合成生物学 ›› 2025, Vol. 6 ›› Issue (3): 566-584.DOI: 10.12211/2096-8280.2024-090
机器学习驱动的基因组规模代谢模型构建与优化
吴柯1,2, 罗家豪1,2, 李斐然1,2
- 1.清华大学深圳国际研究生院,生物医药与健康工程研究院,广东 深圳 518055
2.清华大学化学工程系,工业生物催化教育部重点实验室,北京 100084
-
收稿日期:2024-12-02修回日期:2025-02-12出版日期:2025-06-30发布日期:2025-06-27 -
通讯作者:李斐然 -
作者简介:吴柯 (2000—),男,博士研究生。研究方向为机器学习辅助基因组规模代谢模型开发。 E-mail:wk37@tju.edu.cn李斐然 (1993—),女,助理教授,博士生导师。研究方向为基因组规模代谢模型开发、微生物细胞工厂设计、酶参数预测,致力于构建数字生命模型。 E-mail:feiranli@sz.tsinghua.edu.cn -
基金资助:国家自然科学基金面上项目(22478223)
Applications of machine learning in the reconstruction and curation of genome-scale metabolic models
WU Ke1,2, LUO Jiahao1,2, LI Feiran1,2
- 1.Institute of Biopharmaceutical and Health Engineering,Tsinghua Shenzhen International Graduate School,Tsinghua University,Shenzhen 518055,Guangdong,China
2.Key Laboratory for Industrial Biocatalysis,Ministry of Education,Department of Chemical Engineering,Tsinghua University,Beijing 100084,China
-
Received:2024-12-02Revised:2025-02-12Online:2025-06-30Published:2025-06-27 -
Contact:LI Feiran
摘要:
自1999年首个基因组规模代谢模型(genome-scale metabolic model,GEM)问世以来,GEM已成为解析生物代谢的重要工具。该模型包含代谢基因、代谢物和反应,并结合化学计量矩阵与约束优化,系统描述和模拟生物体内的代谢过程。此外,GEM能够整合热力学参数、动力学参数、多组学数据及多细胞过程,构建更精细且具有更强大预测能力的多约束多过程模型。然而,先验知识的局限成为其发展的瓶颈。机器学习技术凭借强大的数据处理和模式识别能力,为进一步扩展GEM提供了新思路。本综述系统总结了传统GEM及多约束多过程模型的构建流程,并着重探讨了机器学习在其中关键步骤中的应用前景,如基因功能注释、途径解析、空缺填补和生物学参数预测。机器学习技术作为新的驱动力,有望大幅度提升GEM的规模和质量,深化对生物代谢机制的理解,并推动实现数字孪生细胞。
中图分类号:
引用本文
吴柯, 罗家豪, 李斐然. 机器学习驱动的基因组规模代谢模型构建与优化[J]. 合成生物学, 2025, 6(3): 566-584.
WU Ke, LUO Jiahao, LI Feiran. Applications of machine learning in the reconstruction and curation of genome-scale metabolic models[J]. Synthetic Biology Journal, 2025, 6(3): 566-584.
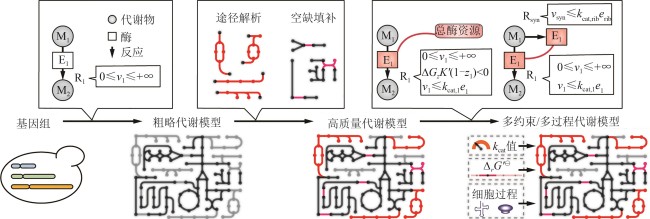
图1 GEM以及多约束多过程模型的构建过程
Fig. 1 Process for reconstructing GEMs and multi-constraint and multi-process models
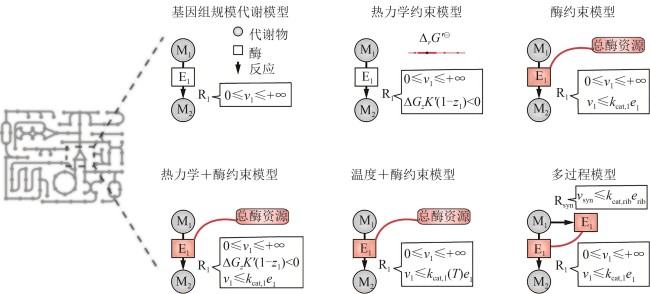
图2 基因组规模代谢模型与多约束多过程模型的构建框架
Fig.2 Framework for constructing GEMs and multi-constraint and multi-process models
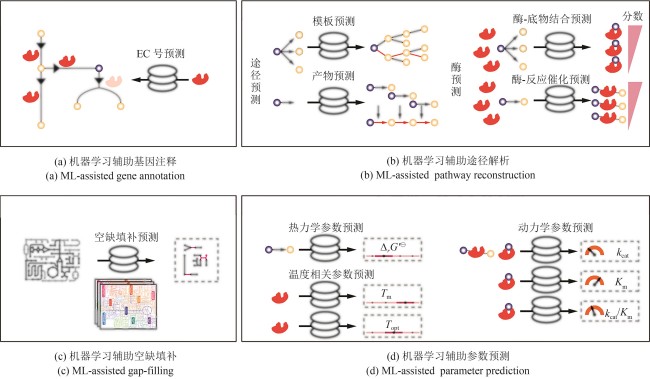
图3 机器学习辅助基因组规模代谢模型与多约束多过程模型构建
Fig. 3 Machine learning-aided reconstruction of GEMs and multi-constraint and multi-process models
| 模型应用 | 方法 | 模型框架 | 输入 | 输出 | 特点 |
|---|---|---|---|---|---|
辅助基因 注释 | DeepEC[ | CNN | 氨基酸序列 | EC编号 | 可区分酶与非酶,无法进行多功能注释 |
| CLEAN[ | 预训练蛋白质大语言模型(ESM-1b),对比学习 | 氨基酸序列 | EC编号 | 无法区分酶与非酶,可进行多功能注释,可用于数据极少的EC编号 | |
| DeepECtransformer[ | 预训练蛋白质大语言模型(ProtBert),Transformer | 氨基酸序列 | EC编号 | 可区分酶与非酶,可进行多功能注释,不可用于数据极少的EC编号 | |
| ECRECer[ | 预训练蛋白质大语言模型(ESM-1b),GRU | 氨基酸序列 | EC编号 | 可区分酶与非酶,可进行多功能注释 | |
| ECPICK[ | One-hot,CNN | 氨基酸序列 | EC编号 | 无法区分酶与非酶,不可进行多功能注释,可输出预测EC编号的置信度 | |
| EnzBert[ | Transformer | 氨基酸序列 | EC编号 | 无法区分酶与非酶,不可进行多功能注释,可推测关键残基 | |
| EnzymeNet[ | CNN,ResNet | 氨基酸序列 | EC编号 | 可区分酶与非酶,无法进行多功能注释 | |
| GraphEC[ | 预训练蛋白质大语言模型(ProtTrans),ESMFold | 氨基酸序列和蛋白质三维结构 | EC编号 | 无法区分酶与非酶,可进行多功能注释,计算资源需求高 | |
| 辅助途径解析-反应预测 | RetroPath RL[ | 基于蒙特卡洛树搜索的强化学习方法 | 化合物SMILES | 合成途径 | 缓解组合爆炸问题,允许探索深度是只基于模板版本的两倍以上 |
| ASKCOS[ | FNN | 化合物SMILES | 合成途径 | 缓解组合爆炸问题,适用于化学合成途径设计 | |
| chemoenzymatic-ASKCOS[ | DNN | 化合物SMILES | 合成途径 | 缓解组合爆炸问题,可进行化学合成与生物合成混合途径设计 | |
| RetroBioCat[ | DNN | 化合物SMILES | 合成途径 | 缓解组合爆炸问题, 处理大分子化合物存在挑战 | |
| Kreutter等开发的方法[ | Transformer | 化合物SMILES和酶功能描述 | 反应产物 | 预测单步反应,无法给出完整的反应,无法推荐酶功能 | |
| Probst等[ | Transformer | 化合物SMILES和EC编号 | 反应产物 | 预测单步反应,无法给出完整的反应,无法推荐EC编号 | |
| BioNavi-NP[ | Transformer | 天然产SMILES | 合成途径 | 针对天然产物及类似物,可预测多步途径 | |
| BioNavi[ | Transformer | 化合物SMILES | 合成途径 | 可进行化学合成与生物合成混合途径设计 | |
| 辅助途径解析-酶挖掘 | ESP[ | 预训练蛋白质大语言模型(ESM-1b),GNN,XGBoost | 氨基酸序列和分子指纹 | 酶-底物结合可能性 | 对于训练集中没出现代谢物的预测性能会有明显下降 |
| EnzRank[ | 分子指纹,CNN | 氨基酸序列和分子指纹 | 酶-底物结合可能性 | 对于天然底物及其相似物具有良好的区分能力 | |
| PU-EPP[ | GNN,正样本和无标签 学习 | 氨基酸序列和化合物SMILES | 酶-底物结合可能性 | 鲁棒性强,可鉴定酶和底物的关键位点 | |
| MEI | 预训练蛋白质大语言模型(ESM-1b),GNN,DNN | 氨基酸序列和化合物SMILES | 酶-底物结合可能性 | 可利用专业数据集微调进行特定任务预测 | |
| REME[ | 集成ESP、DLKcat/TurNuP、DeepET等模型 | 反应SMILES | 酶列表 | 多维度评价与筛选有效提高了推荐酶列表的可信度 | |
| SPEPP[ | Word2Vec,Transformer | 底物、反应物和酶 | 酶催化底物-产物反应的可能性 | 计算效率相较基于相似性的方法显著提高,可大规模使用,需要提供候选酶集合 | |
辅助空缺 填补 | BoostGAPFILL[ | 基于矩阵分解的推荐系统技术 | 代谢模型 | 填补反应 | 融合了拓扑和约束方法,能够识别代谢网络中的潜在模式 |
| CHESHIRE[ | GCN | 代谢模型 | 填补反应 | 计算效率高,可解释性强,可能引入假阳性反应,缺少反应方向性信息 | |
| DSHCNet[ | GCN,MLP | 代谢模型 | 填补反应 | 对反应数据依赖较强,在适应不同反应数据库中存在挑战 | |
| DNNGIOR[ | CNN | 代谢模型 | 填补反应 | 预测性能受系统发育距离影响 |
表1 机器学习辅助基因组规模代谢模型的方法扩展
Table 1 Machine learning assisted expansion of GEMs
| 模型应用 | 方法 | 模型框架 | 输入 | 输出 | 特点 |
|---|---|---|---|---|---|
辅助基因 注释 | DeepEC[ | CNN | 氨基酸序列 | EC编号 | 可区分酶与非酶,无法进行多功能注释 |
| CLEAN[ | 预训练蛋白质大语言模型(ESM-1b),对比学习 | 氨基酸序列 | EC编号 | 无法区分酶与非酶,可进行多功能注释,可用于数据极少的EC编号 | |
| DeepECtransformer[ | 预训练蛋白质大语言模型(ProtBert),Transformer | 氨基酸序列 | EC编号 | 可区分酶与非酶,可进行多功能注释,不可用于数据极少的EC编号 | |
| ECRECer[ | 预训练蛋白质大语言模型(ESM-1b),GRU | 氨基酸序列 | EC编号 | 可区分酶与非酶,可进行多功能注释 | |
| ECPICK[ | One-hot,CNN | 氨基酸序列 | EC编号 | 无法区分酶与非酶,不可进行多功能注释,可输出预测EC编号的置信度 | |
| EnzBert[ | Transformer | 氨基酸序列 | EC编号 | 无法区分酶与非酶,不可进行多功能注释,可推测关键残基 | |
