022: 2207.13921[2022-12-10]. . |
| 38 | WU R D, DING F, WANG R, et al. High-resolution de novo structure prediction from primary sequence[EB/OL]. bioRxiv, 2022[2022-12-10] . |
| 39 | WANG W K, PENG Z L, YANG J Y. Single-sequence protein structure prediction using supervised transformer protein language models[J]. Nature Computational Science, 2022, 2(12): 804-814. |
| 40 | CHOWDHURY R, BOUATTA N, BISWAS S, et al. Single-sequence protein structure prediction using a language model and deep learning[J]. Nature Biotechnology, 2022, 40(11): 1617-1623. |
| 41 | HSU C, VERKUIL R, LIU J, et al. Learning inverse folding from millions of predicted structures[EB/OL]. bioRxiv, 2022[2022-12-10]. . |
| 42 | ELNAGGAR A, HEINZINGER M, DALLAGO C, et al. ProtTrans: towards cracking the language of life's code through self-supervised learning[EB/OL]//IEEE Trans Pattern Analysis & Machine Intelligence, 2021, 4[2022-12-10]. . |
| 43 | BRANDES N, GOLDMAN G, WANG C H, et al. Genome-wide prediction of disease variants with a deep protein language model[EB/OL]. bioRxiv, 2022[2022-12-10]. . |
| 44 | GUO Y Z, WU J X, MA H H, et al. Self-supervised pre-training for protein embeddings using tertiary structures[J]. Proceedings of the AAAI Conference on Artificial Intelligence, 2022, 36(6): 6801-6809. |
| 45 | ZHANG Z, XU M, JAMASB A, et al. Protein representation learning by geometric structure pretraining[EB/OL]. arXiv, 2022: 2203.06125[2023-02-01]. . |
| 46 | ZHOU G, GAO Z, DING Q, et al. Uni-Mol: a universal 3D molecular representation learning framework[EB/OL][2022-12-10]. . |
| 47 | HOPF T A, SCHÄRFE C P I, RODRIGUES J P G L M, et al. Sequence co-evolution gives 3D contacts and structures of protein complexes[J]. eLife, 2014, 3: e03430. |
| 48 | ZENG H, WANG S, ZHOU T M, et al. ComplexContact: a web server for inter-protein contact prediction using deep learning[J]. Nucleic Acids Research, 2018, 46(W1): W432-W437. |
| 49 | CHEN B, XIE Z W, XU J B, et al. Improve the protein complex prediction with protein language models[EB/OL]. bioRxiv, 2022[2022-12-10]. . |
| 50 | ZHENG W, LI Y, ZHANG C X, et al. Protein structure prediction using deep learning distance and hydrogen-bonding restraints in CASP14[J]. Proteins: Structure, Function, and Bioinformatics, 2021, 89(12): 1734-1751. |
| 51 | EVANS R, O'NEILL M, PRITZEL A, et al. Protein complex prediction with AlphaFold-Multimer[EB/OL]. bioRxiv, 2022[2022-12-10]. . |
| 52 | SUN D W, LIU S J, GONG X Q. Review of multimer protein-protein interaction complex topology and structure prediction[J]. Chinese Physics B, 2020, 29(10): 57-66. |
| 53 | YANG Y X, GONG X Q. A new probability method to understand protein-protein interface formation mechanism at amino acid level[J]. Journal of Theoretical Biology, 2018, 436: 18-25. |
| 54 | YANG Y X, WANG W, LOU Y, et al. Geometric and amino acid type determinants for protein-protein interaction interfaces[J]. Quantitative Biology, 2018, 6(2): 163-174. |
| 55 | WANG W, YANG Y X, YIN J X, et al. Different protein-protein interface patterns predicted by different machine learning methods[J]. Scientific Reports, 2017, 7: 16023. |
| 56 | ZHAO Z N, GONG X Q. Trimer protein-protein complex interface interacting residue pairs prediction using deep learning approach[C]//Proceedings of the 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics. September 7-10, 2019, Niagara Falls, NY, USA. New York: ACM, 2019: 580-585. |
