Programming biological operating systems: genome design, assembly and activation
1
2014
... 合成生物学是继DNA双螺旋发现催生分子生物学,“人类基因组计划”实施催生基因组学后的第三次生物技术革命.DNA合成技术是合成生物学的核心使能技术之一.随着我们对生命系统认识的深入,生命体系的重新设计和创造已成为生物学领域最富想象力和活力的研究领域.大规模基因组DNA设计和合成赋予我们改造细胞功能甚至创造人工生命的能力,有助于提高我们对生命体的理解、预测和操控的能力[1-5]. ...
Genome engineering with targetable nucleases
0
2014
Creation of a bacterial cell controlled by a chemically synthesized genome
0
2010
Synthetic biology: applications come of age
0
2010
Foundations for engineering biology
1
2005
... 合成生物学是继DNA双螺旋发现催生分子生物学,“人类基因组计划”实施催生基因组学后的第三次生物技术革命.DNA合成技术是合成生物学的核心使能技术之一.随着我们对生命系统认识的深入,生命体系的重新设计和创造已成为生物学领域最富想象力和活力的研究领域.大规模基因组DNA设计和合成赋予我们改造细胞功能甚至创造人工生命的能力,有助于提高我们对生命体的理解、预测和操控的能力[1-5]. ...
DNA synthesis, assembly and applications in synthetic biology
3
2012
... 自20世纪50年代以来,大量科研工作者尝试通过化学和酶促方法合成DNA.首先获得成功的是化学合成技术.经过多年优化和改进,DNA化学合成经历了从柱式合成到芯片合成的变革发展,并得到了广泛的市场化应用.但是现有化学方法的偶联效率和副反应使寡核苷酸合成长度局限于200~300 nt,难以到达kb级的基因长度[6-8].因此更长片段则需通过组装技术拼接寡核苷酸片段,直至获得基因、染色体或基因组长度的DNA[9].然而,寡核苷酸片段合成和DNA组装过程会产生很多错误,降低长片段DNA的正确率.纠错技术的应用可去除DNA合成和组装过程引入的大量错误,进而降低正确DNA片段的筛选与测序成本[10].本文作者将重点综述DNA合成、组装与纠错技术相关的研究进展,以此期望促进我国DNA合成相关技术创新发展. ...
... 芯片DNA合成的通量高,通过精密的自动化控制,芯片上单个微反应室能以皮升级的反应体系进行合成反应.不同芯片合成通量在3×103~3×106之间,合成密度在105~106/cm2,成本低至0.001~0.1美分/碱基.与柱式合成相比,在进行大规模基因合成时,芯片合成法占有绝对优势.但芯片制作工艺复杂,随着合成密度的增加,对芯片生产技术以及合成仪的自动化控制技术要求也高.此外,芯片合成还存在由定点错位或试剂隔离不良而产生“边缘效应”、脱保护反应不彻底、脱嘌呤等副反应多的问题[6].这些因素直接影响合成序列的完整性和正确性,使DNA芯片合成的效率在90%~99%,合成长度限制在25~200 nt.芯片单个微量反应体系合成的寡核苷酸产量低(约为10-15 mol),错误率也高于柱式合成,难以单独分离纯化.因此,不适用于单基因或小规模的基因、常规探针以及引物合成. ...
... 目前DNA片段从头合成的长度有限,更长的基因或基因组则需要通过寡核苷酸片段的酶促组装或体内组装获得.通常使用的寡核苷酸组装方法有两种:连接酶组装法(ligase chain reaction, LCR)和聚合酶组装法(polymerase cycling assembly, PCA)[6].连接酶组装法通过DNA连接酶将首尾相连、重叠杂交的5' 磷酸化寡核苷酸片段连接成双链DNA.聚合酶组装法则利用DNA聚合酶延伸杂交的重叠寡核苷酸片段获得不同长度的混合物,最后用引物扩增出成功组装的全长片段(图3).PCA具有良好的兼容性,也被应用于芯片合成的寡核苷酸组装[36-37]. ...
The chemical synthesis of DNA/RNA: our gift to science
0
2013
De novo DNA synthesis using polymerase-nucleotide conjugates
2
2018
... 自20世纪50年代以来,大量科研工作者尝试通过化学和酶促方法合成DNA.首先获得成功的是化学合成技术.经过多年优化和改进,DNA化学合成经历了从柱式合成到芯片合成的变革发展,并得到了广泛的市场化应用.但是现有化学方法的偶联效率和副反应使寡核苷酸合成长度局限于200~300 nt,难以到达kb级的基因长度[6-8].因此更长片段则需通过组装技术拼接寡核苷酸片段,直至获得基因、染色体或基因组长度的DNA[9].然而,寡核苷酸片段合成和DNA组装过程会产生很多错误,降低长片段DNA的正确率.纠错技术的应用可去除DNA合成和组装过程引入的大量错误,进而降低正确DNA片段的筛选与测序成本[10].本文作者将重点综述DNA合成、组装与纠错技术相关的研究进展,以此期望促进我国DNA合成相关技术创新发展. ...
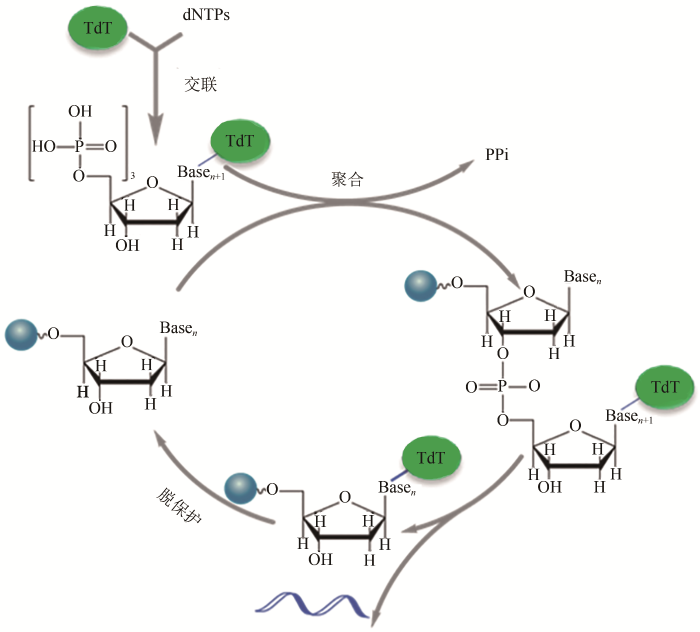
... Bollum首先发现末端脱氧核苷酸转移酶(TdT)[30],并于1962年提出TdT可用于单链寡核苷酸合成[31].1984年,Schott和Schrade用TdT将dNTPs添加到不同长度的引发链[32].研究发现TdT对四种核苷酸的偏好性差异小、偶联效率高,持续合成和延伸单链DNA可产生长达8000 nt的均聚物[29-30,33-34].TdT用于可控酶促DNA合成还需有效的可逆终止方法.使用带有阻断基团的RT-dNTP(RT为可逆终止子,可在dNTP的3'-OH或其他位置)为底物[35],通过偶联-去阻断两步循环迭代,有望将TdT用于长链寡核苷酸的定序合成[24].具有开发潜力的阻断基团包括氨基、烯丙基、磷酸基团、2-硝基苄基、3'-O-(2-氰乙基)等[29].2018年,Keasling团队[8]另辟蹊径在单分子TdT上用可裂解接头连接单个核苷酸,利用TdT将核苷酸添加到引物链后仍保持与DNA链的连接,有效地阻止DNA链的进一步延伸(图2).裂解接头释放TdT后,DNA即可重新进行新一轮的核苷酸添加循环.该方法平均偶联效率可达97.7%,单个循环仅需2~3 min.最近,DNA script公司通过TdT改造结合阻断基团宣称通过酶法以高达99.5%的偶联效率合成了长达280nt的寡核苷酸.而Camena Bioscience所宣传的酶法DNA合成技术gSynth的偶联效率更高达99.9%.同时,gSynth在合成300 nt寡核苷酸片段时,产物中全长序列比例达到了85.3%,远高于亚磷酰胺合成法的22.7%.虽然酶法DNA合成技术至今还未见商业化,但从其所具有的潜力可以预见酶法DNA合成技术将引领新一轮的DNA合成技术革命. ...
Gene synthesis demystified
1
2009
... 自20世纪50年代以来,大量科研工作者尝试通过化学和酶促方法合成DNA.首先获得成功的是化学合成技术.经过多年优化和改进,DNA化学合成经历了从柱式合成到芯片合成的变革发展,并得到了广泛的市场化应用.但是现有化学方法的偶联效率和副反应使寡核苷酸合成长度局限于200~300 nt,难以到达kb级的基因长度[6-8].因此更长片段则需通过组装技术拼接寡核苷酸片段,直至获得基因、染色体或基因组长度的DNA[9].然而,寡核苷酸片段合成和DNA组装过程会产生很多错误,降低长片段DNA的正确率.纠错技术的应用可去除DNA合成和组装过程引入的大量错误,进而降低正确DNA片段的筛选与测序成本[10].本文作者将重点综述DNA合成、组装与纠错技术相关的研究进展,以此期望促进我国DNA合成相关技术创新发展. ...
Error correction in gene synthesis technology
3
2012
... 自20世纪50年代以来,大量科研工作者尝试通过化学和酶促方法合成DNA.首先获得成功的是化学合成技术.经过多年优化和改进,DNA化学合成经历了从柱式合成到芯片合成的变革发展,并得到了广泛的市场化应用.但是现有化学方法的偶联效率和副反应使寡核苷酸合成长度局限于200~300 nt,难以到达kb级的基因长度[6-8].因此更长片段则需通过组装技术拼接寡核苷酸片段,直至获得基因、染色体或基因组长度的DNA[9].然而,寡核苷酸片段合成和DNA组装过程会产生很多错误,降低长片段DNA的正确率.纠错技术的应用可去除DNA合成和组装过程引入的大量错误,进而降低正确DNA片段的筛选与测序成本[10].本文作者将重点综述DNA合成、组装与纠错技术相关的研究进展,以此期望促进我国DNA合成相关技术创新发展. ...
... 经化学合成的寡核苷酸链含有大量错误,对这样的寡核苷酸池的纠错过程主要根据合成错误造成的分子量或基团的差异进行分离纯化.如对柱式合成可通过高效液相色谱法(HPLC)[56]、聚丙烯酰胺凝胶电泳(PAGE)[57]或疏水性纯化柱过滤[10]去除合成不完全的片段.这些方法通量低,对错误区分精确度有限,分离过程损失较大.芯片合成寡核苷酸的纠错可通过直接与序列正确的寡核苷酸捕获探针进行杂交选择[37],或是Tm均一化(热力学参数)严格杂交筛选手段[58],过滤寡核苷酸池中的错误片段.Evonetix的DNA合成平台则通过对温度的控制将合成、组装、纠错进行整合,其纠错过程由精确的温度控制去除非完全匹配的DNA链.还可以使用NGS技术纠错,结合DNA芯片合成与高通量测序平台,将合成、组装、测序纠错一体化[59]. ...