| EnzymeNet[ | CNN,ResNet | 氨基酸序列 | EC编号 | 可区分酶与非酶,无法进行多功能注释 | |
| GraphEC[ | 预训练蛋白质大语言模型(ProtTrans),ESMFold | 氨基酸序列和蛋白质三维结构 | EC编号 | 无法区分酶与非酶,可进行多功能注释,计算资源需求高 | |
| 辅助途径解析-反应预测 | RetroPath RL[ | 基于蒙特卡洛树搜索的强化学习方法 | 化合物SMILES | 合成途径 | 缓解组合爆炸问题,允许探索深度是只基于模板版本的两倍以上 |
| ASKCOS[ | FNN | 化合物SMILES | 合成途径 | 缓解组合爆炸问题,适用于化学合成途径设计 | |
| chemoenzymatic-ASKCOS[ | DNN | 化合物SMILES | 合成途径 | 缓解组合爆炸问题,可进行化学合成与生物合成混合途径设计 | |
| RetroBioCat[ | DNN | 化合物SMILES | 合成途径 | 缓解组合爆炸问题, 处理大分子化合物存在挑战 | |
| Kreutter等开发的方法[ | Transformer | 化合物SMILES和酶功能描述 | 反应产物 | 预测单步反应,无法给出完整的反应,无法推荐酶功能 | |
| Probst等[ | Transformer | 化合物SMILES和EC编号 | 反应产物 | 预测单步反应,无法给出完整的反应,无法推荐EC编号 | |
| BioNavi-NP[ | Transformer | 天然产SMILES | 合成途径 | 针对天然产物及类似物,可预测多步途径 | |
| BioNavi[ | Transformer | 化合物SMILES | 合成途径 | 可进行化学合成与生物合成混合途径设计 | |
| 辅助途径解析-酶挖掘 | ESP[ | 预训练蛋白质大语言模型(ESM-1b),GNN,XGBoost | 氨基酸序列和分子指纹 | 酶-底物结合可能性 | 对于训练集中没出现代谢物的预测性能会有明显下降 |
| EnzRank[ | 分子指纹,CNN | 氨基酸序列和分子指纹 | 酶-底物结合可能性 | 对于天然底物及其相似物具有良好的区分能力 | |
| PU-EPP[ | GNN,正样本和无标签 学习 | 氨基酸序列和化合物SMILES | 酶-底物结合可能性 | 鲁棒性强,可鉴定酶和底物的关键位点 | |
| MEI | 预训练蛋白质大语言模型(ESM-1b),GNN,DNN | 氨基酸序列和化合物SMILES | 酶-底物结合可能性 | 可利用专业数据集微调进行特定任务预测 | |
| REME[ | 集成ESP、DLKcat/TurNuP、DeepET等模型 | 反应SMILES | 酶列表 | 多维度评价与筛选有效提高了推荐酶列表的可信度 | |
| SPEPP[ | Word2Vec,Transformer | 底物、反应物和酶 | 酶催化底物-产物反应的可能性 | 计算效率相较基于相似性的方法显著提高,可大规模使用,需要提供候选酶集合 | |
辅助空缺 填补 | BoostGAPFILL[ | 基于矩阵分解的推荐系统技术 | 代谢模型 | 填补反应 | 融合了拓扑和约束方法,能够识别代谢网络中的潜在模式 |
| CHESHIRE[ | GCN | 代谢模型 | 填补反应 | 计算效率高,可解释性强,可能引入假阳性反应,缺少反应方向性信息 | |
| DSHCNet[ | GCN,MLP | 代谢模型 | 填补反应 | 对反应数据依赖较强,在适应不同反应数据库中存在挑战 | |
| DNNGIOR[ | CNN | 代谢模型 | 填补反应 | 预测性能受系统发育距离影响 |
| 参数类型 | 方法 | 模型框架 | 输入 | 输出 | 特点 |
|---|---|---|---|---|---|
| 动力学参数 | Heckmann等开发的方法[ | 随机森林,MLP | GEM/蛋白质结构/EC号首位/pH等 | kcat | 可预测体内kcat,适用于大肠杆菌 |
| DLKcat[ | GNN,CNN | 氨基酸序列和化合物SMILES | kcat | 预测kcat(R2=0.44),内置于GECKO 3.0 | |
| TurNuP[ | 预训练蛋白质大语言模型(ESM-1b),XGBoost | 氨基酸序列和反应指纹 | kcat | 预测kcat(R2=0.44),无法区分多底物反应中不同底物的kcat | |
| DLTKcat [ | GNN,CNN | 氨基酸序列,化合物SMILES和温度 | kcat | 预测不同温度下的kcat(R2=0.66) | |
| DeepEnzyme[ | GCN | 氨基酸序列,化合物SMILES和蛋白质三维结构 | kcat | 预测kcat(R2=0.58),预测突变型需要具备蛋白质结构预测能力 | |
| Kroll等开发的方法[ | 预训练蛋白质大语言模型(ESM-1b) | 氨基酸序列和分子指纹 | Km | 预测Km(R2=0.53) | |
| GraphKM[ | 预训练蛋白质大语言模型(ESM-2),GNN | 氨基酸序列和化合物SMILES | Km | 预测Km(R2=0.62),模型的预测性能受限于训练数据集的规模和质量 | |
| MLAGO[ | 随机森林 | EC编号,KEGG ID和物种编号 | Km | 预测Km(R2=0.53),泛化能力受限于EC编号、KEGG ID、物种编号信息 | |
| MPEK[ | 预训练蛋白质大语言模型(ProtT5),预训练小分子大语言模型(Mole-BERT),多任务学习 | 氨基酸序列和化合物SMILES | kcat和Km | 支持同时预测kcat(R2=0.64)和Km(R2=0.60) | |
| UniKP[ | 预训练蛋白质大语言模型(UniRef50),预训练小分子大语言模型(SMILES Transformer),极度随机树 | 氨基酸序列和化合物SMILES | kcat、Km和kcat/Km | 支持分别预测kcat(R2=0.67)、Km(R2=0.60)和kcat/Km(R2=0.56),鲁棒性强,支持温度和pH输入 | |
| EITLEM-Kinetics[ | 预训练蛋白质大语言模型(ESM-1v),迁移学习 | 氨基酸序列和化合物SMILES | kcat、Km和kcat/Km | 支持分别预测kcat(R2=0.72),Km(R2=0.69)和kcat/Km(R2=0.68),蛋白突变体的kcat预测性能优异 | |
| 热力学参数 | dGPredictor[ | 线性回归模型 | 分子指纹 | ∆rG′⊖ | 不适用于异构化反应与涉及金属或聚合物结构的反应 |
| Alazmi等开发的方法[ | 线性回归模型 | 分子指纹 | ∆rG′⊖ | 特征提取方式通用性强,可用于非天然反应 | |
| 温度相关参数 | DeepSTABp[ | 预训练蛋白质大语言模型(ProtTrans),MLP | 氨基酸序列、生物体生长温度和实验条件 | Tm | 预测能力对点突变不敏感 |
| DeepTM[ | GCN | 氨基酸序列 | Tm | 特征提取复杂,训练数据未考虑其他对蛋白熔解温度影响的因素 | |
| Tome[ | SVR,随机森林 | 氨基酸序列,OGT | Topt | 训练集高于85 °C的Topt值占比不足5%,限制了Tome对高温稳定性酶的预测能力 | |
| TOMER[ | 集成学习 | 氨基酸序列,OGT | Topt | 重采样缓解了数据分布不平衡,对高于85 ℃的Topt值预测性能显著提升 | |
| DeepET[ | ResNet,迁移学习 | 氨基酸序列 | Tm和Topt | 类似于Tome,数据分布不均衡会限制其对极端温度蛋白质的预测性能 | |
| Preoptem[ | One-hot,CNN | 氨基酸序列 | Topt | Pearson相关系数r=0.58,适用于嗜热蛋白 |
表2 机器学习辅助多约束多过程模型获取参数方法 (温度相关参数)
Table 2 Machine learning assisted obtaining of parameters for multi-constraint and multi-process models
| 参数类型 | 方法 | 模型框架 | 输入 | 输出 | 特点 |
|---|---|---|---|---|---|
| 动力学参数 | Heckmann等开发的方法[ | 随机森林,MLP | GEM/蛋白质结构/EC号首位/pH等 | kcat | 可预测体内kcat,适用于大肠杆菌 |
| DLKcat[ | GNN,CNN | 氨基酸序列和化合物SMILES | kcat | 预测kcat(R2=0.44),内置于GECKO 3.0 | |
| TurNuP[ | 预训练蛋白质大语言模型(ESM-1b),XGBoost | 氨基酸序列和反应指纹 | kcat | 预测kcat(R2=0.44),无法区分多底物反应中不同底物的kcat | |
| DLTKcat [ | GNN,CNN | 氨基酸序列,化合物SMILES和温度 | kcat | 预测不同温度下的kcat(R2=0.66) | |
| DeepEnzyme[ | GCN | 氨基酸序列,化合物SMILES和蛋白质三维结构 | kcat | 预测kcat(R2=0.58),预测突变型需要具备蛋白质结构预测能力 | |
| Kroll等开发的方法[ | 预训练蛋白质大语言模型(ESM-1b) | 氨基酸序列和分子指纹 | Km | 预测Km(R2=0.53) | |
| GraphKM[ | 预训练蛋白质大语言模型(ESM-2),GNN | 氨基酸序列和化合物SMILES | Km | 预测Km(R2=0.62),模型的预测性能受限于训练数据集的规模和质量 | |
| MLAGO[ | 随机森林 | EC编号,KEGG ID和物种编号 | Km | 预测Km(R2=0.53),泛化能力受限于EC编号、KEGG ID、物种编号信息 | |
| MPEK[ | 预训练蛋白质大语言模型(ProtT5),预训练小分子大语言模型(Mole-BERT),多任务学习 | 氨基酸序列和化合物SMILES | kcat和Km | 支持同时预测kcat(R2=0.64)和Km(R2=0.60) | |
| UniKP[ | 预训练蛋白质大语言模型(UniRef50),预训练小分子大语言模型(SMILES Transformer),极度随机树 | 氨基酸序列和化合物SMILES | kcat、Km和kcat/Km | 支持分别预测kcat(R2=0.67)、Km(R2=0.60)和kcat/Km(R2=0.56),鲁棒性强,支持温度和pH输入 | |
| EITLEM-Kinetics[ | 预训练蛋白质大语言模型(ESM-1v),迁移学习 | 氨基酸序列和化合物SMILES | kcat、Km和kcat/Km | 支持分别预测kcat(R2=0.72),Km(R2=0.69)和kcat/Km(R2=0.68),蛋白突变体的kcat预测性能优异 | |