| 57 | SUN D W, GONG X Q. Tetramer protein complex interface residue pairs prediction with LSTM combined with graph representations[J]. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics, 2020, 1868(11): 140504. |
| 58 | LIU J L, GONG X Q. Attention mechanism enhanced LSTM with residual architecture and its application for protein-protein interaction residue pairs prediction[J]. BMC Bioinformatics, 2019, 20(1): 609. |
| 59 | ZHAO Z N, GONG X Q. Protein-protein interaction interface residue pair prediction based on deep learning architecture[J]. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 2019, 16(5): 1753-1759. |
| 60 | LYU Y F, HE R N, HU J J, et al. Prediction of the tetramer protein complex interaction based on CNN and SVM[J]. Frontiers in Genetics, 2023, 14: 1076904. |
| 61 | XIE Z W, XU J B. Deep graph learning of inter-protein contacts[J]. Bioinformatics, 2022, 38(4): 947-953. |
| 62 | MOREHEAD A, CHEN C, CHENG J. Geometric transformers for protein interface contact prediction[EB/OL]. arXiv, 2021: 2110.02423[2022-12-10]. . |
| 63 | GUO Z, LIU J, SKOLNICK J, et al. Prediction of inter-chain distance maps of protein complexes with 2D attention-based deep neural networks[J]. Nature Communications, 2022, 13: 6963. |
| 64 | YAN Y M, HUANG S Y. Accurate prediction of inter-protein residue-residue contacts for homo-oligomeric protein complexes[J]. Briefings in Bioinformatics, 2021, 22(5): bbab038. |
| 65 | LIN P C, YAN Y M, HUANG S Y. DeepHomo2.0: improved protein-protein contact prediction of homodimers by transformer-enhanced deep learning[J]. Briefings in Bioinformatics, 2022, 24(1), bbac499. |
| 66 | ROY R S, QUADIR F, SOLTANIKAZEMI E, et al. A deep dilated convolutional residual network for predicting interchain contacts of protein homodimers[J]. Bioinformatics, 2022, 38(7): 1904-1910. |
| 67 | WU T, HUANG H, LI J S, et al. Inter-chain contact map prediction for protein complex based on graph attention network and triangular multiplication update[C]//2022 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). December 6-8, 2022, Las Vegas, NV, USA. IEEE, 2023: 2143-2148. |
| 68 | GAO M, AN D N, PARKS J M, et al. AF2Complex predicts direct physical interactions in multimeric proteins with deep learning[J]. Nature Communications, 2022, 13: 1744. |
| 69 | HUANG H, ZENG C S, GONG X Q. Inter-protein contact map generated only from intra-monomer by image inpainting[C]//2021 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). December 9-12, 2021. Houston, TX, USA. IEEE, 2021: 131-136. |
| 70 | CHEN Y, WANG W, LIU J L, et al. Protein interface complementarity and gene duplication improve link prediction of protein-protein interaction network[J]. Frontiers in Genetics, 2020, 11: 291. |
| 1 | LENSINK M F, BRYSBAERT G, MAURI T, et al. Prediction of protein assemblies, the next frontier: the CASP14-CAPRI experiment[J]. Proteins: Structure, Function and Bioinformatics, 2021, 89(12): 1800-1823. |
| 2 | JUMPER J, EVANS R, PRITZEL A, et al. Highly accurate protein structure prediction with AlphaFold[J]. Nature, 2021, 596(7873): 583-589. |
| 3 | BADAL V D, KUNDROTAS P J, VAKSER I A. Text mining for protein docking[J]. PLoS Computational Biology, 2015, 11(12): e1004630. |
| 4 | MARKOWETZ F, SPANG R. Inferring cellular networks-a review[J]. BMC Bioinformatics, 2007, 8(): S5. |