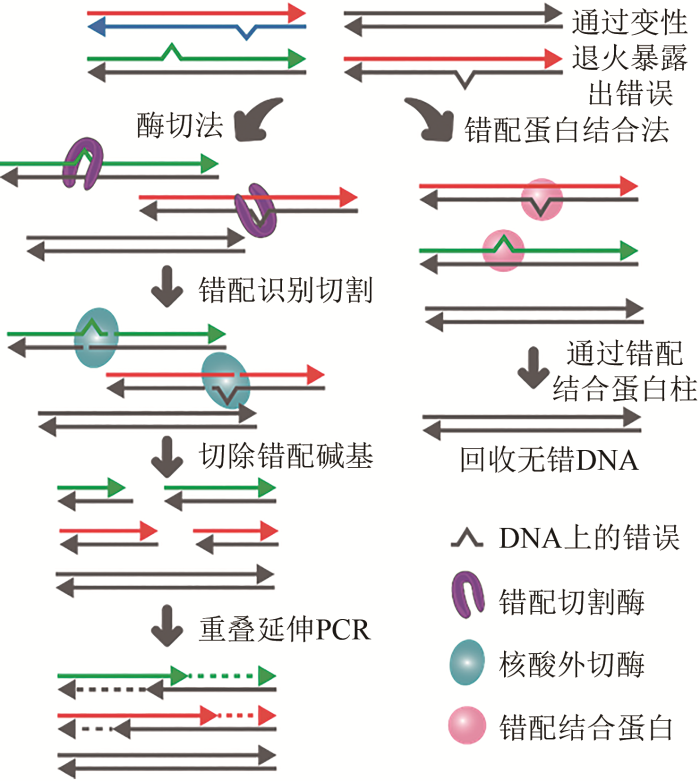
... 经互补配对后的双链DNA片段的核苷酸插入、缺失、取代错误主要表现为错配和凸起等,这些错误的去除更多是借助基于生物体内的DNA修复体系开发出的DNA酶促纠错技术[60-61].通过对互补序列退火,暴露出错配信号,再利用具有错配结合或错配切割活性的酶对DNA双链纠错(图4),从而富集正确序列[10,55]. ...
Deoxynucleoside phosphoramidites—A new class of key intermediates for deoxypolynucleotide synthesis
1
1981
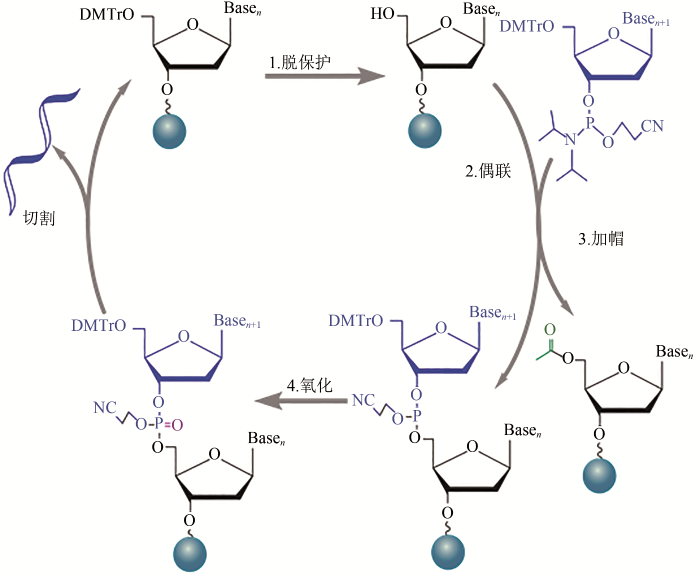
... 寡核苷酸的化学合成始于20世纪50年代,于80年代开发出亚磷酰胺三酯化学合成法[11],并应用于柱式合成,在90年代又应用到基于芯片的高通量合成技术中[12].亚磷酰胺三酯合成法由脱保护(deprotection)、偶联(coupling)、加帽(capping)和氧化(oxidation)四步化学反应组成循环,通过分步活化结合在核苷酸3' 位和5' 位的化学活性保护基团实现可控合成,在固相载体上从3' 到5' 方向逐个延伸合成寡核苷酸链[13](图1). ...
Advancing high-throughput gene synthesis technology
2
2009
... 寡核苷酸的化学合成始于20世纪50年代,于80年代开发出亚磷酰胺三酯化学合成法[11],并应用于柱式合成,在90年代又应用到基于芯片的高通量合成技术中[12].亚磷酰胺三酯合成法由脱保护(deprotection)、偶联(coupling)、加帽(capping)和氧化(oxidation)四步化学反应组成循环,通过分步活化结合在核苷酸3' 位和5' 位的化学活性保护基团实现可控合成,在固相载体上从3' 到5' 方向逐个延伸合成寡核苷酸链[13](图1). ...
... 芯片可作为DNA合成固相载体,以高密度、集成方式在其表面特定位点上进行合成反应,从而在节省试剂的同时实现高通量合成.芯片DNA合成技术仍以亚磷酰胺合成法四步反应循环为基础,但采用不同的“脱保护”定点控制方法,分别发展出了光刻合成、电化学合成和喷墨打印合成等DNA合成平台[12]. ...
Large-scale de novo DNA synthesis: technologies and applications
1
2014
... 寡核苷酸的化学合成始于20世纪50年代,于80年代开发出亚磷酰胺三酯化学合成法[11],并应用于柱式合成,在90年代又应用到基于芯片的高通量合成技术中[12].亚磷酰胺三酯合成法由脱保护(deprotection)、偶联(coupling)、加帽(capping)和氧化(oxidation)四步化学反应组成循环,通过分步活化结合在核苷酸3' 位和5' 位的化学活性保护基团实现可控合成,在固相载体上从3' 到5' 方向逐个延伸合成寡核苷酸链[13](图1). ...
Synthesis of high-quality libraries of long (150mer) oligonucleotides by a novel depurination controlled process
2
2010
... 基于亚磷酰胺三酯合成法的柱式DNA合成技术,以填充多孔玻璃(controlled pore glass, CPG)或聚苯乙烯(polystyrene, PS)筛板的合成柱作为固相载体,带保护的单体经过化学试剂分步活化被逐个按序添加到固定在合成柱的引发剂上.目前商品化的柱式DNA合成反应偶联效率可达98%~99.8%,错误率约为1/600 nt,产量一般在1 µmol以内,单个循环耗时6~8 min.权衡效率与成本后最长合成长度通常控制在100 nt左右[14].若将合成柱固定到多孔装载板上,单轮合成通量可提高到1536根,平均偶联效率为99.5%,错误率约1.53/717 nt,成本大约0.277美分/碱基[15]. ...
... 柱式DNA合成技术发展至今,自动化合成设备成熟.能够方便、灵活地提取用于合成某段基因所需的任意寡核苷酸片段,满足一般实验的要求.随着合成生物学领域对大规模基因和基因组合成需求的日益高涨,柱式合成持续合成能力弱、化学试剂耗费大、副反应多、通量低等不足也日益凸显[14].为突破低效率、低通量、高成本的限制,寻求DNA合成技术持续发展的研究主要致力于:①开发具备高反应通量、多重功能的集成芯片作为固相载体,并行合成寡核苷酸;②开发基于无模板单链DNA合成的酶促寡核苷酸合成技术. ...
Next generation 1536-well oligonucleotide synthesizer with on-the-fly dispense
1
2014
... 基于亚磷酰胺三酯合成法的柱式DNA合成技术,以填充多孔玻璃(controlled pore glass, CPG)或聚苯乙烯(polystyrene, PS)筛板的合成柱作为固相载体,带保护的单体经过化学试剂分步活化被逐个按序添加到固定在合成柱的引发剂上.目前商品化的柱式DNA合成反应偶联效率可达98%~99.8%,错误率约为1/600 nt,产量一般在1 µmol以内,单个循环耗时6~8 min.权衡效率与成本后最长合成长度通常控制在100 nt左右[14].若将合成柱固定到多孔装载板上,单轮合成通量可提高到1536根,平均偶联效率为99.5%,错误率约1.53/717 nt,成本大约0.277美分/碱基[15]. ...
Light-directed, spatially addressable parallel chemical synthesis
1
1991
... 最早出现的光刻合成通过精确控制光在芯片表面指定位点的投射,分解光敏保护基团或光敏催化剂产酸进行脱保护,实现核苷酸在不同合成位点的有序添加.根据光照控制方式又分为掩模光刻合成[16-17](为Affymetrix采用)与无掩模光刻合成[18-20](为NimbleGen和LC Sciences采用).电化学合成利用电化学反应产生酸,控制聚合物膜上常规亚磷酰胺单体的加成[21],被CustomArray(已被Genscript收购)所采用.Agilent的喷墨打印合成则通过将酸溶液或试剂喷射在反应位点催化脱保护[22-23]. ...
Photolithographic synthesis of high-density oligonucleotide probe arrays
1
2001
... 最早出现的光刻合成通过精确控制光在芯片表面指定位点的投射,分解光敏保护基团或光敏催化剂产酸进行脱保护,实现核苷酸在不同合成位点的有序添加.根据光照控制方式又分为掩模光刻合成[16-17](为Affymetrix采用)与无掩模光刻合成[18-20](为NimbleGen和LC Sciences采用).电化学合成利用电化学反应产生酸,控制聚合物膜上常规亚磷酰胺单体的加成[21],被CustomArray(已被Genscript收购)所采用.Agilent的喷墨打印合成则通过将酸溶液或试剂喷射在反应位点催化脱保护[22-23]. ...
Maskless fabrication of light-directed oligonucleotide microarrays using a digital micromirror array
1
1999
... 最早出现的光刻合成通过精确控制光在芯片表面指定位点的投射,分解光敏保护基团或光敏催化剂产酸进行脱保护,实现核苷酸在不同合成位点的有序添加.根据光照控制方式又分为掩模光刻合成[16-17](为Affymetrix采用)与无掩模光刻合成[18-20](为NimbleGen和LC Sciences采用).电化学合成利用电化学反应产生酸,控制聚合物膜上常规亚磷酰胺单体的加成[21],被CustomArray(已被Genscript收购)所采用.Agilent的喷墨打印合成则通过将酸溶液或试剂喷射在反应位点催化脱保护[22-23]. ...
A flexible light-directed DNA chip synthesis gated by deprotection using solution photogenerated acids
0
2001
Efficiency, error and yield in light-directed maskless synthesis of DNA microarrays
1
2011
... 最早出现的光刻合成通过精确控制光在芯片表面指定位点的投射,分解光敏保护基团或光敏催化剂产酸进行脱保护,实现核苷酸在不同合成位点的有序添加.根据光照控制方式又分为掩模光刻合成[16-17](为Affymetrix采用)与无掩模光刻合成[18-20](为NimbleGen和LC Sciences采用).电化学合成利用电化学反应产生酸,控制聚合物膜上常规亚磷酰胺单体的加成[21],被CustomArray(已被Genscript收购)所采用.Agilent的喷墨打印合成则通过将酸溶液或试剂喷射在反应位点催化脱保护[22-23]. ...
Electrochemical detection of short DNA oligomer hybridization using the CombiMatrix ElectraSense microarray reader
1
18
... 最早出现的光刻合成通过精确控制光在芯片表面指定位点的投射,分解光敏保护基团或光敏催化剂产酸进行脱保护,实现核苷酸在不同合成位点的有序添加.根据光照控制方式又分为掩模光刻合成[16-17](为Affymetrix采用)与无掩模光刻合成[18-20](为NimbleGen和LC Sciences采用).电化学合成利用电化学反应产生酸,控制聚合物膜上常规亚磷酰胺单体的加成[21],被CustomArray(已被Genscript收购)所采用.Agilent的喷墨打印合成则通过将酸溶液或试剂喷射在反应位点催化脱保护[22-23]. ...
Expression profiling using microarrays fabricated by an ink-jet oligonucleotide synthesizer
1
2001
... 最早出现的光刻合成通过精确控制光在芯片表面指定位点的投射,分解光敏保护基团或光敏催化剂产酸进行脱保护,实现核苷酸在不同合成位点的有序添加.根据光照控制方式又分为掩模光刻合成[16-17](为Affymetrix采用)与无掩模光刻合成[18-20](为NimbleGen和LC Sciences采用).电化学合成利用电化学反应产生酸,控制聚合物膜上常规亚磷酰胺单体的加成[21],被CustomArray(已被Genscript收购)所采用.Agilent的喷墨打印合成则通过将酸溶液或试剂喷射在反应位点催化脱保护[22-23]. ...