| 热力学参数 | dGPredictor[ | 线性回归模型 | 分子指纹 | ∆rG′⊖ | 不适用于异构化反应与涉及金属或聚合物结构的反应 |
| Alazmi等开发的方法[ | 线性回归模型 | 分子指纹 | ∆rG′⊖ | 特征提取方式通用性强,可用于非天然反应 | |
| 温度相关参数 | DeepSTABp[ | 预训练蛋白质大语言模型(ProtTrans),MLP | 氨基酸序列、生物体生长温度和实验条件 | Tm | 预测能力对点突变不敏感 |
| DeepTM[ | GCN | 氨基酸序列 | Tm | 特征提取复杂,训练数据未考虑其他对蛋白熔解温度影响的因素 | |
| Tome[ | SVR,随机森林 | 氨基酸序列,OGT | Topt | 训练集高于85 °C的Topt值占比不足5%,限制了Tome对高温稳定性酶的预测能力 | |
| TOMER[ | 集成学习 | 氨基酸序列,OGT | Topt | 重采样缓解了数据分布不平衡,对高于85 ℃的Topt值预测性能显著提升 | |
| DeepET[ | ResNet,迁移学习 | 氨基酸序列 | Tm和Topt | 类似于Tome,数据分布不均衡会限制其对极端温度蛋白质的预测性能 | |
| Preoptem[ | One-hot,CNN | 氨基酸序列 | Topt | Pearson相关系数r=0.58,适用于嗜热蛋白 |
| 1 | GONG Z J, CHEN J Y, JIAO X Y, et al. Genome-scale metabolic network models for industrial microorganisms metabolic engineering: Current advances and future prospects[J]. Biotechnology Advances, 2024, 72: 108319. |
| 2 | ORTH J D, THIELE I, PALSSON B Ø. What is flux balance analysis?[J]. Nature Biotechnology, 2010, 28(3): 245-248. |
| 3 | WAGNER A, WANG C, FESSLER J, et al. Metabolic modeling of single Th17 cells reveals regulators of autoimmunity[J]. Cell, 2021, 184(16): 4168-4185. e21. |
| 4 | KIM B J, KIM W J, KIM D I, et al. Applications of genome-scale metabolic network model in metabolic engineering[J]. Journal of Industrial Microbiology & Biotechnology, 2015, 42(3): 339-348. |
| 5 | RYU J Y, KIM H U, LEE S Y. Reconstruction of genome-scale human metabolic models using omics data[J]. Integrative Biology, 2015, 7(8): 859-868. |
| 6 | GU C D, KIM G B, KIM W J, et al. Current status and applications of genome-scale metabolic models[J]. Genome Biology, 2019, 20(1): 121. |
| 7 | EDWARDS J S, PALSSON B O. Systems properties of the Haemophilus influenzae Rd metabolic genotype[J]. Journal of Biological Chemistry, 1999, 274(25): 17410-17416. |
| 8 | ABRAMSON J, ADLER J, DUNGER J, et al. Accurate structure prediction of biomolecular interactions with AlphaFold3[J]. Nature, 2024, 630(8016): 493-500. |
| 9 | DAUPARAS J, ANISHCHENKO I, BENNETT N, et al. Robust deep learning-based protein sequence design using ProteinMPNN[J]. Science, 2022, 378(6615): 49-56. |
| 10 | JUMPER J, EVANS R, PRITZEL A, et al. Highly accurate protein structure prediction with AlphaFold[J]. Nature, 2021, 596(7873): 583-589. |
| 11 | YU T H, CUI H Y, LI J C, et al. Enzyme function prediction using contrastive learning[J]. Science, 2023, 379(6639): 1358-1363. |
| 12 | FINNIGAN W, HEPWORTH L J, FLITSCH S L, et al. RetroBioCat as a computer-aided synthesis planning tool for biocatalytic reactions and cascades[J]. Nature Catalysis, 2021, 4(2): 98-104. |
| 13 | 曾涛, 巫瑞波. 数据驱动的酶反应预测与设计[J]. 合成生物学, 2023, 4(3): 535-550. |
| ZENG T, WU R B. Data-driven prediction and design for enzymatic reactions[J]. Synthetic Biology Journal, 2023, 4(3): 535-550. | |
| 14 | SABZEVARI M, SZEDMAK S, PENTTILÄ M, et al. Strain design optimization using reinforcement learning[J]. PLoS Computational Biology, 2022, 18(6): e1010177. |
| 15 | 禹伟, 高教琪, 周雍进.一碳生物转化合成有机酸的研究进展[J]. 合成生物学, 2024, 5(5): 1169-1188. |
| YU W, GAO J Q, ZHOU Y J. Bioconversion of one carbon feedstocks for producing organic acids[J]. Synthetic Biology Journal, 2024, 5(5): 1169-1188. | |
| 16 | KUNDU P, BEURA S, MONDAL S, et al. Machine learning for the advancement of genome-scale metabolic modeling[J]. Biotechnology Advances, 2024, 74: 108400. |
| 17 | KANEHISA M, FURUMICHI M, SATO Y, et al. KEGG: biological systems database as a model of the real world[J]. Nucleic Acids Research, 2025, 53(D1): D672-D677. |
| 18 | The UniProt Consortium. UniProt: the universal protein knowledgebase[J]. Nucleic Acids Research, 2018, 46(5): 2699. |
| 19 | CHANG A, JESKE L, ULBRICH S, et al. BRENDA, the ELIXIR core data resource in 2021: new developments and updates[J]. Nucleic Acids Research, 2021, 49(D1): D498-D508. |
| 20 | DUVAUD S, GABELLA C, LISACEK F, et al. Expasy, the Swiss bioinformatics resource portal, as designed by its users[J]. Nucleic Acids Research, 2021, 49(W1): W216-W227. |
| 21 | WITTIG U, REY M, WEIDEMANN A, et al. SABIO-RK: an updated resource for manually curated biochemical reaction kinetics[J]. Nucleic Acids Research, 2018, 46(D1): D656-D660. |
| 22 | BANSAL P, MORGAT A, AXELSEN K B, et al. Rhea, the reaction knowledgebase in 2022[J]. Nucleic Acids Research, 2022, 50(D1): D693-D700. |
| 23 | KARP P D, BILLINGTON R, CASPI R, et al. The BioCyc collection of microbial genomes and metabolic pathways[J]. Briefings in Bioinformatics, 2019, 20(4): 1085-1093. |
| 24 | MORETTI S, TRAN V D T, MEHL F, et al. MetaNetX/MNXref: unified namespace for metabolites and biochemical reactions in the context of metabolic models[J]. Nucleic Acids Research, 2021, 49(D1): D570-D574. |
| 25 | KING Z A, LU J, DRÄGER A, et al. BiGG Models: a platform for integrating, standardizing and sharing genome-scale models[J]. Nucleic Acids Research, 2016, 44(D1): D515-D522. |