| 5 | MORCOS F, PAGNANI A, LUNT B, et al. Direct-coupling analysis of residue coevolution captures native contacts across many protein families[J]. Proceedings of the National Academy of Sciences of the United States of America, 2011, 108(49): E1293-E1301. |
| 6 | BALAKRISHNAN S, KAMISETTY H, CARBONELL J G, et al. Learning generative models for protein fold families[J]. Proteins: Structure, Function, and Bioinformatics, 2011, 79(4): 1061-1078. |
| 7 | HUANG H, GONG X Q. A review of protein inter-residue distance prediction[J]. Current Bioinformatics, 2020, 15(8): 821-830. |
| 8 | 张海仓, 高玉娟, 邓明华, 等. 蛋白质中残基远程相互作用预测算法研究综述[J]. 计算机研究与发展, 2017, 54(1): 1-19. |
| ZHANG H C, GAO Y J, DENG M H, et al. A survey on algorithms for protein contact prediction[J]. Journal of Computer Research and Development, 2017, 54(1): 1-19. | |
| 9 | 於东军, 李阳. 蛋白质残基接触图预测[J]. 南京理工大学学报, 2019, 43(1): 1-12. |
| YU D J, LI Y. Protein residue-residue contact map prediction[J]. Journal of Nanjing University of Science and Technology, 2019, 43(1): 1-12. | |
| 10 | WEIGT M, WHITE R A, SZURMANT H, et al. Identification of direct residue contacts in protein-protein interaction by message passing[J]. Proceedings of the National Academy of Sciences of the United States of America, 2009, 106(1): 67-72. |
| 11 | JONES D T, BUCHAN D W A, COZZETTO D, et al. PSICOV: precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments[J]. Bioinformatics, 2012, 28(2): 184-190. |
| 12 | EKEBERG M, LÖVKVIST C, LAN Y H, et al. Improved contact prediction in proteins: using pseudolikelihoods to infer Potts models[J]. Physical Review E-Statistical, Nonlinear, and Soft Matter Physics, 2013, 87(1): 012707. |
| 13 | ZHANG H C, ZHANG Q, JU F S, et al. Predicting protein inter-residue contacts using composite likelihood maximization and deep learning[J]. BMC Bioinformatics, 2019, 20(1): 537. |
| 14 | KAMISETTY H, OVCHINNIKOV S, BAKER D. Assessing the utility of coevolution-based residue-residue contact predictions in a sequence- and structure-rich era[J]. Proceedings of the National Academy of Sciences of the United States of America, 2013, 110(39): 15674-15679. |
| 15 | OVCHINNIKOV S, KAMISETTY H, BAKER D. Robust and accurate prediction of residue-residue interactions across protein interfaces using evolutionary information[J]. eLife, 2014, 3: e02030. |
| 16 | WANG S, SUN S Q, LI Z, et al. Accurate de novo prediction of protein contact map by ultra-deep learning model[J]. PLoS Computational Biology, 2017, 13(1): e1005324. |
| 17 | YANG J Y, ANISHCHENKO I, PARK H, et al. Improved protein structure prediction using predicted interresidue orientations[J]. Proceedings of the National Academy of Sciences, 2020, 117(3): 1496-1503. |
| 18 | SEEMAYER S, GRUBER M, SÖDING J. CCMpred—fast and precise prediction of protein residue-residue contacts from correlated mutations[J]. Bioinformatics, 2014, 30(21): 3128-3130. |
| 19 | HE K M, ZHANG X Y, REN S Q, et al. Deep residual learning for image recognition[C/OL]// 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), VegasLas, NV, USA, 2016, 770-778 [2022-12-10]. . |
| 20 | SENIOR A W, EVANS R, JUMPER J, et al. Improved protein structure prediction using potentials from deep learning[J]. Nature, 2020, 577(7792): 706-710. |
| 21 | MAO W, DING W, GONG H. AmoebaContact and GDFold: a new pipeline for rapid prediction of protein structures[EB/OL]. arXiv, 2019: 1905.11640[2022-12-10]. . |