High density synthetic oligonucleotide arrays
1
1999
... 最早出现的光刻合成通过精确控制光在芯片表面指定位点的投射,分解光敏保护基团或光敏催化剂产酸进行脱保护,实现核苷酸在不同合成位点的有序添加.根据光照控制方式又分为掩模光刻合成[16-17](为Affymetrix采用)与无掩模光刻合成[18-20](为NimbleGen和LC Sciences采用).电化学合成利用电化学反应产生酸,控制聚合物膜上常规亚磷酰胺单体的加成[21],被CustomArray(已被Genscript收购)所采用.Agilent的喷墨打印合成则通过将酸溶液或试剂喷射在反应位点催化脱保护[22-23]. ...
A theoretical model for template-free synthesis of long DNA sequence
2
2008
... DNA化学合成中大量使用有毒、易燃且不稳定的有机试剂,环境友好度低,因而生物合成又重新受到人们的关注.常规的DNA聚合酶具有模板依赖性,无法用于DNA的从头合成.寻找非模板依赖性的DNA合成酶成为酶法DNA合成技术开发的首要任务.此外,使酶在受控条件下逐个添加指定的核苷酸是酶促寡核苷酸合成技术的另一挑战[24]. ...
... Bollum首先发现末端脱氧核苷酸转移酶(TdT)[30],并于1962年提出TdT可用于单链寡核苷酸合成[31].1984年,Schott和Schrade用TdT将dNTPs添加到不同长度的引发链[32].研究发现TdT对四种核苷酸的偏好性差异小、偶联效率高,持续合成和延伸单链DNA可产生长达8000 nt的均聚物[29-30,33-34].TdT用于可控酶促DNA合成还需有效的可逆终止方法.使用带有阻断基团的RT-dNTP(RT为可逆终止子,可在dNTP的3'-OH或其他位置)为底物[35],通过偶联-去阻断两步循环迭代,有望将TdT用于长链寡核苷酸的定序合成[24].具有开发潜力的阻断基团包括氨基、烯丙基、磷酸基团、2-硝基苄基、3'-O-(2-氰乙基)等[29].2018年,Keasling团队[8]另辟蹊径在单分子TdT上用可裂解接头连接单个核苷酸,利用TdT将核苷酸添加到引物链后仍保持与DNA链的连接,有效地阻止DNA链的进一步延伸(图2).裂解接头释放TdT后,DNA即可重新进行新一轮的核苷酸添加循环.该方法平均偶联效率可达97.7%,单个循环仅需2~3 min.最近,DNA script公司通过TdT改造结合阻断基团宣称通过酶法以高达99.5%的偶联效率合成了长达280nt的寡核苷酸.而Camena Bioscience所宣传的酶法DNA合成技术gSynth的偶联效率更高达99.9%.同时,gSynth在合成300 nt寡核苷酸片段时,产物中全长序列比例达到了85.3%,远高于亚磷酰胺合成法的22.7%.虽然酶法DNA合成技术至今还未见商业化,但从其所具有的潜力可以预见酶法DNA合成技术将引领新一轮的DNA合成技术革命. ...
New approach to the synthesis of polyribonucleotides of defined sequence
1
1971
... Mackey和Gilham[25]使用核苷酸磷酸化酶(PNPase)将引入2' 末端封闭的5'-二磷酸-2'-O-(α-甲氧基乙基)核苷酸偶联到寡腺苷酸引物的3' 末端,定序合成了寡聚核糖核苷酸链.Gillam和Smith[26]采用相同的方法合成了寡聚脱氧核糖核苷酸链.England和Uhlenbeck[27]则是首先将连接法用于DNA合成,使用T4 RNA连接酶将5',3'-核糖核苷二磷酸底物偶联到引发链的3' 末端合成寡核糖核苷酸链.1999年,T4 RNA连接酶首次被用于固相酶法DNA合成[28].尽管PNPase和T4 RNA连接酶能够合成RNA和DNA,但两者偶联效率低,单轮循环耗时长,用于DNA合成技术的局限性大于可用性[29]. ...
Enzymatic synthesis of deoxyribo-oligonucleotides of defined sequence. Deoxyribo-oligonucleotide synthesis
1
1974
... Mackey和Gilham[25]使用核苷酸磷酸化酶(PNPase)将引入2' 末端封闭的5'-二磷酸-2'-O-(α-甲氧基乙基)核苷酸偶联到寡腺苷酸引物的3' 末端,定序合成了寡聚核糖核苷酸链.Gillam和Smith[26]采用相同的方法合成了寡聚脱氧核糖核苷酸链.England和Uhlenbeck[27]则是首先将连接法用于DNA合成,使用T4 RNA连接酶将5',3'-核糖核苷二磷酸底物偶联到引发链的3' 末端合成寡核糖核苷酸链.1999年,T4 RNA连接酶首次被用于固相酶法DNA合成[28].尽管PNPase和T4 RNA连接酶能够合成RNA和DNA,但两者偶联效率低,单轮循环耗时长,用于DNA合成技术的局限性大于可用性[29]. ...
Enzymatic oligoribonucleotide synthesis with T4 RNA ligase
1
1978
... Mackey和Gilham[25]使用核苷酸磷酸化酶(PNPase)将引入2' 末端封闭的5'-二磷酸-2'-O-(α-甲氧基乙基)核苷酸偶联到寡腺苷酸引物的3' 末端,定序合成了寡聚核糖核苷酸链.Gillam和Smith[26]采用相同的方法合成了寡聚脱氧核糖核苷酸链.England和Uhlenbeck[27]则是首先将连接法用于DNA合成,使用T4 RNA连接酶将5',3'-核糖核苷二磷酸底物偶联到引发链的3' 末端合成寡核糖核苷酸链.1999年,T4 RNA连接酶首次被用于固相酶法DNA合成[28].尽管PNPase和T4 RNA连接酶能够合成RNA和DNA,但两者偶联效率低,单轮循环耗时长,用于DNA合成技术的局限性大于可用性[29]. ...
Solid-phase enzymatic synthesis of oligonucleotides
1
1999
... Mackey和Gilham[25]使用核苷酸磷酸化酶(PNPase)将引入2' 末端封闭的5'-二磷酸-2'-O-(α-甲氧基乙基)核苷酸偶联到寡腺苷酸引物的3' 末端,定序合成了寡聚核糖核苷酸链.Gillam和Smith[26]采用相同的方法合成了寡聚脱氧核糖核苷酸链.England和Uhlenbeck[27]则是首先将连接法用于DNA合成,使用T4 RNA连接酶将5',3'-核糖核苷二磷酸底物偶联到引发链的3' 末端合成寡核糖核苷酸链.1999年,T4 RNA连接酶首次被用于固相酶法DNA合成[28].尽管PNPase和T4 RNA连接酶能够合成RNA和DNA,但两者偶联效率低,单轮循环耗时长,用于DNA合成技术的局限性大于可用性[29]. ...
Template-independent enzymatic oligonucleotide synthesis (TiEOS): its history, prospects, and challenges
3
2018
... Mackey和Gilham[25]使用核苷酸磷酸化酶(PNPase)将引入2' 末端封闭的5'-二磷酸-2'-O-(α-甲氧基乙基)核苷酸偶联到寡腺苷酸引物的3' 末端,定序合成了寡聚核糖核苷酸链.Gillam和Smith[26]采用相同的方法合成了寡聚脱氧核糖核苷酸链.England和Uhlenbeck[27]则是首先将连接法用于DNA合成,使用T4 RNA连接酶将5',3'-核糖核苷二磷酸底物偶联到引发链的3' 末端合成寡核糖核苷酸链.1999年,T4 RNA连接酶首次被用于固相酶法DNA合成[28].尽管PNPase和T4 RNA连接酶能够合成RNA和DNA,但两者偶联效率低,单轮循环耗时长,用于DNA合成技术的局限性大于可用性[29]. ...
... Bollum首先发现末端脱氧核苷酸转移酶(TdT)[30],并于1962年提出TdT可用于单链寡核苷酸合成[31].1984年,Schott和Schrade用TdT将dNTPs添加到不同长度的引发链[32].研究发现TdT对四种核苷酸的偏好性差异小、偶联效率高,持续合成和延伸单链DNA可产生长达8000 nt的均聚物[29-30,33-34].TdT用于可控酶促DNA合成还需有效的可逆终止方法.使用带有阻断基团的RT-dNTP(RT为可逆终止子,可在dNTP的3'-OH或其他位置)为底物[35],通过偶联-去阻断两步循环迭代,有望将TdT用于长链寡核苷酸的定序合成[24].具有开发潜力的阻断基团包括氨基、烯丙基、磷酸基团、2-硝基苄基、3'-O-(2-氰乙基)等[29].2018年,Keasling团队[8]另辟蹊径在单分子TdT上用可裂解接头连接单个核苷酸,利用TdT将核苷酸添加到引物链后仍保持与DNA链的连接,有效地阻止DNA链的进一步延伸(图2).裂解接头释放TdT后,DNA即可重新进行新一轮的核苷酸添加循环.该方法平均偶联效率可达97.7%,单个循环仅需2~3 min.最近,DNA script公司通过TdT改造结合阻断基团宣称通过酶法以高达99.5%的偶联效率合成了长达280nt的寡核苷酸.而Camena Bioscience所宣传的酶法DNA合成技术gSynth的偶联效率更高达99.9%.同时,gSynth在合成300 nt寡核苷酸片段时,产物中全长序列比例达到了85.3%,远高于亚磷酰胺合成法的22.7%.虽然酶法DNA合成技术至今还未见商业化,但从其所具有的潜力可以预见酶法DNA合成技术将引领新一轮的DNA合成技术革命. ...
... [29].2018年,Keasling团队[8]另辟蹊径在单分子TdT上用可裂解接头连接单个核苷酸,利用TdT将核苷酸添加到引物链后仍保持与DNA链的连接,有效地阻止DNA链的进一步延伸(图2).裂解接头释放TdT后,DNA即可重新进行新一轮的核苷酸添加循环.该方法平均偶联效率可达97.7%,单个循环仅需2~3 min.最近,DNA script公司通过TdT改造结合阻断基团宣称通过酶法以高达99.5%的偶联效率合成了长达280nt的寡核苷酸.而Camena Bioscience所宣传的酶法DNA合成技术gSynth的偶联效率更高达99.9%.同时,gSynth在合成300 nt寡核苷酸片段时,产物中全长序列比例达到了85.3%,远高于亚磷酰胺合成法的22.7%.虽然酶法DNA合成技术至今还未见商业化,但从其所具有的潜力可以预见酶法DNA合成技术将引领新一轮的DNA合成技术革命. ...
Thermal conversion of nonpriming deoxyribonucleic acid to primer
2
1959
... Bollum首先发现末端脱氧核苷酸转移酶(TdT)[30],并于1962年提出TdT可用于单链寡核苷酸合成[31].1984年,Schott和Schrade用TdT将dNTPs添加到不同长度的引发链[32].研究发现TdT对四种核苷酸的偏好性差异小、偶联效率高,持续合成和延伸单链DNA可产生长达8000 nt的均聚物[29-30,33-34].TdT用于可控酶促DNA合成还需有效的可逆终止方法.使用带有阻断基团的RT-dNTP(RT为可逆终止子,可在dNTP的3'-OH或其他位置)为底物[35],通过偶联-去阻断两步循环迭代,有望将TdT用于长链寡核苷酸的定序合成[24].具有开发潜力的阻断基团包括氨基、烯丙基、磷酸基团、2-硝基苄基、3'-O-(2-氰乙基)等[29].2018年,Keasling团队[8]另辟蹊径在单分子TdT上用可裂解接头连接单个核苷酸,利用TdT将核苷酸添加到引物链后仍保持与DNA链的连接,有效地阻止DNA链的进一步延伸(图2).裂解接头释放TdT后,DNA即可重新进行新一轮的核苷酸添加循环.该方法平均偶联效率可达97.7%,单个循环仅需2~3 min.最近,DNA script公司通过TdT改造结合阻断基团宣称通过酶法以高达99.5%的偶联效率合成了长达280nt的寡核苷酸.而Camena Bioscience所宣传的酶法DNA合成技术gSynth的偶联效率更高达99.9%.同时,gSynth在合成300 nt寡核苷酸片段时,产物中全长序列比例达到了85.3%,远高于亚磷酰胺合成法的22.7%.虽然酶法DNA合成技术至今还未见商业化,但从其所具有的潜力可以预见酶法DNA合成技术将引领新一轮的DNA合成技术革命. ...
