合成生物学 ›› 2022, Vol. 3 ›› Issue (3): 567-586.DOI: 10.12211/2096-8280.2021-013
预反应态模型浅析:催化活性和近过渡态分子模拟
SIM Byuri, 赵一雷
- 上海交通大学生命科学技术学院,微生物代谢国家重点实验室,上海 200240
-
收稿日期:2021-01-27修回日期:2021-02-03出版日期:2022-06-30发布日期:2022-07-13 -
通讯作者:赵一雷 -
作者简介:SIM Byuri (1990—),男,硕士研究生。研究方向为醇脱氢酶CpRCR的共进化突变效应。E-mail:byurisim@sjtu.edu.cn赵一雷 (1972—),男,教授,博士生导师。研究方向为酶催化反应分子机制、蛋白质与核酸化学修饰,计算化学与分子生物信息学在蛋白质工程和生物医学中应用。E-mail:yileizhao@sjtu.edu.cn -
基金资助:国家自然科学基金(31970041);国家重点研发计划(2020YFA0907700)
Assessment on the pre-reaction state of enzyme: could we understand catalytic activity with near transition-state molecular dynamic simulation?-a review
SIM Byuri, ZHAO Yilei
- State Key Laboratory of Microbial Metabolism,School of Life Sciences and Biotechnology,Shanghai Jiao Tong University,Shanghai 200240,China
-
Received:2021-01-27Revised:2021-02-03Online:2022-06-30Published:2022-07-13 -
Contact:ZHAO Yilei
摘要:
当今生物合成催化元件超进化分子理性设计的瓶颈在于有限的计算资源、研究时间与催化反应复杂势能面接近无穷无尽的计算需求之间的矛盾。然而,两个前所未有的数据集合有望拓新蛋白质工程人工智能化分子设计,其一是高通量定向进化实验带来的巨量高效突变体序列信息,其二是基于结构生物学的高阶量子力学计算所揭示的全原子飞秒精度反应机制。本文从催化基本理论、米氏复合物近进攻构象、催化循环效率控制点的角度浅析预反应态模型的基本概念和应用。预反应态模型尝试利用在低反应势垒生物化学反应中内禀的近进攻构象与过渡态具有相近的物理化学稳定性,弹性地选择与催化元件进化目标相关的关键过渡态,利用经典分子动力学模拟分析近过渡态的活性构象布居数与远端突变、底物结构、实验条件的关系。预反应态分析的基本流程为:首先,基于高阶量子力学反应势能面提取催化中心关键过渡态的结构特征;其次,从高精度蛋白质三维结构出发,结合氨基酸质子化生物信息学预测工具构建出关键过渡态对应的近进攻态活性构象;最后,利用过渡态结构特征设定分子动力学模拟初始约束条件,并逐步取消约束条件测试预反应态随氨基酸突变和底物变化的稳定性变化,以近进攻构象在预反应态轨迹中布居数作为“预反应态-酶活”半定量相关系数,从预反应态稳定性中挖掘酶与底物的适配图谱。当前在预反应态动态结构与酶活的定量关系分析上还有诸多难题亟待突破,利用高通量高阶量子化学再采样计算、结合机器学习人工智能分析代表了预反应态模型的发展方向。
中图分类号:
引用本文
SIM Byuri, 赵一雷. 预反应态模型浅析:催化活性和近过渡态分子模拟[J]. 合成生物学, 2022, 3(3): 567-586.
SIM Byuri, ZHAO Yilei. Assessment on the pre-reaction state of enzyme: could we understand catalytic activity with near transition-state molecular dynamic simulation?-a review[J]. Synthetic Biology Journal, 2022, 3(3): 567-586.