| 22 | JU F S, ZHU J W, SHAO B, et al. CopulaNet: learning residue co-evolution directly from multiple sequence alignment for protein structure prediction[J]. Nature Communications, 2021, 12: 2535. |
| 23 | ZHENG W, ZHANG C, LI Y, et al. Folding non-homologous proteins by coupling deep-learning contact maps with I-TASSER assembly simulations[J]. Cell Reports Methods, 2021, 1(3): 100014. |
| 71 | WEN J, CHAN R H F, YAU S C, et al. K-mer natural vector and its application to the phylogenetic analysis of genetic sequences[J]. Gene, 2014, 546(1): 25-34. |
| 72 | ZHAO N, ZHUO M J, TIAN K, et al. Protein-protein interaction and non-interaction predictions using gene sequence natural vector[J]. Communications Biology, 2022, 5: 652. |
| 73 | GE F Q, PENG C X, CUI X Y, et al. Inter-domain distance prediction based on deep learning for domain assembly[J]. Briefings in Bioinformatics, 2023: bbad100. |
| 74 | SEN N, MADHUSUDHAN M S. A structural database of chain-chain and domain-domain interfaces of proteins[J]. Protein Science, 2022, 31(9): e4406. |
| 75 | 谢腾宇, 周晓根, 胡俊, 等. 基于接触图残基对距离约束的蛋白质结构预测算法[J]. 计算机科学, 2020, 47(1): 59-65. |
| XIE T Y, ZHOU X G, HU J, et al. Contact map-based residue-pair distances restrained protein structure prediction algorithm[J]. Computer Science, 2020, 47(1): 59-65. | |
| 76 | QUADIR F, ROY R S, SOLTANIKAZEMI E, et al. Deep Complex: a web server of predicting protein complex structures by deep learning inter-chain contact prediction and distance-based modelling[J]. Frontiers in Molecular Biosciences, 2021, 8: 716973. |
| 77 | SOLTANIKAZEMI E, ROY R S, QUADIR F, et al. DRL Complex: reconstruction of protein quaternary structures using deep reinforcement learning[EB/OL]. arXiv, 2022: 2205.13594[2022-12-10]. . |
| 78 | BAEK M, ANISHCHENKO I, PARK H, et al. Protein oligomer modeling guided by predicted interchain contacts in CASP14[J]. Proteins: Structure, Function, and Bioinformatics, 2021, 89(12): 1824-1833. |
| 79 | DE VRIES S J, VAN DIJK M, BONVIN A M J J. The HADDOCK web server for data-driven biomolecular docking[J]. Nature Protocols, 2010, 5(5): 883-897. |
| 80 | ESQUIVEL-RODRÍGUEZ J, YANG Y D, KIHARA D. Multi-LZerD: multiple protein docking for asymmetric complexes[J]. Proteins: Structure, Function, and Bioinformatics, 2012, 80(7): 1818-1833. |
| 81 | RITCHIE D W, GRUDININ S. Spherical polar Fourier assembly of protein complexes with arbitrary point group symmetry[J]. Journal of Applied Crystallography, 2016, 49(1): 158-167. |
| 24 | MORTUZA S M, ZHENG W, ZHANG C X, et al. Improving fragment-based ab initio protein structure assembly using low-accuracy contact-map predictions[J]. Nature Communications, 2021, 12: 5011. |
| 25 | DING W Z, GONG H P. Predicting the real-valued inter-residue distances for proteins[J]. Advanced Science, 2020, 7(19): 2001314. |
| 26 | WU T Q, GUO Z Y, HOU J, et al. DeepDist: real-value inter-residue distance prediction with deep residual convolutional network[J]. BMC Bioinformatics, 2021, 22(1): 30. |
| 27 | CHAUDHURY S, LYSKOV S, GRAY J J. PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta[J]. Bioinformatics, 2010, 26(5): 689-691. |
| 28 | BRUNGER A T. Version 1.2 of the crystallography and NMR system[J]. Nature Protocols, 2007, 2(11): 2728-2733. |