... -30,33-34].TdT用于可控酶促DNA合成还需有效的可逆终止方法.使用带有阻断基团的RT-dNTP(RT为可逆终止子,可在dNTP的3'-OH或其他位置)为底物[35],通过偶联-去阻断两步循环迭代,有望将TdT用于长链寡核苷酸的定序合成[24].具有开发潜力的阻断基团包括氨基、烯丙基、磷酸基团、2-硝基苄基、3'-O-(2-氰乙基)等[29].2018年,Keasling团队[8]另辟蹊径在单分子TdT上用可裂解接头连接单个核苷酸,利用TdT将核苷酸添加到引物链后仍保持与DNA链的连接,有效地阻止DNA链的进一步延伸(图2).裂解接头释放TdT后,DNA即可重新进行新一轮的核苷酸添加循环.该方法平均偶联效率可达97.7%,单个循环仅需2~3 min.最近,DNA script公司通过TdT改造结合阻断基团宣称通过酶法以高达99.5%的偶联效率合成了长达280nt的寡核苷酸.而Camena Bioscience所宣传的酶法DNA合成技术gSynth的偶联效率更高达99.9%.同时,gSynth在合成300 nt寡核苷酸片段时,产物中全长序列比例达到了85.3%,远高于亚磷酰胺合成法的22.7%.虽然酶法DNA合成技术至今还未见商业化,但从其所具有的潜力可以预见酶法DNA合成技术将引领新一轮的DNA合成技术革命. ...
Oligodeoxyribonucleotide-primed reactions catalyzed by calf thymus polymerase
1
1962
... Bollum首先发现末端脱氧核苷酸转移酶(TdT)[30],并于1962年提出TdT可用于单链寡核苷酸合成[31].1984年,Schott和Schrade用TdT将dNTPs添加到不同长度的引发链[32].研究发现TdT对四种核苷酸的偏好性差异小、偶联效率高,持续合成和延伸单链DNA可产生长达8000 nt的均聚物[29-30,33-34].TdT用于可控酶促DNA合成还需有效的可逆终止方法.使用带有阻断基团的RT-dNTP(RT为可逆终止子,可在dNTP的3'-OH或其他位置)为底物[35],通过偶联-去阻断两步循环迭代,有望将TdT用于长链寡核苷酸的定序合成[24].具有开发潜力的阻断基团包括氨基、烯丙基、磷酸基团、2-硝基苄基、3'-O-(2-氰乙基)等[29].2018年,Keasling团队[8]另辟蹊径在单分子TdT上用可裂解接头连接单个核苷酸,利用TdT将核苷酸添加到引物链后仍保持与DNA链的连接,有效地阻止DNA链的进一步延伸(图2).裂解接头释放TdT后,DNA即可重新进行新一轮的核苷酸添加循环.该方法平均偶联效率可达97.7%,单个循环仅需2~3 min.最近,DNA script公司通过TdT改造结合阻断基团宣称通过酶法以高达99.5%的偶联效率合成了长达280nt的寡核苷酸.而Camena Bioscience所宣传的酶法DNA合成技术gSynth的偶联效率更高达99.9%.同时,gSynth在合成300 nt寡核苷酸片段时,产物中全长序列比例达到了85.3%,远高于亚磷酰胺合成法的22.7%.虽然酶法DNA合成技术至今还未见商业化,但从其所具有的潜力可以预见酶法DNA合成技术将引领新一轮的DNA合成技术革命. ...
Single-step elongation of oligodeoxynucleotides using terminal deoxynucleotidyl transferase
1
1984
... Bollum首先发现末端脱氧核苷酸转移酶(TdT)[30],并于1962年提出TdT可用于单链寡核苷酸合成[31].1984年,Schott和Schrade用TdT将dNTPs添加到不同长度的引发链[32].研究发现TdT对四种核苷酸的偏好性差异小、偶联效率高,持续合成和延伸单链DNA可产生长达8000 nt的均聚物[29-30,33-34].TdT用于可控酶促DNA合成还需有效的可逆终止方法.使用带有阻断基团的RT-dNTP(RT为可逆终止子,可在dNTP的3'-OH或其他位置)为底物[35],通过偶联-去阻断两步循环迭代,有望将TdT用于长链寡核苷酸的定序合成[24].具有开发潜力的阻断基团包括氨基、烯丙基、磷酸基团、2-硝基苄基、3'-O-(2-氰乙基)等[29].2018年,Keasling团队[8]另辟蹊径在单分子TdT上用可裂解接头连接单个核苷酸,利用TdT将核苷酸添加到引物链后仍保持与DNA链的连接,有效地阻止DNA链的进一步延伸(图2).裂解接头释放TdT后,DNA即可重新进行新一轮的核苷酸添加循环.该方法平均偶联效率可达97.7%,单个循环仅需2~3 min.最近,DNA script公司通过TdT改造结合阻断基团宣称通过酶法以高达99.5%的偶联效率合成了长达280nt的寡核苷酸.而Camena Bioscience所宣传的酶法DNA合成技术gSynth的偶联效率更高达99.9%.同时,gSynth在合成300 nt寡核苷酸片段时,产物中全长序列比例达到了85.3%,远高于亚磷酰胺合成法的22.7%.虽然酶法DNA合成技术至今还未见商业化,但从其所具有的潜力可以预见酶法DNA合成技术将引领新一轮的DNA合成技术革命. ...
Amplified on-chip fluorescence detection of DNA hybridization by surface-initiated enzymatic polymerization
1
2011
... Bollum首先发现末端脱氧核苷酸转移酶(TdT)[30],并于1962年提出TdT可用于单链寡核苷酸合成[31].1984年,Schott和Schrade用TdT将dNTPs添加到不同长度的引发链[32].研究发现TdT对四种核苷酸的偏好性差异小、偶联效率高,持续合成和延伸单链DNA可产生长达8000 nt的均聚物[29-30,33-34].TdT用于可控酶促DNA合成还需有效的可逆终止方法.使用带有阻断基团的RT-dNTP(RT为可逆终止子,可在dNTP的3'-OH或其他位置)为底物[35],通过偶联-去阻断两步循环迭代,有望将TdT用于长链寡核苷酸的定序合成[24].具有开发潜力的阻断基团包括氨基、烯丙基、磷酸基团、2-硝基苄基、3'-O-(2-氰乙基)等[29].2018年,Keasling团队[8]另辟蹊径在单分子TdT上用可裂解接头连接单个核苷酸,利用TdT将核苷酸添加到引物链后仍保持与DNA链的连接,有效地阻止DNA链的进一步延伸(图2).裂解接头释放TdT后,DNA即可重新进行新一轮的核苷酸添加循环.该方法平均偶联效率可达97.7%,单个循环仅需2~3 min.最近,DNA script公司通过TdT改造结合阻断基团宣称通过酶法以高达99.5%的偶联效率合成了长达280nt的寡核苷酸.而Camena Bioscience所宣传的酶法DNA合成技术gSynth的偶联效率更高达99.9%.同时,gSynth在合成300 nt寡核苷酸片段时,产物中全长序列比例达到了85.3%,远高于亚磷酰胺合成法的22.7%.虽然酶法DNA合成技术至今还未见商业化,但从其所具有的潜力可以预见酶法DNA合成技术将引领新一轮的DNA合成技术革命. ...
Terminal deoxynucleotidyl transferase: the story of a misguided DNA polymerase
1
2010
... Bollum首先发现末端脱氧核苷酸转移酶(TdT)[30],并于1962年提出TdT可用于单链寡核苷酸合成[31].1984年,Schott和Schrade用TdT将dNTPs添加到不同长度的引发链[32].研究发现TdT对四种核苷酸的偏好性差异小、偶联效率高,持续合成和延伸单链DNA可产生长达8000 nt的均聚物[29-30,33-34].TdT用于可控酶促DNA合成还需有效的可逆终止方法.使用带有阻断基团的RT-dNTP(RT为可逆终止子,可在dNTP的3'-OH或其他位置)为底物[35],通过偶联-去阻断两步循环迭代,有望将TdT用于长链寡核苷酸的定序合成[24].具有开发潜力的阻断基团包括氨基、烯丙基、磷酸基团、2-硝基苄基、3'-O-(2-氰乙基)等[29].2018年,Keasling团队[8]另辟蹊径在单分子TdT上用可裂解接头连接单个核苷酸,利用TdT将核苷酸添加到引物链后仍保持与DNA链的连接,有效地阻止DNA链的进一步延伸(图2).裂解接头释放TdT后,DNA即可重新进行新一轮的核苷酸添加循环.该方法平均偶联效率可达97.7%,单个循环仅需2~3 min.最近,DNA script公司通过TdT改造结合阻断基团宣称通过酶法以高达99.5%的偶联效率合成了长达280nt的寡核苷酸.而Camena Bioscience所宣传的酶法DNA合成技术gSynth的偶联效率更高达99.9%.同时,gSynth在合成300 nt寡核苷酸片段时,产物中全长序列比例达到了85.3%,远高于亚磷酰胺合成法的22.7%.虽然酶法DNA合成技术至今还未见商业化,但从其所具有的潜力可以预见酶法DNA合成技术将引领新一轮的DNA合成技术革命. ...
3'-O-modified nucleotides as reversible terminators for pyrosequencing
1
2007
... Bollum首先发现末端脱氧核苷酸转移酶(TdT)[30],并于1962年提出TdT可用于单链寡核苷酸合成[31].1984年,Schott和Schrade用TdT将dNTPs添加到不同长度的引发链[32].研究发现TdT对四种核苷酸的偏好性差异小、偶联效率高,持续合成和延伸单链DNA可产生长达8000 nt的均聚物[29-30,33-34].TdT用于可控酶促DNA合成还需有效的可逆终止方法.使用带有阻断基团的RT-dNTP(RT为可逆终止子,可在dNTP的3'-OH或其他位置)为底物[35],通过偶联-去阻断两步循环迭代,有望将TdT用于长链寡核苷酸的定序合成[24].具有开发潜力的阻断基团包括氨基、烯丙基、磷酸基团、2-硝基苄基、3'-O-(2-氰乙基)等[29].2018年,Keasling团队[8]另辟蹊径在单分子TdT上用可裂解接头连接单个核苷酸,利用TdT将核苷酸添加到引物链后仍保持与DNA链的连接,有效地阻止DNA链的进一步延伸(图2).裂解接头释放TdT后,DNA即可重新进行新一轮的核苷酸添加循环.该方法平均偶联效率可达97.7%,单个循环仅需2~3 min.最近,DNA script公司通过TdT改造结合阻断基团宣称通过酶法以高达99.5%的偶联效率合成了长达280nt的寡核苷酸.而Camena Bioscience所宣传的酶法DNA合成技术gSynth的偶联效率更高达99.9%.同时,gSynth在合成300 nt寡核苷酸片段时,产物中全长序列比例达到了85.3%,远高于亚磷酰胺合成法的22.7%.虽然酶法DNA合成技术至今还未见商业化,但从其所具有的潜力可以预见酶法DNA合成技术将引领新一轮的DNA合成技术革命. ...
Parallel gene synthesis in a microfluidic device
1
2007
... 目前DNA片段从头合成的长度有限,更长的基因或基因组则需要通过寡核苷酸片段的酶促组装或体内组装获得.通常使用的寡核苷酸组装方法有两种:连接酶组装法(ligase chain reaction, LCR)和聚合酶组装法(polymerase cycling assembly, PCA)[6].连接酶组装法通过DNA连接酶将首尾相连、重叠杂交的5' 磷酸化寡核苷酸片段连接成双链DNA.聚合酶组装法则利用DNA聚合酶延伸杂交的重叠寡核苷酸片段获得不同长度的混合物,最后用引物扩增出成功组装的全长片段(图3).PCA具有良好的兼容性,也被应用于芯片合成的寡核苷酸组装[36-37]. ...