| 26 | MALIK-SHERIFF R S, GLONT M, NGUYEN T V N, et al. BioModels-15 years of sharing computational models in life science[J]. Nucleic Acids Research, 2020, 48(D1): D407-D415. |
| 27 | ARKIN A P, COTTINGHAM R W, HENRY C S, et al. KBase: the United States Department of Energy systems biology knowledgebase[J]. Nature Biotechnology, 2018, 36(7): 566-569. |
| 28 | LIEVEN C, BEBER M E, OLIVIER B G, et al. Publisher Correction: MEMOTE for standardized genome-scale metabolic model testing[J]. Nature Biotechnology, 2020, 38(4): 504. |
| 29 | THIELE I, PALSSON B Ø. A protocol for generating a high-quality genome-scale metabolic reconstruction[J]. Nature Protocols, 2010, 5(1): 93-121. |
| 30 | HEIRENDT L, ARRECKX S, PFAU T, et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0[J]. Nature Protocols, 2019, 14(3): 639-702. |
| 31 | WANG H, MARCIŠAUSKAS S, SÁNCHEZ B J, et al. RAVEN 2.0: a versatile toolbox for metabolic network reconstruction and a case study on Streptomyces coelicolor [J]. PLoS Computational Biology, 2018, 14(10): e1006541. |
| 32 | DEVOID S, OVERBEEK R, DEJONGH M, et al. Automated genome annotation and metabolic model reconstruction in the SEED and Model SEED[J]. Methods in Molecular Biology, 2013, 985: 17-45. |
| 33 | MACHADO D, ANDREJEV S, TRAMONTANO M, et al. Fast automated reconstruction of genome-scale metabolic models for microbial species and communities[J]. Nucleic Acids Research, 2018, 46(15): 7542-7553. |
| 34 | CAPELA J, LAGOA D, RODRIGUES R, et al. Merlin, an improved framework for the reconstruction of high-quality genome-scale metabolic models[J]. Nucleic Acids Research, 2022, 50(11): 6052-6066. |
| 35 | ZIMMERMANN J, KALETA C, WASCHINA S. Gapseq: informed prediction of bacterial metabolic pathways and reconstruction of accurate metabolic models[J]. Genome Biology, 2021, 22(1): 81. |
| 36 | HEINKEN A, HERTEL J, ACHARYA G, et al. Genome-scale metabolic reconstruction of 7302 human microorganisms for personalized medicine[J]. Nature Biotechnology, 2023, 41(9): 1320-1331. |
| 37 | FANG X, LLOYD C J, PALSSON B O. Reconstructing organisms in silico: genome-scale models and their emerging applications[J]. Nature Reviews Microbiology, 2020, 18(12): 731-743. |
| 38 | LU H Z, XIAO L C, LIAO W B, et al. Cell factory design with advanced metabolic modelling empowered by artificial intelligence[J]. Metabolic Engineering, 2024, 85: 61-72. |
| 39 | CARRASCO MURIEL J, LONG C, SONNENSCHEIN N. Simultaneous application of enzyme and thermodynamic constraints to metabolic models using an updated Python implementation of GECKO[J]. Microbiology Spectrum, 2023, 11(6): e0170523. |
| 40 | BI X Y, CHENG Y, XU X H, et al. etiBsu1209: a comprehensive multiscale metabolic model for Bacillus subtilis [J]. Biotechnology and Bioengineering, 2023, 120(6): 1623-1639. |
| 41 | SCHROEDER W L, SUTHERS P F, WILLIS T C, et al. Current state, challenges, and opportunities in genome-scale resource allocation models: a mathematical perspective[J]. Metabolites, 2024, 14(7): 365. |
| 42 | HENRY C S, JANKOWSKI M D, BROADBELT L J, et al. Genome-scale thermodynamic analysis of Escherichia coli metabolism[J]. Biophysical Journal, 2006, 90(4): 1453-1461. |
| 43 | HENRY C S, BROADBELT L J, HATZIMANIKATIS V. Thermodynamics-based metabolic flux analysis[J]. Biophysical Journal, 2007, 92(5): 1792-1805. |
| 44 | DASH S, OLSON D G, JOSHUA CHAN S H, et al. Thermodynamic analysis of the pathway for ethanol production from cellobiose in Clostridium thermocellum [J]. Metabolic Engineering, 2019, 55: 161-169. |
| 45 | NOOR E, BAR-EVEN A, FLAMHOLZ A, et al. Pathway thermodynamics highlights kinetic obstacles in central metabolism[J]. PLoS Computational Biology, 2014, 10(2): e1003483. |
| 46 | HÄDICKE O, VON KAMP A, AYDOGAN T, et al. OptMDFpathway: identification of metabolic pathways with maximal thermodynamic driving force and its application for analyzing the endogenous CO2 fixation potential of Escherichia coli [J]. PLoS Computational Biology, 2018, 14(9): e1006492. |
| 47 | FLAMHOLZ A, NOOR E, BAR-EVEN A, et al. Glycolytic strategy as a tradeoff between energy yield and protein cost[J]. Proceedings of the National Academy of Sciences of the United States of America, 2013, 110(24): 10039-10044. |
| 48 | BEKIARIS P S, KLAMT S. Automatic construction of metabolic models with enzyme constraints[J]. BMC Bioinformatics, 2020, 21(1): 19. |
| 49 | BEG Q K, VAZQUEZ A, ERNST J, et al. Intracellular crowding defines the mode and sequence of substrate uptake by Escherichia coli and constrains its metabolic activity[J]. Proceedings of the National Academy of Sciences of the United States of America, 2007, 104(31): 12663-12668. |
| 50 | ADADI R, VOLKMER B, MILO R, et al. Prediction of microbial growth rate versus biomass yield by a metabolic network with kinetic parameters[J]. PLoS Computational Biology, 2012, 8(7): e1002575. |