图1 酶催化历程所涉及的运动时间尺度,包括了高活化能过程及多种微观运动
Fig. 1 Timescale in catalytic reactions, including the high-activation energy process and various motions
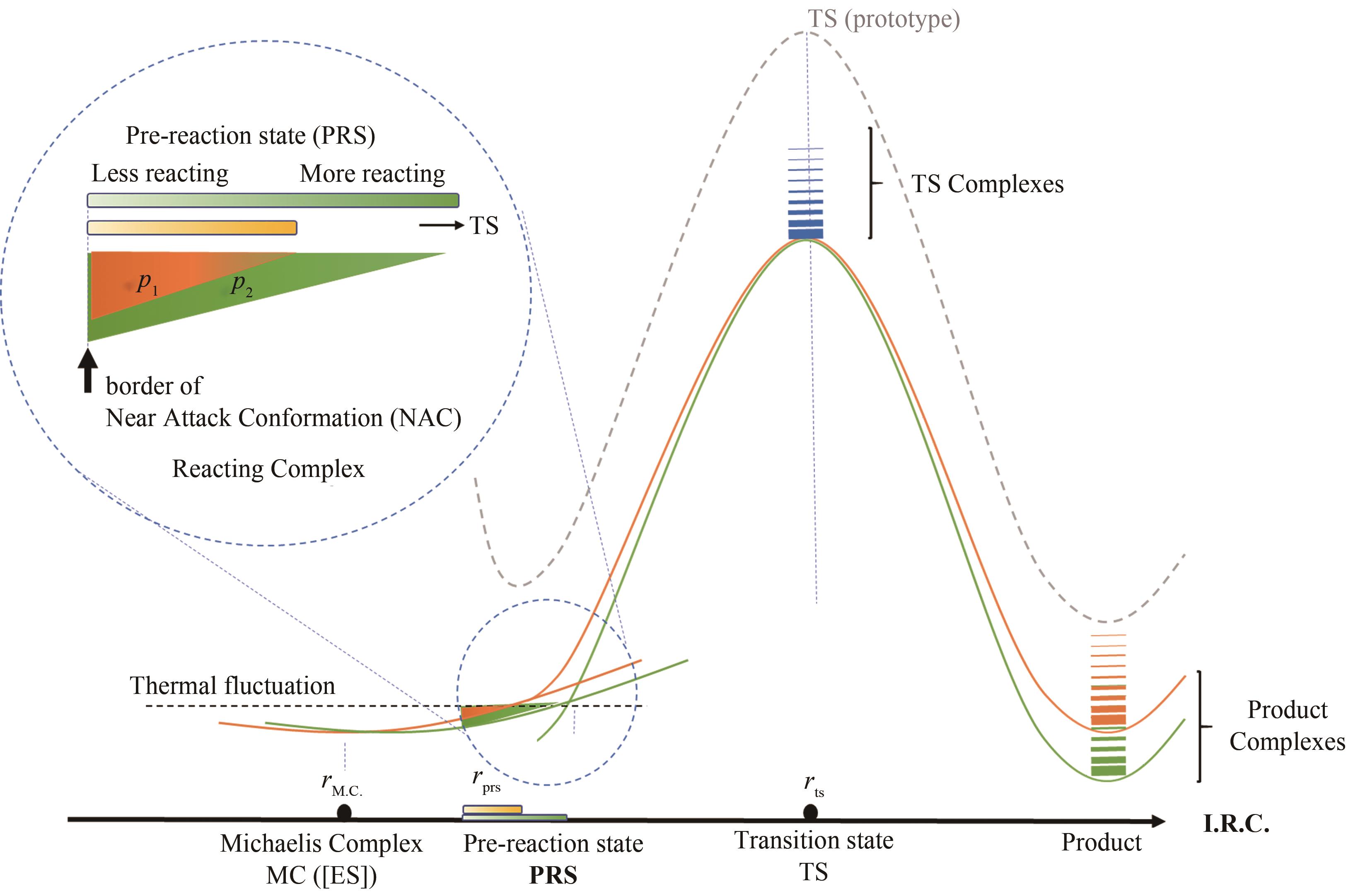
图2 催化元件突变导致反应势能面微扰的示意图[在酶催化单步反应米氏模型中,预反应态与近进攻构象定义相同,米氏复合物热运动中接近反应过渡态(近进攻构象)活性构象的布居数p与催化活性正相关]
Fig. 2 Diagram for the perturbation of energy profile caused by the mutation of catalytic elements(In the simplest Michaelis's model, pre-reaction state and near attack conformation are identical, using the correlation of active conformation population and enzyme proficiency.)

图3 催化循环周转效率TOF与催化反应势能面的关系[在酶催化多步催化循环中,预反应态定义为关键决速步过渡态相关的活性构象的布居数p,它的变化与催化元件优化目标直接相关(如突变效应或底物适配)]
Fig. 3 Correlation of the turn-over frequency (TOF) and reaction potential energy surface(In multiple steps of a catalytic cycle, pre-reaction state is defined as the active conformation near rate-determining transition state, in which rate is dependent on the protein engineering target such as mutation effect and substrate diversity.)