Accurate multiplex gene synthesis from programmable DNA microchips
2
2004
... 目前DNA片段从头合成的长度有限,更长的基因或基因组则需要通过寡核苷酸片段的酶促组装或体内组装获得.通常使用的寡核苷酸组装方法有两种:连接酶组装法(ligase chain reaction, LCR)和聚合酶组装法(polymerase cycling assembly, PCA)[6].连接酶组装法通过DNA连接酶将首尾相连、重叠杂交的5' 磷酸化寡核苷酸片段连接成双链DNA.聚合酶组装法则利用DNA聚合酶延伸杂交的重叠寡核苷酸片段获得不同长度的混合物,最后用引物扩增出成功组装的全长片段(图3).PCA具有良好的兼容性,也被应用于芯片合成的寡核苷酸组装[36-37]. ...
... 经化学合成的寡核苷酸链含有大量错误,对这样的寡核苷酸池的纠错过程主要根据合成错误造成的分子量或基团的差异进行分离纯化.如对柱式合成可通过高效液相色谱法(HPLC)[56]、聚丙烯酰胺凝胶电泳(PAGE)[57]或疏水性纯化柱过滤[10]去除合成不完全的片段.这些方法通量低,对错误区分精确度有限,分离过程损失较大.芯片合成寡核苷酸的纠错可通过直接与序列正确的寡核苷酸捕获探针进行杂交选择[37],或是Tm均一化(热力学参数)严格杂交筛选手段[58],过滤寡核苷酸池中的错误片段.Evonetix的DNA合成平台则通过对温度的控制将合成、组装、纠错进行整合,其纠错过程由精确的温度控制去除非完全匹配的DNA链.还可以使用NGS技术纠错,结合DNA芯片合成与高通量测序平台,将合成、组装、测序纠错一体化[59]. ...
Parallel on-chip gene synthesis and application to optimization of protein expression
1
2011
... 为了提高基因合成通量并降低成本,整合了合成和组装的微型化和自动化基因合成技术也取得了新进展.2011年,Tian等[38]开发了一种采用多功能芯片和组合酶技术的基因合成方法,将整个基因合成过程从寡核苷酸库合成、库扩增、纠错到基因组装等所有步骤整合到同一块芯片上,中途无需更换反应体系,极大地简化了基因合成流程.Twist Bioscience公司以Agilent的寡核苷酸原位合成技术为基础,开发了一套对接式硅片反应器用于自动化基因合成.整合了合成和组装的酶促基因合成技术也取得了新进展.据报道,gSynth酶法DNA合成技术通过合成与组装的循环实现了2.7 kb的pUC19质粒的从头合成. ...
A one pot, one step, precision cloning method with high throughput capability
1
2008
... 对于寡核苷酸组装后双链DNA的进一步拼接,早期的方法依靠限制性内切酶产生的黏性末端来串联DNA片段.由此发展出来的有BioBrick与BglBrick系统以及采用ⅡS型限制性内切酶切割产生黏性末端实现组装的Golden Gate技术[39](图3).但序列依赖性和DNA残痕的引入以及烦琐的操作过程限制了这类方法的应用.利用核酸外切酶、DNA聚合酶与连接酶的单独或协同作用开发的组装方法则摆脱了对限制性内切酶的依赖.这类方法通过产生同源单链互补末端进行组装,包括SLIC[40]、SLiCE[41]、LCR[42]、CPEC[43] 和Gibson组装[44](图3)等多种高效简单的组装方法.其中Gibson组装通过体外一步拼接可以无缝组装长达几十万碱基对的基因组水平的片段. ...
Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC
1
2007
... 对于寡核苷酸组装后双链DNA的进一步拼接,早期的方法依靠限制性内切酶产生的黏性末端来串联DNA片段.由此发展出来的有BioBrick与BglBrick系统以及采用ⅡS型限制性内切酶切割产生黏性末端实现组装的Golden Gate技术[39](图3).但序列依赖性和DNA残痕的引入以及烦琐的操作过程限制了这类方法的应用.利用核酸外切酶、DNA聚合酶与连接酶的单独或协同作用开发的组装方法则摆脱了对限制性内切酶的依赖.这类方法通过产生同源单链互补末端进行组装,包括SLIC[40]、SLiCE[41]、LCR[42]、CPEC[43] 和Gibson组装[44](图3)等多种高效简单的组装方法.其中Gibson组装通过体外一步拼接可以无缝组装长达几十万碱基对的基因组水平的片段. ...
SLiCE: a novel bacterial cell extract-based DNA cloning method
1
2012
... 对于寡核苷酸组装后双链DNA的进一步拼接,早期的方法依靠限制性内切酶产生的黏性末端来串联DNA片段.由此发展出来的有BioBrick与BglBrick系统以及采用ⅡS型限制性内切酶切割产生黏性末端实现组装的Golden Gate技术[39](图3).但序列依赖性和DNA残痕的引入以及烦琐的操作过程限制了这类方法的应用.利用核酸外切酶、DNA聚合酶与连接酶的单独或协同作用开发的组装方法则摆脱了对限制性内切酶的依赖.这类方法通过产生同源单链互补末端进行组装,包括SLIC[40]、SLiCE[41]、LCR[42]、CPEC[43] 和Gibson组装[44](图3)等多种高效简单的组装方法.其中Gibson组装通过体外一步拼接可以无缝组装长达几十万碱基对的基因组水平的片段. ...
Rapid and reliable DNA assembly via ligase cycling reaction
1
2014
... 对于寡核苷酸组装后双链DNA的进一步拼接,早期的方法依靠限制性内切酶产生的黏性末端来串联DNA片段.由此发展出来的有BioBrick与BglBrick系统以及采用ⅡS型限制性内切酶切割产生黏性末端实现组装的Golden Gate技术[39](图3).但序列依赖性和DNA残痕的引入以及烦琐的操作过程限制了这类方法的应用.利用核酸外切酶、DNA聚合酶与连接酶的单独或协同作用开发的组装方法则摆脱了对限制性内切酶的依赖.这类方法通过产生同源单链互补末端进行组装,包括SLIC[40]、SLiCE[41]、LCR[42]、CPEC[43] 和Gibson组装[44](图3)等多种高效简单的组装方法.其中Gibson组装通过体外一步拼接可以无缝组装长达几十万碱基对的基因组水平的片段. ...
Circular polymerase extension cloning of complex gene libraries and pathways
1
2009
... 对于寡核苷酸组装后双链DNA的进一步拼接,早期的方法依靠限制性内切酶产生的黏性末端来串联DNA片段.由此发展出来的有BioBrick与BglBrick系统以及采用ⅡS型限制性内切酶切割产生黏性末端实现组装的Golden Gate技术[39](图3).但序列依赖性和DNA残痕的引入以及烦琐的操作过程限制了这类方法的应用.利用核酸外切酶、DNA聚合酶与连接酶的单独或协同作用开发的组装方法则摆脱了对限制性内切酶的依赖.这类方法通过产生同源单链互补末端进行组装,包括SLIC[40]、SLiCE[41]、LCR[42]、CPEC[43] 和Gibson组装[44](图3)等多种高效简单的组装方法.其中Gibson组装通过体外一步拼接可以无缝组装长达几十万碱基对的基因组水平的片段. ...
Enzymatic assembly of DNA molecules up to several hundred kilobases
1
2009
... 对于寡核苷酸组装后双链DNA的进一步拼接,早期的方法依靠限制性内切酶产生的黏性末端来串联DNA片段.由此发展出来的有BioBrick与BglBrick系统以及采用ⅡS型限制性内切酶切割产生黏性末端实现组装的Golden Gate技术[39](图3).但序列依赖性和DNA残痕的引入以及烦琐的操作过程限制了这类方法的应用.利用核酸外切酶、DNA聚合酶与连接酶的单独或协同作用开发的组装方法则摆脱了对限制性内切酶的依赖.这类方法通过产生同源单链互补末端进行组装,包括SLIC[40]、SLiCE[41]、LCR[42]、CPEC[43] 和Gibson组装[44](图3)等多种高效简单的组装方法.其中Gibson组装通过体外一步拼接可以无缝组装长达几十万碱基对的基因组水平的片段. ...
High molecular weight DNA assembly in vivo for synthetic biology applications
1
2017
... 随着片段长度的增加,DNA在体外很容易受常规操作影响而变得不稳定,超过20 kb片段的拼接更多借助生物体内的重组系统进行(图3).大肠杆菌、枯草芽孢杆菌和酿酒酵母是体内DNA长片段组装的主要宿主细胞[45],经重组系统与细菌人工染色体(BAC)[46]、枯草芽孢杆菌基因组(BGM)[47]或酵母人工染色体(YAC)[48]重组后,这些宿主细胞可稳定携带大片段DNA,其中BGM具有超过3 Mb的克隆能力[49].相较于大肠杆菌和枯草芽孢杆菌,酿酒酵母拥有更高的同源重组率,对长片段的兼容性好,是同时装配多个DNA片段的首选底盘,基于该系统开发的应用方法也更多[50-52].Gibson等[53]在酿酒酵母中一步装配25个DNA片段,形成一个长592 kb的环状支原体基因组.中国科学院的研究人员甚至将接近12 Mb的酿酒酵母完整基因组拼接成单一的染色体[54]. ...
Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector
1
1992
... 随着片段长度的增加,DNA在体外很容易受常规操作影响而变得不稳定,超过20 kb片段的拼接更多借助生物体内的重组系统进行(图3).大肠杆菌、枯草芽孢杆菌和酿酒酵母是体内DNA长片段组装的主要宿主细胞[45],经重组系统与细菌人工染色体(BAC)[46]、枯草芽孢杆菌基因组(BGM)[47]或酵母人工染色体(YAC)[48]重组后,这些宿主细胞可稳定携带大片段DNA,其中BGM具有超过3 Mb的克隆能力[49].相较于大肠杆菌和枯草芽孢杆菌,酿酒酵母拥有更高的同源重组率,对长片段的兼容性好,是同时装配多个DNA片段的首选底盘,基于该系统开发的应用方法也更多[50-52].Gibson等[53]在酿酒酵母中一步装配25个DNA片段,形成一个长592 kb的环状支原体基因组.中国科学院的研究人员甚至将接近12 Mb的酿酒酵母完整基因组拼接成单一的染色体[54]. ...
Conversion of sub-megasized DNA to desired structures using a novel Bacillus subtilis genome vector
1
2003
... 随着片段长度的增加,DNA在体外很容易受常规操作影响而变得不稳定,超过20 kb片段的拼接更多借助生物体内的重组系统进行(图3).大肠杆菌、枯草芽孢杆菌和酿酒酵母是体内DNA长片段组装的主要宿主细胞[45],经重组系统与细菌人工染色体(BAC)[46]、枯草芽孢杆菌基因组(BGM)[47]或酵母人工染色体(YAC)[48]重组后,这些宿主细胞可稳定携带大片段DNA,其中BGM具有超过3 Mb的克隆能力[49].相较于大肠杆菌和枯草芽孢杆菌,酿酒酵母拥有更高的同源重组率,对长片段的兼容性好,是同时装配多个DNA片段的首选底盘,基于该系统开发的应用方法也更多[50-52].Gibson等[53]在酿酒酵母中一步装配25个DNA片段,形成一个长592 kb的环状支原体基因组.中国科学院的研究人员甚至将接近12 Mb的酿酒酵母完整基因组拼接成单一的染色体[54]. ...