| 51 | MAO Z T, ZHAO X, YANG X, et al. ECMpy, a simplified workflow for constructing enzymatic constrained metabolic network model[J]. Biomolecules, 2022, 12(1): 65. |
| 52 | MAO Z T, NIU J H, ZHAO J X, et al. ECMpy 2.0: a Python package for automated construction and analysis of enzyme-constrained models[J]. Synthetic and Systems Biotechnology, 2024, 9(3): 494-502. |
| 53 | SÁNCHEZ B J, ZHANG C, NILSSON A, et al. Improving the phenotype predictions of a yeast genome-scale metabolic model by incorporating enzymatic constraints[J]. Molecular Systems Biology, 2017, 13(8): 935. |
| 54 | DOMENZAIN I, SÁNCHEZ B, ANTON M, et al. Reconstruction of a catalogue of genome-scale metabolic models with enzymatic constraints using GECKO 2.0[J]. Nature Communications, 2022, 13(1): 3766. |
| 55 | CHEN Y, GUSTAFSSON J, TAFUR RANGEL A, et al. Reconstruction, simulation and analysis of enzyme-constrained metabolic models using GECKO Toolbox 3.0[J]. Nature Protocols, 2024, 19(3): 629-667. |
| 56 | KERKHOVEN E J. Advances in constraint-based models: methods for improved predictive power based on resource allocation constraints[J]. Current Opinion in Microbiology, 2022, 68: 102168. |
| 57 | YANG X, MAO Z T, ZHAO X, et al. Integrating thermodynamic and enzymatic constraints into genome-scale metabolic models[J]. Metabolic Engineering, 2021, 67: 133-144. |
| 58 | LI G, HU Y T, ZRIMEC J, et al. Bayesian genome scale modelling identifies thermal determinants of yeast metabolism[J]. Nature Communications, 2021, 12(1): 190. |
| 59 | O’BRIEN E J, LERMAN J A, CHANG R L, et al. Genome-scale models of metabolism and gene expression extend and refine growth phenotype prediction[J]. Molecular Systems Biology, 2013, 9: 693. |
| 60 | ALSIYABI A, CHOWDHURY N B, LONG D N, et al. Enhancing in silico strain design predictions through next generation metabolic modeling approaches[J]. Biotechnology Advances, 2022, 54: 107806. |
| 61 | CHEN K, GAO Y, MIH N, et al. Thermosensitivity of growth is determined by chaperone-mediated proteome reallocation[J]. Proceedings of the National Academy of Sciences of the United States of America, 2017, 114(43): 11548-11553. |
| 62 | DU B, YANG L, LLOYD C J, et al. Genome-scale model of metabolism and gene expression provides a multi-scale description of acid stress responses in Escherichia coli [J]. PLoS Computational Biology, 2019, 15(12): e1007525. |
| 63 | YANG L, MIH N, ANAND A, et al. Cellular responses to reactive oxygen species are predicted from molecular mechanisms[J]. Proceedings of the National Academy of Sciences of the United States of America, 2019, 116(28): 14368-14373. |
| 64 | ZHAO J, CHEN K, PALSSON B O, et al. StressME: unified computing framework of Escherichia coli metabolism, gene expression, and stress responses[J]. PLoS Computational Biology, 2024, 20(2): e1011865. |
| 65 | CHEN Y, VAN PELT-KLEINJAN E, VAN OLST B, et al. Proteome constraints reveal targets for improving microbial fitness in nutrient-rich environments[J]. Molecular Systems Biology, 2021, 17(4): e10093. |
| 66 | ELSEMMAN I E, RODRIGUEZ PRADO A, GRIGAITIS P, et al. Whole-cell modeling in yeast predicts compartment-specific proteome constraints that drive metabolic strategies[J]. Nature Communications, 2022, 13(1): 801. |
| 67 | LI F R, CHEN Y, QI Q, et al. Improving recombinant protein production by yeast through genome-scale modeling using proteome constraints[J]. Nature Communications, 2022, 13(1): 2969. |
| 68 | CHEN Y, LI F R, MAO J W, et al. Yeast optimizes metal utilization based on metabolic network and enzyme kinetics[J]. Proceedings of the National Academy of Sciences of the United States of America, 2021, 118(12): e2020154118. |
| 69 | SALVY P, HATZIMANIKATIS V. The ETFL formulation allows multi-omics integration in thermodynamics-compliant metabolism and expression models[J]. Nature Communications, 2020, 11(1): 30. |
| 70 | OFTADEH O, SALVY P, MASID M, et al. A genome-scale metabolic model of Saccharomyces cerevisiae that integrates expression constraints and reaction thermodynamics[J]. Nature Communications, 2021, 12(1): 4790. |
| 71 | RYU J Y, KIM H U, LEE S Y. Deep learning enables high-quality and high-throughput prediction of enzyme commission numbers[J]. Proceedings of the National Academy of Sciences of the United States of America, 2019, 116(28): 13996-14001. |
| 72 | KIM G B, KIM J Y, LEE J A, et al. Functional annotation of enzyme-encoding genes using deep learning with transformer layers[J]. Nature Communications, 2023, 14(1): 7370. |
| 73 | SHI Z K, DENG R, YUAN Q Q, et al. Enzyme commission number prediction and benchmarking with hierarchical dual-core multitask learning framework[J]. Research, 2023, 6: 0153. |
| 74 | HAN S R, PARK M, KOSARAJU S, et al. Evidential deep learning for trustworthy prediction of enzyme commission number[J]. Briefings in Bioinformatics, 2023, 25(1): bbad401. |