| 29 | VASWANI A, SHAZEER N, PARMAR N, et al. Attention is all you need[EB/OL]. arXiv, 2017: 1706.03762[2022-12-10]. . |
| 30 | ALQURAISHI M. End-to-end differentiable learning of protein structure[J]. Cell Systems, 2019, 8(4): 292-301.e3. |
| 31 | VARADI M, ANYANGO S, DESHPANDE M, et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models[J]. Nucleic Acids Research, 2022, 50(D1): D439-D444. |
| 32 | LIN Z, AKIN H, RAO R, et al. Evolutionary-scale prediction of atomic-level protein structure with a language model[J]. Science, 2023, 379(6637): 1123-1130. |
| 33 | DEVLIN J, CHANG M, LEE K, et al. BERT: Pre-training of deep bidirectional transformers for language understanding[EB/OL]. arXiv, 2018: 1810.04805[2022-12-10]. . |
| 34 | RAO R, MEIER J, SERCU T, et al. Transformer protein language models are unsupervised structure learners[EB/OL].bioRxiv, 2020[2022-12-10]. . |
| 82 | YAN Y M, TAO H Y, HUANG S Y. HSYMDOCK: a docking web server for predicting the structure of protein homo-oligomers with C n or D n symmetry[J]. Nucleic Acids Research, 2018, 46(W1): W423-W431. |
| 83 | PARK T, WOO H, YANG J, et al. Protein oligomer structure prediction using GALAXY in CASP14[J]. Proteins: Structure, Function, and Bioinformatics, 2021, 89(12): 1844-1851. |
| 84 | ROY BURMAN S S, YOVANNO R A, GRAY J J. Flexible backbone assembly and refinement of symmetrical homomeric complexes[J]. Structure, 2019, 27(6): 1041-1051.e8. |
| 85 | PIERCE B, TONG W W, WENG Z P. M-ZDOCK: a grid-based approach for C n symmetric multimer docking[J]. Bioinformatics, 2005, 21(8): 1472-1478. |
| 86 | PIERCE B G, WIEHE K, HWANG H, et al. ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers[J]. Bioinformatics, 2014, 30(12): 1771-1773. |
| 87 | JIMÉNEZ-GARCÍA B, PONS C, FERNÁNDEZ-RECIO J. pyDockWEB: a web server for rigid-body protein-protein docking using electrostatics and desolvation scoring[J]. Bioinformatics, 2013, 29(13): 1698-1699. |
| 88 | KOZAKOV D, HALL D R, XIA B, et al. The ClusPro web server for protein-protein docking[J]. Nature Protocols, 2017, 12(2): 255-278. |
| 89 | XU X J, QIU L M, YAN C F, et al. Performance of MDockPP in CAPRI rounds 28-29 and 31-35 including the prediction of water-mediated interactions[J]. Proteins: Structure, Function, and Bioinformatics, 2017, 85(3): 424-434. |
| 90 | KONG R, LIU R R, XU X M, et al. Template-based modeling and ab-initio docking using CoDock in CAPRI[J]. Proteins: Structure, Function, and Bioinformatics, 2020, 88(8): 1100-1109. |
| 91 | MARZE N A, ROY BURMAN S S, SHEFFLER W, et al. Efficient flexible backbone protein-protein docking for challenging targets[J]. Bioinformatics, 2018, 34(20): 3461-3469. |
| 92 | TORCHALA M, MOAL I H, CHALEIL R A G, et al. SwarmDock: a server for flexible protein-protein docking[J]. Bioinformatics, 2013, 29(6): 807-809. |
| 93 | QUIGNOT C, REY J, YU J C, et al. InterEvDock2: an expanded server for protein docking using evolutionary and biological information from homology models and multimeric inputs[J]. Nucleic Acids Research, 2018, 46(W1): W408-W416. |
| 94 | BAEK M, PARK T, HEO L, et al. GalaxyHomomer: a web server for protein homo-oligomer structure prediction from a monomer sequence or structure[J]. Nucleic Acids Research, 2017, 45(W1): W320-W324. |
| 95 | GANEA O E, HUANG X, ZURICH E, et al. Independent SE(3)- equivariant models for end-to-end rigid protein docking[EB/OL]. arXiv, 2022: 2111.07786[2022-12-10]. . |