Cloning of large segments of exogenous DNA into yeast by means of artificial chromosome vectors
1
1987
... 随着片段长度的增加,DNA在体外很容易受常规操作影响而变得不稳定,超过20 kb片段的拼接更多借助生物体内的重组系统进行(图3).大肠杆菌、枯草芽孢杆菌和酿酒酵母是体内DNA长片段组装的主要宿主细胞[45],经重组系统与细菌人工染色体(BAC)[46]、枯草芽孢杆菌基因组(BGM)[47]或酵母人工染色体(YAC)[48]重组后,这些宿主细胞可稳定携带大片段DNA,其中BGM具有超过3 Mb的克隆能力[49].相较于大肠杆菌和枯草芽孢杆菌,酿酒酵母拥有更高的同源重组率,对长片段的兼容性好,是同时装配多个DNA片段的首选底盘,基于该系统开发的应用方法也更多[50-52].Gibson等[53]在酿酒酵母中一步装配25个DNA片段,形成一个长592 kb的环状支原体基因组.中国科学院的研究人员甚至将接近12 Mb的酿酒酵母完整基因组拼接成单一的染色体[54]. ...
An inducible recA expression Bacillus subtilis genome vector for stable manipulation of large DNA fragments
1
2015
... 随着片段长度的增加,DNA在体外很容易受常规操作影响而变得不稳定,超过20 kb片段的拼接更多借助生物体内的重组系统进行(图3).大肠杆菌、枯草芽孢杆菌和酿酒酵母是体内DNA长片段组装的主要宿主细胞[45],经重组系统与细菌人工染色体(BAC)[46]、枯草芽孢杆菌基因组(BGM)[47]或酵母人工染色体(YAC)[48]重组后,这些宿主细胞可稳定携带大片段DNA,其中BGM具有超过3 Mb的克隆能力[49].相较于大肠杆菌和枯草芽孢杆菌,酿酒酵母拥有更高的同源重组率,对长片段的兼容性好,是同时装配多个DNA片段的首选底盘,基于该系统开发的应用方法也更多[50-52].Gibson等[53]在酿酒酵母中一步装配25个DNA片段,形成一个长592 kb的环状支原体基因组.中国科学院的研究人员甚至将接近12 Mb的酿酒酵母完整基因组拼接成单一的染色体[54]. ...
Synthesis of DNA fragments in yeast by one-step assembly of overlapping oligonucleotides
1
2009
... 随着片段长度的增加,DNA在体外很容易受常规操作影响而变得不稳定,超过20 kb片段的拼接更多借助生物体内的重组系统进行(图3).大肠杆菌、枯草芽孢杆菌和酿酒酵母是体内DNA长片段组装的主要宿主细胞[45],经重组系统与细菌人工染色体(BAC)[46]、枯草芽孢杆菌基因组(BGM)[47]或酵母人工染色体(YAC)[48]重组后,这些宿主细胞可稳定携带大片段DNA,其中BGM具有超过3 Mb的克隆能力[49].相较于大肠杆菌和枯草芽孢杆菌,酿酒酵母拥有更高的同源重组率,对长片段的兼容性好,是同时装配多个DNA片段的首选底盘,基于该系统开发的应用方法也更多[50-52].Gibson等[53]在酿酒酵母中一步装配25个DNA片段,形成一个长592 kb的环状支原体基因组.中国科学院的研究人员甚至将接近12 Mb的酿酒酵母完整基因组拼接成单一的染色体[54]. ...
RADOM, an efficient in vivo method for assembling designed DNA fragments up to 10 kb long in Saccharomyces cerevisiae
0
2015
CasEMBLR: Cas9-facilitated multiloci genomic integration of in vivo assembled DNA parts in Saccharomyces cerevisiae
1
2015
... 随着片段长度的增加,DNA在体外很容易受常规操作影响而变得不稳定,超过20 kb片段的拼接更多借助生物体内的重组系统进行(图3).大肠杆菌、枯草芽孢杆菌和酿酒酵母是体内DNA长片段组装的主要宿主细胞[45],经重组系统与细菌人工染色体(BAC)[46]、枯草芽孢杆菌基因组(BGM)[47]或酵母人工染色体(YAC)[48]重组后,这些宿主细胞可稳定携带大片段DNA,其中BGM具有超过3 Mb的克隆能力[49].相较于大肠杆菌和枯草芽孢杆菌,酿酒酵母拥有更高的同源重组率,对长片段的兼容性好,是同时装配多个DNA片段的首选底盘,基于该系统开发的应用方法也更多[50-52].Gibson等[53]在酿酒酵母中一步装配25个DNA片段,形成一个长592 kb的环状支原体基因组.中国科学院的研究人员甚至将接近12 Mb的酿酒酵母完整基因组拼接成单一的染色体[54]. ...
One-step assembly in yeast of 25 overlapping DNA fragments to form a complete synthetic Mycoplasma genitalium genome
1
2008
... 随着片段长度的增加,DNA在体外很容易受常规操作影响而变得不稳定,超过20 kb片段的拼接更多借助生物体内的重组系统进行(图3).大肠杆菌、枯草芽孢杆菌和酿酒酵母是体内DNA长片段组装的主要宿主细胞[45],经重组系统与细菌人工染色体(BAC)[46]、枯草芽孢杆菌基因组(BGM)[47]或酵母人工染色体(YAC)[48]重组后,这些宿主细胞可稳定携带大片段DNA,其中BGM具有超过3 Mb的克隆能力[49].相较于大肠杆菌和枯草芽孢杆菌,酿酒酵母拥有更高的同源重组率,对长片段的兼容性好,是同时装配多个DNA片段的首选底盘,基于该系统开发的应用方法也更多[50-52].Gibson等[53]在酿酒酵母中一步装配25个DNA片段,形成一个长592 kb的环状支原体基因组.中国科学院的研究人员甚至将接近12 Mb的酿酒酵母完整基因组拼接成单一的染色体[54]. ...
Creating a functional single-chromosome yeast
1
2018
... 随着片段长度的增加,DNA在体外很容易受常规操作影响而变得不稳定,超过20 kb片段的拼接更多借助生物体内的重组系统进行(图3).大肠杆菌、枯草芽孢杆菌和酿酒酵母是体内DNA长片段组装的主要宿主细胞[45],经重组系统与细菌人工染色体(BAC)[46]、枯草芽孢杆菌基因组(BGM)[47]或酵母人工染色体(YAC)[48]重组后,这些宿主细胞可稳定携带大片段DNA,其中BGM具有超过3 Mb的克隆能力[49].相较于大肠杆菌和枯草芽孢杆菌,酿酒酵母拥有更高的同源重组率,对长片段的兼容性好,是同时装配多个DNA片段的首选底盘,基于该系统开发的应用方法也更多[50-52].Gibson等[53]在酿酒酵母中一步装配25个DNA片段,形成一个长592 kb的环状支原体基因组.中国科学院的研究人员甚至将接近12 Mb的酿酒酵母完整基因组拼接成单一的染色体[54]. ...
A systematic comparison of error correction enzymes by next-generation sequencing
2
2017
... 寡核苷酸合成与酶促组装过程都不可避免地产生多种类型的错误.常见的错误包括核苷酸的插入、缺失和取代.纠错技术的使用可有效地去除不同类型的错误从而提高合成产物的正确率[55]. ...
... 经互补配对后的双链DNA片段的核苷酸插入、缺失、取代错误主要表现为错配和凸起等,这些错误的去除更多是借助基于生物体内的DNA修复体系开发出的DNA酶促纠错技术[60-61].通过对互补序列退火,暴露出错配信号,再利用具有错配结合或错配切割活性的酶对DNA双链纠错(图4),从而富集正确序列[10,55]. ...
Analysis and purification of synthetic nucleic acids using HPLC
1
2015
... 经化学合成的寡核苷酸链含有大量错误,对这样的寡核苷酸池的纠错过程主要根据合成错误造成的分子量或基团的差异进行分离纯化.如对柱式合成可通过高效液相色谱法(HPLC)[56]、聚丙烯酰胺凝胶电泳(PAGE)[57]或疏水性纯化柱过滤[10]去除合成不完全的片段.这些方法通量低,对错误区分精确度有限,分离过程损失较大.芯片合成寡核苷酸的纠错可通过直接与序列正确的寡核苷酸捕获探针进行杂交选择[37],或是Tm均一化(热力学参数)严格杂交筛选手段[58],过滤寡核苷酸池中的错误片段.Evonetix的DNA合成平台则通过对温度的控制将合成、组装、纠错进行整合,其纠错过程由精确的温度控制去除非完全匹配的DNA链.还可以使用NGS技术纠错,结合DNA芯片合成与高通量测序平台,将合成、组装、测序纠错一体化[59]. ...
Introduction to the synthesis and purification of oligonucleotides
1
2000
... 经化学合成的寡核苷酸链含有大量错误,对这样的寡核苷酸池的纠错过程主要根据合成错误造成的分子量或基团的差异进行分离纯化.如对柱式合成可通过高效液相色谱法(HPLC)[56]、聚丙烯酰胺凝胶电泳(PAGE)[57]或疏水性纯化柱过滤[10]去除合成不完全的片段.这些方法通量低,对错误区分精确度有限,分离过程损失较大.芯片合成寡核苷酸的纠错可通过直接与序列正确的寡核苷酸捕获探针进行杂交选择[37],或是Tm均一化(热力学参数)严格杂交筛选手段[58],过滤寡核苷酸池中的错误片段.Evonetix的DNA合成平台则通过对温度的控制将合成、组装、纠错进行整合,其纠错过程由精确的温度控制去除非完全匹配的DNA链.还可以使用NGS技术纠错,结合DNA芯片合成与高通量测序平台,将合成、组装、测序纠错一体化[59]. ...
High-quality gene assembly directly from unpurified mixtures of microarray-synthesized oligonucleotides
1
2010
... 经化学合成的寡核苷酸链含有大量错误,对这样的寡核苷酸池的纠错过程主要根据合成错误造成的分子量或基团的差异进行分离纯化.如对柱式合成可通过高效液相色谱法(HPLC)[56]、聚丙烯酰胺凝胶电泳(PAGE)[57]或疏水性纯化柱过滤[10]去除合成不完全的片段.这些方法通量低,对错误区分精确度有限,分离过程损失较大.芯片合成寡核苷酸的纠错可通过直接与序列正确的寡核苷酸捕获探针进行杂交选择[37],或是Tm均一化(热力学参数)严格杂交筛选手段[58],过滤寡核苷酸池中的错误片段.Evonetix的DNA合成平台则通过对温度的控制将合成、组装、纠错进行整合,其纠错过程由精确的温度控制去除非完全匹配的DNA链.还可以使用NGS技术纠错,结合DNA芯片合成与高通量测序平台,将合成、组装、测序纠错一体化[59]. ...
High-fidelity gene synthesis by retrieval of sequence-verified DNA identified using high-throughput pyrosequencing
1
2010
... 经化学合成的寡核苷酸链含有大量错误,对这样的寡核苷酸池的纠错过程主要根据合成错误造成的分子量或基团的差异进行分离纯化.如对柱式合成可通过高效液相色谱法(HPLC)[56]、聚丙烯酰胺凝胶电泳(PAGE)[57]或疏水性纯化柱过滤[10]去除合成不完全的片段.这些方法通量低,对错误区分精确度有限,分离过程损失较大.芯片合成寡核苷酸的纠错可通过直接与序列正确的寡核苷酸捕获探针进行杂交选择[37],或是Tm均一化(热力学参数)严格杂交筛选手段[58],过滤寡核苷酸池中的错误片段.Evonetix的DNA合成平台则通过对温度的控制将合成、组装、纠错进行整合,其纠错过程由精确的温度控制去除非完全匹配的DNA链.还可以使用NGS技术纠错,结合DNA芯片合成与高通量测序平台,将合成、组装、测序纠错一体化[59]. ...
Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints
1
2004
... 经互补配对后的双链DNA片段的核苷酸插入、缺失、取代错误主要表现为错配和凸起等,这些错误的去除更多是借助基于生物体内的DNA修复体系开发出的DNA酶促纠错技术[60-61].通过对互补序列退火,暴露出错配信号,再利用具有错配结合或错配切割活性的酶对DNA双链纠错(图4),从而富集正确序列[10,55]. ...