| 75 | BUTON N, COSTE F, LE CUNFF Y. Predicting enzymatic function of protein sequences with attention[J]. Bioinformatics, 2023, 39(10): btad620. |
| 76 | WATANABE N, YAMAMOTO M, MURATA M, et al. EnzymeNet: residual neural networks model for Enzyme Commission number prediction[J]. Bioinformatics Advances, 2023, 3(1): vbad173. |
| 77 | SONG Y D, YUAN Q M, CHEN S, et al. Accurately predicting enzyme functions through geometric graph learning on ESMFold-predicted structures[J]. Nature Communications, 2024, 15(1): 8180. |
| 78 | KOCH M, DUIGOU T, FAULON J L. Reinforcement learning for bioretrosynthesis[J]. ACS Synthetic Biology, 2020, 9(1): 157-168. |
| 79 | COLEY C W, THOMAS D A, LUMMISS J A M, et al. A robotic platform for flow synthesis of organic compounds informed by AI planning[J]. Science, 2019, 365(6453): eaax1566. |
| 80 | LEVIN I, LIU M J, VOIGT C A, et al. Merging enzymatic and synthetic chemistry with computational synthesis planning[J]. Nature Communications, 2022, 13(1): 7747. |
| 81 | KREUTTER D, SCHWALLER P, REYMOND J L. Predicting enzymatic reactions with a molecular transformer[J]. Chemical Science, 2021, 12(25): 8648-8659. |
| 82 | PROBST D, MANICA M, NANA TEUKAM Y G, et al. Biocatalysed synthesis planning using data-driven learning[J]. Nature Communications, 2022, 13(1): 964. |
| 83 | ZHENG S J, ZENG T, LI C T, et al. Deep learning driven biosynthetic pathways navigation for natural products with BioNavi-NP[J]. Nature Communications, 2022, 13(1): 3342. |
| 84 | ZENG T, JIN Z H, ZHENG S J, et al. Developing BioNavi for hybrid retrosynthesis planning[J]. JACS Au, 2024, 4(7): 2492-2502. |
| 85 | KROLL A, RANJAN S, ENGQVIST M K M, et al. A general model to predict small molecule substrates of enzymes based on machine and deep learning[J]. Nature Communications, 2023, 14(1): 2787. |
| 86 | UPADHYAY V, BOORLA V S, MARANAS C D. Rank-ordering of known enzymes as starting points for re-engineering novel substrate activity using a convolutional neural network[J]. Metabolic Engineering, 2023, 78: 171-182. |
| 87 | ZHANG D C, XING H D, LIU D L, et al. Discovery of toxin-degrading enzymes with positive unlabeled deep learning[J]. ACS Catalysis, 2024, 14(5): 3336-3348. |
| 88 | SHI Z K, WANG D H, LI Y, et al. REME: an integrated platform for reaction enzyme mining and evaluation[J]. Nucleic Acids Research, 2024, 52(W1): W299-W305. |
| 89 | XING H D, CAI P L, LIU D L, et al. High-throughput prediction of enzyme promiscuity based on substrate-product pairs[J]. Briefings in Bioinformatics, 2024, 25(2): bbae089. |
| 90 | OYETUNDE T, ZHANG M H, CHEN Y X, et al. BoostGAPFILL: improving the fidelity of metabolic network reconstructions through integrated constraint and pattern-based methods[J]. Bioinformatics, 2017, 33(4): 608-611. |
| 91 | CHEN C, LIAO C, LIU Y Y. Teasing out missing reactions in genome-scale metabolic networks through hypergraph learning[J]. Nature Communications, 2023, 14(1): 2375. |
| 92 | HUANG W H, YANG F, ZHANG Q, et al. A dual-scale fused hypergraph convolution-based hyperedge prediction model for predicting missing reactions in genome-scale metabolic networks[J]. Briefings in Bioinformatics, 2024, 25(5): bbae383. |
| 93 | BOER M D, MELKONIAN C, ZAFEIROPOULOS H, et al. Improving genome-scale metabolic models of incomplete genomes with deep learning[J]. iScience, 2024, 27(12): 111349. |
| 94 | ASHBURNER M, BALL C A, BLAKE J A, et al. Gene ontology: tool for the unification of biology. The gene ontology consortium[J]. Nature Genetics, 2000, 25(1): 25-29. |
| 95 | SHEN H B, CHOU K C. EzyPred: a top-down approach for predicting enzyme functional classes and subclasses[J]. Biochemical and Biophysical Research Communications, 2007, 364(1): 53-59. |
| 96 | LI Y H, XU J Y, TAO L, et al. SVM-prot 2016: a web-server for machine learning prediction of protein functional families from sequence irrespective of similarity[J]. PLoS One, 2016, 11(8): e0155290. |
| 97 | LI Y, WANG S, UMAROV R, et al. DEEPre: sequence-based enzyme EC number prediction by deep learning[J]. Bioinformatics, 2018, 34(5): 760-769. |
| 98 | NURSIMULU N, XU L L, WASMUTH J D, et al. Improved enzyme annotation with EC-specific cutoffs using DETECT v2[J]. Bioinformatics, 2018, 34(19): 3393-3395. |
| 99 | DALKIRAN A, RIFAIOGLU A S, MARTIN M J, et al. ECPred: a tool for the prediction of the enzymatic functions of protein sequences based on the EC nomenclature[J]. BMC Bioinformatics, 2018, 19(1): 334. |
| 100 | RIVES A, MEIER J, SERCU T, et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences[J]. Proceedings of the National Academy of Sciences of the United States of America, 2021, 118(15): e2016239118. |
| 101 | LIN Z M, AKIN H, RAO R, et al. Evolutionary-scale prediction of atomic-level protein structure with a language model[J]. Science, 2023, 379(6637): 1123-1130. |