| 96 | VREVEN T, MOAL I H, VANGONE A, et al. Updates to the integrated protein-protein interaction benchmarks: Docking benchmark version 5 and affinity benchmark version 2[J]. Journal of Molecular Biology, 2015, 427(19): 3031-3041. |
| 97 | YU J C, GUEROIS R. PPI4DOCK: large scale assessment of the use of homology models in free docking over more than 1000 realistic targets[J]. Bioinformatics, 2016, 32(24): 3760-3767. |
| 98 | YAN Y M, HUANG S Y. A non-redundant benchmark for symmetric protein docking[J]. Big Data Mining and Analytics, 2019, 2(2): 92-99. |
| 99 | MIRDITA M, SCHÜTZE K, MORIWAKI Y, et al. ColabFold: making protein folding accessible to all[J]. Nature Methods, 2022, 19(6): 679-682. |
| 100 | BRYANT P, POZZATI G, ELOFSSON A. Improved prediction of protein-protein interactions using AlphaFold2[J]. Nature Communications, 2022, 13: 1265. |
| 101 | ZHU W, SHENOY A, KUNDROTAS P, et al. Evaluation of AlphaFold-Multimer prediction on multi-chain protein complexes[EB/OL]. 2022[2022-12-29]. . |
| 102 | TSUCHIYA Y, YAMAMORI Y, TOMII K. Protein-protein interaction prediction methods: from docking-based to AI-based approaches[J].Biophysical Reviews, 2022, 14(6): 1341-1348. |
| 103 | LI Z Y, LIU X Y, CHEN W J, et al. Uni-fold: an open-source platform for developing protein folding models beyond AlphaFold[EB/OL]. bioRxiv, 2022[2022-12-10]. . |
| 104 | LI Z Y, YANG S W, LIU X Y, et al. Uni-fold symmetry: harnessing symmetry in folding large protein complexes[EB/OL]. bioRxiv, 2022[2022-12-10]. . |
| 105 | BRYANT P, POZZATI G, ZHU W S, et al. Predicting the structure of large protein complexes using AlphaFold and Monte Carlo tree search[J]. Nature Communications, 2022, 13: 6028. |
| 106 | GHANI U, DESTA I, JINDAL A, et al. Improved docking of protein models by a combination of AlphaFold2 and ClusPro[EB/OL]. bioRxiv, 2022[2022-12-10]. . |
| 107 | HUMPHREYS I R, PEI J M, BAEK M, et al. Computed structures of core eukaryotic protein complexes[J]. Science, 2021, 374(6573): eabm4805. |
| 108 | JOHANSSON-ÅKHE I, WALLNER B. Improving peptide-protein docking with AlphaFold-Multimer using forced sampling[J]. Frontiers in Bioinformatics, 2022, 2: 959160. |
| 109 | LEE C E, SU B H, TSENG Y J. Comparative studies of AlphaFold, RoseTTAFold and Modeller: a case study involving the use of G-protein-coupled receptors[J]. Briefings in Bioinformatics, 2022, 23(5): bbac308. |
| 110 | YIN R, FENG B Y, VARSHNEY A, et al. Benchmarking AlphaFold for protein complex modeling reveals accuracy determinants[J]. Protein Science, 2022, 31(8): e4379. |
| 111 | BRYANT P. Deep learning for protein complex structure prediction[J]. Current Opinion in Structural Biology, 2023, 79: 102529. |
| 112 | HAN B Q, REN C J, WANG W D, et al. Computational prediction of protein intrinsically disordered region related interactions and functions[J]. Genes, 2023, 14(2): 432. |
| [1] | Qiaozhen MENG, Fei GUO. Applications of foldability in intelligent enzyme engineering and design: take AlphaFold2 for example [J]. Synthetic Biology Journal, 2023, 4(3): 571-589. |
| [2] | Yiming TANG, Yifei YAO, Zhongyuan YANG, Yun ZHOU, Zichao WANG, Guanghong WEI. Pathological aggregation and liquid-liquid phase separation of proteins associated with neurodegenerative diseases [J]. Synthetic Biology Journal, 2023, 4(3): 590-610. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||