The multifaceted mismatch-repair system
1
2006
... 经互补配对后的双链DNA片段的核苷酸插入、缺失、取代错误主要表现为错配和凸起等,这些错误的去除更多是借助基于生物体内的DNA修复体系开发出的DNA酶促纠错技术[60-61].通过对互补序列退火,暴露出错配信号,再利用具有错配结合或错配切割活性的酶对DNA双链纠错(图4),从而富集正确序列[10,55]. ...
Single-molecule views of MutS on mismatched DNA
1
2014
... 参与DNA修复的错配结合酶MutS及其同源蛋白可以识别并结合各种含错配碱基与单链环的DNA[62-63],然后可通过凝胶电泳、毛细管电泳、亲和磁珠或吸附树脂等方法使MutS结合含错配的异源双链与未被结合的同质双链分离.这样两轮重复后,可将错误率降低到1/10 kb,与传统的基因合成技术相比错误降低超过15倍[64-65].针对错配结合酶MutS的工程改造,在提其高稳定性的同时也有助于市场化应用[66].对于错误率高的寡核苷酸池,Binkowski等[67]利用MutS进一步开发了同序改组法,通过引入限制性内切酶对DNA双链进行片段化,使得MutS不需要去除整条错误的双链DNA,从而保留大量含有正确序列的短片段,最后通过OE-PCR组装回收全长序列.测试发现3.5 kb的片段经过两轮同序改组后错误率降低至1/3.5 kb,正确率提高了3.5~4.3倍.MutS纠错方法本质是对含错DNA双链的物理分离.为保证MutS处理后样品中有足够的正确片段,寡核苷酸池中需要有相当一部分序列正确的片段. ...
DNA mismatch repair
1
2005
... 参与DNA修复的错配结合酶MutS及其同源蛋白可以识别并结合各种含错配碱基与单链环的DNA[62-63],然后可通过凝胶电泳、毛细管电泳、亲和磁珠或吸附树脂等方法使MutS结合含错配的异源双链与未被结合的同质双链分离.这样两轮重复后,可将错误率降低到1/10 kb,与传统的基因合成技术相比错误降低超过15倍[64-65].针对错配结合酶MutS的工程改造,在提其高稳定性的同时也有助于市场化应用[66].对于错误率高的寡核苷酸池,Binkowski等[67]利用MutS进一步开发了同序改组法,通过引入限制性内切酶对DNA双链进行片段化,使得MutS不需要去除整条错误的双链DNA,从而保留大量含有正确序列的短片段,最后通过OE-PCR组装回收全长序列.测试发现3.5 kb的片段经过两轮同序改组后错误率降低至1/3.5 kb,正确率提高了3.5~4.3倍.MutS纠错方法本质是对含错DNA双链的物理分离.为保证MutS处理后样品中有足够的正确片段,寡核苷酸池中需要有相当一部分序列正确的片段. ...
Protein-mediated error correction for de novo DNA synthesis
1
2004
... 参与DNA修复的错配结合酶MutS及其同源蛋白可以识别并结合各种含错配碱基与单链环的DNA[62-63],然后可通过凝胶电泳、毛细管电泳、亲和磁珠或吸附树脂等方法使MutS结合含错配的异源双链与未被结合的同质双链分离.这样两轮重复后,可将错误率降低到1/10 kb,与传统的基因合成技术相比错误降低超过15倍[64-65].针对错配结合酶MutS的工程改造,在提其高稳定性的同时也有助于市场化应用[66].对于错误率高的寡核苷酸池,Binkowski等[67]利用MutS进一步开发了同序改组法,通过引入限制性内切酶对DNA双链进行片段化,使得MutS不需要去除整条错误的双链DNA,从而保留大量含有正确序列的短片段,最后通过OE-PCR组装回收全长序列.测试发现3.5 kb的片段经过两轮同序改组后错误率降低至1/3.5 kb,正确率提高了3.5~4.3倍.MutS纠错方法本质是对含错DNA双链的物理分离.为保证MutS处理后样品中有足够的正确片段,寡核苷酸池中需要有相当一部分序列正确的片段. ...
Error removal in microchip-synthesized DNA using immobilized MutS
1
2014
... 参与DNA修复的错配结合酶MutS及其同源蛋白可以识别并结合各种含错配碱基与单链环的DNA[62-63],然后可通过凝胶电泳、毛细管电泳、亲和磁珠或吸附树脂等方法使MutS结合含错配的异源双链与未被结合的同质双链分离.这样两轮重复后,可将错误率降低到1/10 kb,与传统的基因合成技术相比错误降低超过15倍[64-65].针对错配结合酶MutS的工程改造,在提其高稳定性的同时也有助于市场化应用[66].对于错误率高的寡核苷酸池,Binkowski等[67]利用MutS进一步开发了同序改组法,通过引入限制性内切酶对DNA双链进行片段化,使得MutS不需要去除整条错误的双链DNA,从而保留大量含有正确序列的短片段,最后通过OE-PCR组装回收全长序列.测试发现3.5 kb的片段经过两轮同序改组后错误率降低至1/3.5 kb,正确率提高了3.5~4.3倍.MutS纠错方法本质是对含错DNA双链的物理分离.为保证MutS处理后样品中有足够的正确片段,寡核苷酸池中需要有相当一部分序列正确的片段. ...
Efficient and low-cost error removal in DNA synthesis by a high-durability MutS
1
2020
... 参与DNA修复的错配结合酶MutS及其同源蛋白可以识别并结合各种含错配碱基与单链环的DNA[62-63],然后可通过凝胶电泳、毛细管电泳、亲和磁珠或吸附树脂等方法使MutS结合含错配的异源双链与未被结合的同质双链分离.这样两轮重复后,可将错误率降低到1/10 kb,与传统的基因合成技术相比错误降低超过15倍[64-65].针对错配结合酶MutS的工程改造,在提其高稳定性的同时也有助于市场化应用[66].对于错误率高的寡核苷酸池,Binkowski等[67]利用MutS进一步开发了同序改组法,通过引入限制性内切酶对DNA双链进行片段化,使得MutS不需要去除整条错误的双链DNA,从而保留大量含有正确序列的短片段,最后通过OE-PCR组装回收全长序列.测试发现3.5 kb的片段经过两轮同序改组后错误率降低至1/3.5 kb,正确率提高了3.5~4.3倍.MutS纠错方法本质是对含错DNA双链的物理分离.为保证MutS处理后样品中有足够的正确片段,寡核苷酸池中需要有相当一部分序列正确的片段. ...
Correcting errors in synthetic DNA through consensus shuffling
1
2005
... 参与DNA修复的错配结合酶MutS及其同源蛋白可以识别并结合各种含错配碱基与单链环的DNA[62-63],然后可通过凝胶电泳、毛细管电泳、亲和磁珠或吸附树脂等方法使MutS结合含错配的异源双链与未被结合的同质双链分离.这样两轮重复后,可将错误率降低到1/10 kb,与传统的基因合成技术相比错误降低超过15倍[64-65].针对错配结合酶MutS的工程改造,在提其高稳定性的同时也有助于市场化应用[66].对于错误率高的寡核苷酸池,Binkowski等[67]利用MutS进一步开发了同序改组法,通过引入限制性内切酶对DNA双链进行片段化,使得MutS不需要去除整条错误的双链DNA,从而保留大量含有正确序列的短片段,最后通过OE-PCR组装回收全长序列.测试发现3.5 kb的片段经过两轮同序改组后错误率降低至1/3.5 kb,正确率提高了3.5~4.3倍.MutS纠错方法本质是对含错DNA双链的物理分离.为保证MutS处理后样品中有足够的正确片段,寡核苷酸池中需要有相当一部分序列正确的片段. ...
Mismatch cleavage by single-strand specific nucleases
1
2004
... 运用错配切割酶能达到在原处理池中纠正错误的目的.错配切割酶是识别DNA双链错配位点并在错配位点附近切割的一组错配特异性核酸内切酶.主要包括识别单碱基错配的核酸内切酶和单链特异性核酸酶[68-70],这些酶与聚合酶共同作用,利用具有核酸外切酶活性的聚合酶水解切割错误区域,然后通过OE-PCR组装回收全长序列.这种方法可以消除单碱基水平的错误,同时保留大部分序列正确的区域,并且可以进行多次“纠错-组装”循环,直到获得所需纯度的产品.T4核酸内切酶Ⅶ,T7核酸内切酶Ⅰ和E.coli核酸内切酶Ⅴ等可识别并切割双链DNA中单碱基错配、单核苷酸凸起等类型的错误,能有效减少合成基因产物中错配、缺失和插入等错误[69,71-73].单链特异性核酸酶中CEL可在中性pH值下特异性切割不同类型的碱基错配和DNA畸变[74-76].在芯片的基因合成纠错中结合使用CEL核酸酶可以将合成基因产物的错误率从1/526 bp减少到1/3883 bp.两次酶解错误切割反应可进一步将错误率降低至1/8700 bp,错误减少了16倍以上[77]. ...
Removal of mismatched bases from synthetic genes by enzymatic mismatch cleavage
1
2005
... 运用错配切割酶能达到在原处理池中纠正错误的目的.错配切割酶是识别DNA双链错配位点并在错配位点附近切割的一组错配特异性核酸内切酶.主要包括识别单碱基错配的核酸内切酶和单链特异性核酸酶[68-70],这些酶与聚合酶共同作用,利用具有核酸外切酶活性的聚合酶水解切割错误区域,然后通过OE-PCR组装回收全长序列.这种方法可以消除单碱基水平的错误,同时保留大部分序列正确的区域,并且可以进行多次“纠错-组装”循环,直到获得所需纯度的产品.T4核酸内切酶Ⅶ,T7核酸内切酶Ⅰ和E.coli核酸内切酶Ⅴ等可识别并切割双链DNA中单碱基错配、单核苷酸凸起等类型的错误,能有效减少合成基因产物中错配、缺失和插入等错误[69,71-73].单链特异性核酸酶中CEL可在中性pH值下特异性切割不同类型的碱基错配和DNA畸变[74-76].在芯片的基因合成纠错中结合使用CEL核酸酶可以将合成基因产物的错误率从1/526 bp减少到1/3883 bp.两次酶解错误切割反应可进一步将错误率降低至1/8700 bp,错误减少了16倍以上[77]. ...
Single-strand-specific nucleases
1
2003
... 运用错配切割酶能达到在原处理池中纠正错误的目的.错配切割酶是识别DNA双链错配位点并在错配位点附近切割的一组错配特异性核酸内切酶.主要包括识别单碱基错配的核酸内切酶和单链特异性核酸酶[68-70],这些酶与聚合酶共同作用,利用具有核酸外切酶活性的聚合酶水解切割错误区域,然后通过OE-PCR组装回收全长序列.这种方法可以消除单碱基水平的错误,同时保留大部分序列正确的区域,并且可以进行多次“纠错-组装”循环,直到获得所需纯度的产品.T4核酸内切酶Ⅶ,T7核酸内切酶Ⅰ和E.coli核酸内切酶Ⅴ等可识别并切割双链DNA中单碱基错配、单核苷酸凸起等类型的错误,能有效减少合成基因产物中错配、缺失和插入等错误[69,71-73].单链特异性核酸酶中CEL可在中性pH值下特异性切割不同类型的碱基错配和DNA畸变[74-76].在芯片的基因合成纠错中结合使用CEL核酸酶可以将合成基因产物的错误率从1/526 bp减少到1/3883 bp.两次酶解错误切割反应可进一步将错误率降低至1/8700 bp,错误减少了16倍以上[77]. ...