| 102 | HADADI N, HAFNER J, SHAJKOFCI A, et al. ATLAS of biochemistry: a repository of all possible biochemical reactions for synthetic biology and metabolic engineering studies[J]. ACS Synthetic Biology, 2016, 5(10): 1155-1166. |
| 103 | KHERSONSKY O, TAWFIK D S. Enzyme promiscuity: a mechanistic and evolutionary perspective[J]. Annual Review of Biochemistry, 2010, 79: 471-505. |
| 104 | PONTRELLI S, FRICKE R C B, TEOH S T, et al. Metabolic repair through emergence of new pathways in Escherichia coli [J]. Nature Chemical Biology, 2018, 14(11): 1005-1009. |
| 105 | NI Z F, STINE A E, TYO K E J, et al. Curating a comprehensive set of enzymatic reaction rules for efficient novel biosynthetic pathway design[J]. Metabolic Engineering, 2021, 65: 79-87. |
| 106 | NAM H, LEWIS N E, LERMAN J A, et al. Network context and selection in the evolution to enzyme specificity[J]. Science, 2012, 337(6098): 1101-1104. |
| 107 | MORIYA Y, SHIGEMIZU D, HATTORI M, et al. PathPred: an enzyme-catalyzed metabolic pathway prediction server[J]. Nucleic Acids Research, 2010, 38(S2): W138-W143. |
| 108 | DELÉPINE B, DUIGOU T, CARBONELL P, et al. RetroPath2.0: a retrosynthesis workflow for metabolic engineers[J]. Metabolic Engineering, 2018, 45: 158-170. |
| 109 | DING S Z, TIAN Y, CAI P L, et al. novoPathFinder: a webserver of designing novel-pathway with integrating GEM-model[J]. Nucleic Acids Research, 2020, 48(W1): W477-W487. |
| 110 | GENHEDEN S, THAKKAR A, CHADIMOVÁ V, et al. AiZynthFinder: a fast, robust and flexible open-source software for retrosynthetic planning[J]. Journal of Cheminformatics, 2020, 12(1): 70. |
| 111 | GRICOURT G, MEYER P, DUIGOU T, et al. Artificial intelligence methods and models for retro-biosynthesis: a scoping review[J]. ACS Synthetic Biology, 2024, 13(8): 2276-2294. |
| 112 | COLEY C W, ROGERS L, GREEN W H, et al. SCScore: synthetic complexity learned from a reaction corpus[J]. Journal of Chemical Information and Modeling, 2018, 58(2): 252-261. |
| 113 | CARBONELL P, WONG J, SWAINSTON N, et al. Selenzyme: enzyme selection tool for pathway design[J]. Bioinformatics, 2018, 34(12): 2153-2154. |
| 114 | RAHMAN S A, CUESTA S M, FURNHAM N, et al. EC-BLAST: a tool to automatically search and compare enzyme reactions[J]. Nature Methods, 2014, 11(2): 171-174. |
| 115 | GIRI V, SIVAKUMAR T V, CHO K M, et al. RxnSim: a tool to compare biochemical reactions[J]. Bioinformatics, 2015, 31(22): 3712-3714. |
| 116 | STONEY R A, HANKO E K R, CARBONELL P, et al. SelenzymeRF: updated enzyme suggestion software for unbalanced biochemical reactions[J]. Computational and Structural Biotechnology Journal, 2023, 21: 5868-5876. |
| 117 | WILLETT P. Searching techniques for databases of two- and three-dimensional chemical structures[J]. Journal of Medicinal Chemistry, 2005, 48(13): 4183-4199. |
| 118 | SCHWALLER P, HOOVER B, REYMOND J L, et al. Extraction of organic chemistry grammar from unsupervised learning of chemical reactions[J]. Science Advances, 2021, 7(15): eabe4166. |
| 119 | QIAN W J, WANG X R, HUANG Y S, et al. Deep learning-driven insights into enzyme-substrate interaction discovery[J]. Journal of Chemical Information and Modeling, 2025, 65(1): 187-200. |
| 120 | ORTH J D, PALSSON B Ø. Systematizing the generation of missing metabolic knowledge[J]. Biotechnology and Bioengineering, 2010, 107(3): 403-412. |
| 121 | THIELE I, VLASSIS N, FLEMING R M T. fastGapFill: efficient gap filling in metabolic networks[J]. Bioinformatics, 2014, 30(17): 2529-2531. |
| 122 | KUMAR V S, DASIKA M S, MARANAS C D. Optimization based automated curation of metabolic reconstructions[J]. BMC Bioinformatics, 2007, 8: 212. |
| 123 | HECKMANN D, LLOYD C J, MIH N, et al. Machine learning applied to enzyme turnover numbers reveals protein structural correlates and improves metabolic models[J]. Nature Communications, 2018, 9(1): 5252. |
| 124 | LI F R, YUAN L, LU H Z, et al. Deep learning-based kcat prediction enables improved enzyme-constrained model reconstruction[J]. Nature Catalysis, 2022, 5(8): 662-672. |
| 125 | KROLL A, ROUSSET Y, HU X P, et al. Turnover number predictions for kinetically uncharacterized enzymes using machine and deep learning[J]. Nature Communications, 2023, 14(1): 4139. |
| 126 | QIU S Z, ZHAO S M, YANG A D. DLTKcat: deep learning-based prediction of temperature-dependent enzyme turnover rates[J]. Briefings in Bioinformatics, 2023, 25(1): bbad506. |
| 127 | WANG T, XIANG G M, HE S W, et al. DeepEnzyme: a robust deep learning model for improved enzyme turnover number prediction by utilizing features of protein 3D-structures[J]. Briefings in Bioinformatics, 2024, 25(5): bbae409. |
| 128 | KROLL A, ENGQVIST M K M, HECKMANN D, et al. Deep learning allows genome-scale prediction of Michaelis constants from structural features[J]. PLoS Biology, 2021, 19(10): e3001402. |
| 129 | HE X, YAN M. GraphKM: machine and deep learning for Km prediction of wildtype and mutant enzymes[J]. BMC Bioinformatics, 2024, 25(1): 135. |
| 130 | MAEDA K, HATAE A, SAKAI Y, et al. MLAGO: machine learning-aided global optimization for Michaelis constant estimation of kinetic modeling[J]. BMC Bioinformatics, 2022, 23(1): 455. |
| 131 | WANG J J, YANG Z J, CHEN C, et al. MPEK: a multitask deep learning framework based on pretrained language models for enzymatic reaction kinetic parameters prediction[J]. Briefings in Bioinformatics, 2024, 25(5): bbae387. |
| 132 | YU H, DENG H X, HE J H, et al. UniKP: a unified framework for the prediction of enzyme kinetic parameters[J]. Nature Communications, 2023, 14(1): 8211. |
| 133 | SHEN X W, CUI Z H, LONG J Y, et al. EITLEM-Kinetics: a deep-learning framework for kinetic parameter prediction of mutant enzymes[J]. Chem Catalysis, 2024, 4(9): 101094. |
| 134 | WANG L, UPADHYAY V, MARANAS C D. dGPredictor: automated fragmentation method for metabolic reaction free energy prediction and de novo pathway design[J]. PLoS Computational Biology, 2021, 17(9): e1009448. |
| 135 | ALAZMI M, KUWAHARA H, SOUFAN O, et al. Systematic selection of chemical fingerprint features improves the Gibbs energy prediction of biochemical reactions[J]. Bioinformatics, 2019, 35(15): 2634-2643. |
| 136 | JUNG F, FREY K, ZIMMER D, et al. DeepSTABp: a deep learning approach for the prediction of thermal protein stability[J]. International Journal of Molecular Sciences, 2023, 24(8): 7444. |
| 137 | LI M Y, WANG H Z, YANG Z W, et al. DeepTM: a deep learning algorithm for prediction of melting temperature of thermophilic proteins directly from sequences[J]. Computational and Structural Biotechnology Journal, 2023, 21: 5544-5560. |
| 138 | LI G, RABE K S, NIELSEN J, et al. Machine learning applied to predicting microorganism growth temperatures and enzyme catalytic optima[J]. ACS Synthetic Biology, 2019, 8(6): 1411-1420. |
| 139 | GADO J E, BECKHAM G T, PAYNE C M. Improving enzyme optimum temperature prediction with resampling strategies and ensemble learning[J]. Journal of Chemical Information and Modeling, 2020, 60(8): 4098-4107. |
| 140 | LI G, BURIC F, ZRIMEC J, et al. Learning deep representations of enzyme thermal adaptation[J]. Protein Science, 2022, 31(12): e4480. |
| 141 | ZHANG Y, GUAN F F, XU G S, et al. A novel thermophilic chitinase directly mined from the marine metagenome using the deep learning tool Preoptem[J]. Bioresources and Bioprocessing, 2022, 9(1): 54. |
| 142 | NILSSON A, NIELSEN J, PALSSON B O. Metabolic models of protein allocation call for the kinetome[J]. Cell Systems, 2017, 5(6): 538-541. |
| 143 | SUTHERS P F, FOSTER C J, SARKAR D, et al. Recent advances in constraint and machine learning-based metabolic modeling by leveraging stoichiometric balances, thermodynamic feasibility and kinetic law formalisms[J]. Metabolic Engineering,2021, 63: 13-33. |
| 144 | JANKOWSKI M D, HENRY C S, BROADBELT L J, et al. Group contribution method for thermodynamic analysis of complex metabolic networks[J]. Biophysical Journal, 2008, 95(3): 1487-1499. |
| 145 | NOOR E, HARALDSDÓTTIR H S, MILO R, et al. Consistent estimation of Gibbs energy using component contributions[J]. PLoS Computational Biology, 2013, 9(7): e1003098. |
| 146 | BEBER M E, GOLLUB M G, MOZAFFARI D, et al. eQuilibrator 3.0: a database solution for thermodynamic constant estimation[J]. Nucleic Acids Research, 2022, 50(D1): D603-D609. |
| 147 | LEUENBERGER P, GANSCHA S, KAHRAMAN A, et al. Cell-wide analysis of protein thermal unfolding reveals determinants of thermostability[J]. Science, 2017, 355(6327): eaai7825. |
| [1] | 田晓军, 张日新. 合成基因回路面临的细胞“经济学窘境”[J]. 合成生物学, 2025, 6(3): 532-546. |
| [2] | 章益蜻, 刘高雯. 合成生物学视角下的基因功能探索与酵母工程菌株文库构建[J]. 合成生物学, 2025, 6(3): 685-700. |
| [3] | 黄怡, 司同, 陆安静. 生物制造标准体系建设的现状、问题与建议[J]. 合成生物学, 2025, 6(3): 701-714. |
| [4] | 宋成治, 林一瀚. AI+定向进化赋能蛋白改造及优化[J]. 合成生物学, 2025, 6(3): 617-635. |
| [5] | 张梦瑶, 蔡鹏, 周雍进. 合成生物学助力萜类香精香料可持续生产[J]. 合成生物学, 2025, 6(2): 334-356. |
| [6] | 张璐鸥, 徐丽, 胡晓旭, 杨滢. 合成生物学助力化妆品走进生物制造新时代[J]. 合成生物学, 2025, 6(2): 479-491. |
| [7] | 伊进行, 唐宇琳, 李春雨, 吴鹤云, 马倩, 谢希贤. 氨基酸衍生物在化妆品中的应用及其生物合成研究进展[J]. 合成生物学, 2025, 6(2): 254-289. |
| [8] | 韦灵珍, 王佳, 孙新晓, 袁其朋, 申晓林. 黄酮类化合物生物合成及其在化妆品中应用的研究[J]. 合成生物学, 2025, 6(2): 373-390. |
| [9] | 肖森, 胡立涛, 石智诚, 王发银, 余思婷, 堵国成, 陈坚, 康振. 可控分子量透明质酸的生物合成研究进展[J]. 合成生物学, 2025, 6(2): 445-460. |
| [10] | 王倩, 果士婷, 辛波, 钟成, 王钰. L-精氨酸的微生物合成研究进展[J]. 合成生物学, 2025, 6(2): 290-305. |
| [11] | 左一萌, 张姣姣, 连佳长. 酿酒酵母使能技术在化妆品原料合成中的应用[J]. 合成生物学, 2025, 6(2): 233-253. |
| [12] | 汤传根, 王璟, 张烁, 张昊宁, 康振. 功能肽合成和挖掘策略研究进展[J]. 合成生物学, 2025, 6(2): 461-478. |
| [13] | 郭婷婷, 韩湘凝, 黄熙婷, 张婷婷, 孔健. 乳酸菌的合成生物学工具及在合成益肤因子中的应用[J]. 合成生物学, 2025, 6(2): 320-333. |
| [14] | 张萍, 张维娇, 胥睿睿, 李江华, 陈坚, 康振. 防晒化合物类菌孢素氨基酸的生物合成[J]. 合成生物学, 2025, 6(2): 306-319. |
| [15] | 黄姝涵, 马赫, 罗云孜. 生物合成红景天苷的研究进展[J]. 合成生物学, 2025, 6(2): 391-407. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