T7 Endonuclease I mediates error correction in artificial gene synthesis
1
2016
... 运用错配切割酶能达到在原处理池中纠正错误的目的.错配切割酶是识别DNA双链错配位点并在错配位点附近切割的一组错配特异性核酸内切酶.主要包括识别单碱基错配的核酸内切酶和单链特异性核酸酶[68-70],这些酶与聚合酶共同作用,利用具有核酸外切酶活性的聚合酶水解切割错误区域,然后通过OE-PCR组装回收全长序列.这种方法可以消除单碱基水平的错误,同时保留大部分序列正确的区域,并且可以进行多次“纠错-组装”循环,直到获得所需纯度的产品.T4核酸内切酶Ⅶ,T7核酸内切酶Ⅰ和E.coli核酸内切酶Ⅴ等可识别并切割双链DNA中单碱基错配、单核苷酸凸起等类型的错误,能有效减少合成基因产物中错配、缺失和插入等错误[69,71-73].单链特异性核酸酶中CEL可在中性pH值下特异性切割不同类型的碱基错配和DNA畸变[74-76].在芯片的基因合成纠错中结合使用CEL核酸酶可以将合成基因产物的错误率从1/526 bp减少到1/3883 bp.两次酶解错误切割反应可进一步将错误率降低至1/8700 bp,错误减少了16倍以上[77]. ...
Gene synthesis by circular assembly amplification
0
2008
Mutation detection using fluorescent enzyme mismatch cleavage with T4 endonuclease Ⅶ
1
1999
... 运用错配切割酶能达到在原处理池中纠正错误的目的.错配切割酶是识别DNA双链错配位点并在错配位点附近切割的一组错配特异性核酸内切酶.主要包括识别单碱基错配的核酸内切酶和单链特异性核酸酶[68-70],这些酶与聚合酶共同作用,利用具有核酸外切酶活性的聚合酶水解切割错误区域,然后通过OE-PCR组装回收全长序列.这种方法可以消除单碱基水平的错误,同时保留大部分序列正确的区域,并且可以进行多次“纠错-组装”循环,直到获得所需纯度的产品.T4核酸内切酶Ⅶ,T7核酸内切酶Ⅰ和E.coli核酸内切酶Ⅴ等可识别并切割双链DNA中单碱基错配、单核苷酸凸起等类型的错误,能有效减少合成基因产物中错配、缺失和插入等错误[69,71-73].单链特异性核酸酶中CEL可在中性pH值下特异性切割不同类型的碱基错配和DNA畸变[74-76].在芯片的基因合成纠错中结合使用CEL核酸酶可以将合成基因产物的错误率从1/526 bp减少到1/3883 bp.两次酶解错误切割反应可进一步将错误率降低至1/8700 bp,错误减少了16倍以上[77]. ...
Enzymatic mutation detection technologies
1
2005
... 运用错配切割酶能达到在原处理池中纠正错误的目的.错配切割酶是识别DNA双链错配位点并在错配位点附近切割的一组错配特异性核酸内切酶.主要包括识别单碱基错配的核酸内切酶和单链特异性核酸酶[68-70],这些酶与聚合酶共同作用,利用具有核酸外切酶活性的聚合酶水解切割错误区域,然后通过OE-PCR组装回收全长序列.这种方法可以消除单碱基水平的错误,同时保留大部分序列正确的区域,并且可以进行多次“纠错-组装”循环,直到获得所需纯度的产品.T4核酸内切酶Ⅶ,T7核酸内切酶Ⅰ和E.coli核酸内切酶Ⅴ等可识别并切割双链DNA中单碱基错配、单核苷酸凸起等类型的错误,能有效减少合成基因产物中错配、缺失和插入等错误[69,71-73].单链特异性核酸酶中CEL可在中性pH值下特异性切割不同类型的碱基错配和DNA畸变[74-76].在芯片的基因合成纠错中结合使用CEL核酸酶可以将合成基因产物的错误率从1/526 bp减少到1/3883 bp.两次酶解错误切割反应可进一步将错误率降低至1/8700 bp,错误减少了16倍以上[77]. ...
Mutation detection using a novel plant endonuclease
0
1998
Purification, cloning, and characterization of the CEL I nuclease
1
2000
... 运用错配切割酶能达到在原处理池中纠正错误的目的.错配切割酶是识别DNA双链错配位点并在错配位点附近切割的一组错配特异性核酸内切酶.主要包括识别单碱基错配的核酸内切酶和单链特异性核酸酶[68-70],这些酶与聚合酶共同作用,利用具有核酸外切酶活性的聚合酶水解切割错误区域,然后通过OE-PCR组装回收全长序列.这种方法可以消除单碱基水平的错误,同时保留大部分序列正确的区域,并且可以进行多次“纠错-组装”循环,直到获得所需纯度的产品.T4核酸内切酶Ⅶ,T7核酸内切酶Ⅰ和E.coli核酸内切酶Ⅴ等可识别并切割双链DNA中单碱基错配、单核苷酸凸起等类型的错误,能有效减少合成基因产物中错配、缺失和插入等错误[69,71-73].单链特异性核酸酶中CEL可在中性pH值下特异性切割不同类型的碱基错配和DNA畸变[74-76].在芯片的基因合成纠错中结合使用CEL核酸酶可以将合成基因产物的错误率从1/526 bp减少到1/3883 bp.两次酶解错误切割反应可进一步将错误率降低至1/8700 bp,错误减少了16倍以上[77]. ...
Error correction of microchip synthesized genes using Surveyor nuclease
1
2012
... 运用错配切割酶能达到在原处理池中纠正错误的目的.错配切割酶是识别DNA双链错配位点并在错配位点附近切割的一组错配特异性核酸内切酶.主要包括识别单碱基错配的核酸内切酶和单链特异性核酸酶[68-70],这些酶与聚合酶共同作用,利用具有核酸外切酶活性的聚合酶水解切割错误区域,然后通过OE-PCR组装回收全长序列.这种方法可以消除单碱基水平的错误,同时保留大部分序列正确的区域,并且可以进行多次“纠错-组装”循环,直到获得所需纯度的产品.T4核酸内切酶Ⅶ,T7核酸内切酶Ⅰ和E.coli核酸内切酶Ⅴ等可识别并切割双链DNA中单碱基错配、单核苷酸凸起等类型的错误,能有效减少合成基因产物中错配、缺失和插入等错误[69,71-73].单链特异性核酸酶中CEL可在中性pH值下特异性切割不同类型的碱基错配和DNA畸变[74-76].在芯片的基因合成纠错中结合使用CEL核酸酶可以将合成基因产物的错误率从1/526 bp减少到1/3883 bp.两次酶解错误切割反应可进一步将错误率降低至1/8700 bp,错误减少了16倍以上[77]. ...
Deep functional analysis of synⅡ, a 770-kilobase synthetic yeast chromosome
1
2017
... 近年来,DNA合成、组装与错误纠正技术的不断发展,使染色体或基因组的合成、人工设计基因组的创造、可控细胞工厂与人工生物的构建都成为可能[78-82].这些研究也推动了合成生物学的快速发展,其中,设计构建新功能基因、遗传网络甚至基因组,以实现从头合成、按需合成和生物大分子的定向改造的时代已经到来.各界对高效保真的DNA合成技术,高通量的组装与错误校正体系开发等基因合成相关技术的需求更为强烈.DNA合成技术在生物医疗、生物制造、DNA存储等诸多领域具有广阔应用前景与巨大的市场潜力[83-84],预计2030年全球DNA合成市场将增长到1.6万亿美元.然而2018年11月,合成生物学被美国列为拟限制出口的前沿生物技术领域之一,高性能DNA合成仪已禁止向中国销售,这将严重影响我国合成生物学的健康发展.因此,推动DNA合成、组装、纠错等技术的发展,开发长片段高效无差错的微量DNA合成技术,利用计算机设计序列优化组装过程,结合酶促或NGS测序技术开发DNA合成纠错平台,以绿色、高通量、自动化和一体化的方式低成本高质量地合成DNA,实现大规模基因和基因组合成,具有重要科学意义和重大应用价值. ...
Bug mapping and fitness testing of chemically synthesized chromosome Ⅹ
0
2017
‘Perfect’ designer chromosome V and behavior of a ring derivative
0
2017
Engineering the ribosomal DNA in a megabase synthetic chromosome
0
2017
Design of a synthetic yeast genome
1
2017
... 近年来,DNA合成、组装与错误纠正技术的不断发展,使染色体或基因组的合成、人工设计基因组的创造、可控细胞工厂与人工生物的构建都成为可能[78-82].这些研究也推动了合成生物学的快速发展,其中,设计构建新功能基因、遗传网络甚至基因组,以实现从头合成、按需合成和生物大分子的定向改造的时代已经到来.各界对高效保真的DNA合成技术,高通量的组装与错误校正体系开发等基因合成相关技术的需求更为强烈.DNA合成技术在生物医疗、生物制造、DNA存储等诸多领域具有广阔应用前景与巨大的市场潜力[83-84],预计2030年全球DNA合成市场将增长到1.6万亿美元.然而2018年11月,合成生物学被美国列为拟限制出口的前沿生物技术领域之一,高性能DNA合成仪已禁止向中国销售,这将严重影响我国合成生物学的健康发展.因此,推动DNA合成、组装、纠错等技术的发展,开发长片段高效无差错的微量DNA合成技术,利用计算机设计序列优化组装过程,结合酶促或NGS测序技术开发DNA合成纠错平台,以绿色、高通量、自动化和一体化的方式低成本高质量地合成DNA,实现大规模基因和基因组合成,具有重要科学意义和重大应用价值. ...
DNA nanotechnology
1
2017
... 近年来,DNA合成、组装与错误纠正技术的不断发展,使染色体或基因组的合成、人工设计基因组的创造、可控细胞工厂与人工生物的构建都成为可能[78-82].这些研究也推动了合成生物学的快速发展,其中,设计构建新功能基因、遗传网络甚至基因组,以实现从头合成、按需合成和生物大分子的定向改造的时代已经到来.各界对高效保真的DNA合成技术,高通量的组装与错误校正体系开发等基因合成相关技术的需求更为强烈.DNA合成技术在生物医疗、生物制造、DNA存储等诸多领域具有广阔应用前景与巨大的市场潜力[83-84],预计2030年全球DNA合成市场将增长到1.6万亿美元.然而2018年11月,合成生物学被美国列为拟限制出口的前沿生物技术领域之一,高性能DNA合成仪已禁止向中国销售,这将严重影响我国合成生物学的健康发展.因此,推动DNA合成、组装、纠错等技术的发展,开发长片段高效无差错的微量DNA合成技术,利用计算机设计序列优化组装过程,结合酶促或NGS测序技术开发DNA合成纠错平台,以绿色、高通量、自动化和一体化的方式低成本高质量地合成DNA,实现大规模基因和基因组合成,具有重要科学意义和重大应用价值. ...
Towards practical, high-capacity, low-maintenance information storage in synthesized DNA
1
2013
... 近年来,DNA合成、组装与错误纠正技术的不断发展,使染色体或基因组的合成、人工设计基因组的创造、可控细胞工厂与人工生物的构建都成为可能[78-82].这些研究也推动了合成生物学的快速发展,其中,设计构建新功能基因、遗传网络甚至基因组,以实现从头合成、按需合成和生物大分子的定向改造的时代已经到来.各界对高效保真的DNA合成技术,高通量的组装与错误校正体系开发等基因合成相关技术的需求更为强烈.DNA合成技术在生物医疗、生物制造、DNA存储等诸多领域具有广阔应用前景与巨大的市场潜力[83-84],预计2030年全球DNA合成市场将增长到1.6万亿美元.然而2018年11月,合成生物学被美国列为拟限制出口的前沿生物技术领域之一,高性能DNA合成仪已禁止向中国销售,这将严重影响我国合成生物学的健康发展.因此,推动DNA合成、组装、纠错等技术的发展,开发长片段高效无差错的微量DNA合成技术,利用计算机设计序列优化组装过程,结合酶促或NGS测序技术开发DNA合成纠错平台,以绿色、高通量、自动化和一体化的方式低成本高质量地合成DNA,实现大规模基因和基因组合成,具有重要科学意义和重大应用价值. ...







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}