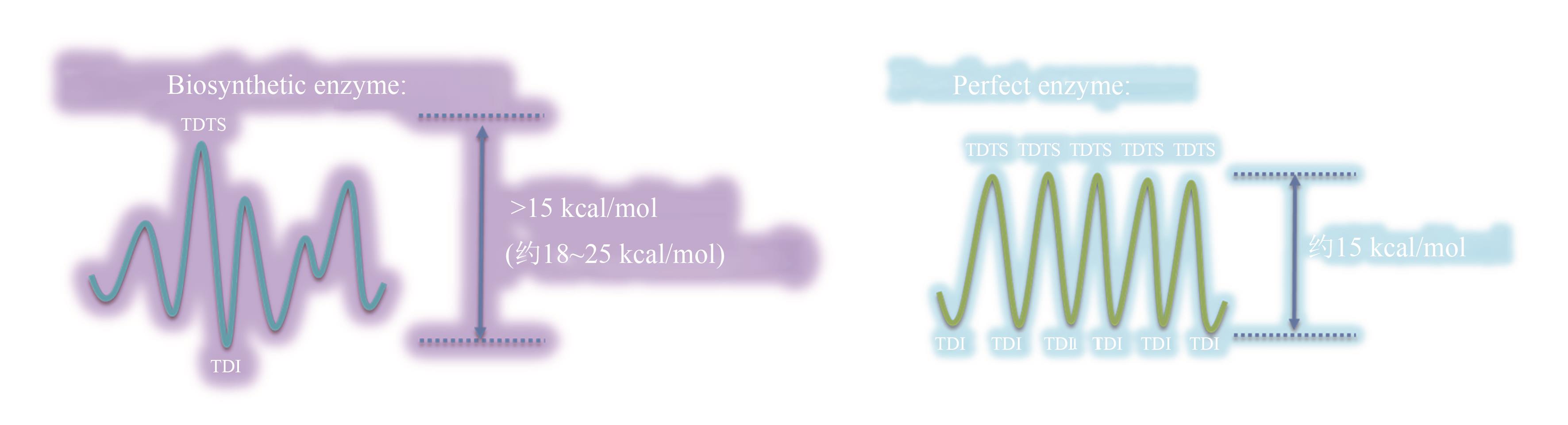
图4 催化元件分子进化前后的反应势能面[进化完成前通常存在阶段性的最大控制点(左图),而进化后反应势能面各过渡态的控制度相近(右图)]
Fig. 4 Imaginary reaction potential energy surface for molecular evolution(Before evolving to a "perfect" enzyme, one point on the reaction potential energy surface controls the overall rate, but many other points are equivalently important for the perfect enzyme.)
| 研究对象 | 预反应态所选择的控制点 | 实例 |
|---|---|---|
| 红霉素DEBS硫酯酶 | 底物上载后共价复合物水解和环化途径选择 |
与反应位点间隔4~5个化学键的碳原子对反应中心环化和水解活性结构的远程作用[ PRS结构参数:NH259-OH距离、OH-C距离 |
| 苦霉素硫酯酶 | 底物上载后共价复合物水解和环化途径选择 |
12元和14元大环成环对反应中心环化和水解活性结构的影响[ PRS结构参数:NH268-OH距离、OH-C距离 |
| 变构霉素硫酯酶/苦霉素硫酯酶 | 底物上载后共价复合物水解和环化途径选择 |
变构霉素生物合成时环化释放受阻,利用异源硫酯酶调整环化水解选择性,适配性分析[ 变构霉素硫酯酶PRS结构参数:NH255-OH距离、OH-C距离 苦霉素硫酯酶PRS结构参数:NH268-OH距离、OH-C距离 |
| 匹马霉素硫酯酶 | 底物上载后共价复合物水解和环化途径选择 |
与反应位点间隔11~13个化学键的碳原子修饰对反应中心环化和水解活性结构的远程作用[ PRS结构参数:NH261-OH距离、OH-C距离 |
| 醇脱氢酶KpADH | 质子偶合的负氢原子转移关键过渡态 |
高效突变体与野生型反应中心的活性构象分布变化[ PRS结构参数:HY164-O距离、HNADPH-C距离 |
| 醇脱氢酶CpRCR | 质子偶合的负氢原子转移关键过渡态 |
高效突变体与野生型反应中心的活性构象分布变化[ PRS结构参数:HS46-O距离、HNADPH-C距离 |
| DNA甲基化酶3A | SAM甲基转移到DNA-酶共价结合中间体 |
白血病高发的R882H远端突变对活性中心SAM甲基转移的影响[ PRS结构参数:CSAM-C距离 |
| 脱羧酶TyDC | PLP辅因子与底物的共价加合物C-C键断裂 |
通过H98、H251控制共价加合物的反应前线轨道布局[ PRS结构参数:CCαNSB角度,CCαNSBC4’二面角 |
| 酰胺水解酶NiHyuC | 碳四面体氧负离子关键过渡态 |
高效突变体对活化后巯基与被进攻的羧基反应前线轨道布局的影响[ PRS结构参数:SC171-C距离、SC171CO进攻角 |
| 普鲁兰多糖酶 | 取代反应SN2过渡态(碳氧键断裂) |
高效突变体对[O-C-O]取代反应前线轨道布局的影响[ PRS结构参数:OD619-C距离、OD619CO夹角 |
| MERS和SARS冠状病毒主蛋白水解酶 | 碳四面体氧负离子关键过渡态 |
两种病毒主蛋白水解酶的关键反应前线轨道布局[ MERS病毒PRS结构参数:SC148-C距离、SC148CO进攻角 SARS病毒PRS结构参数:SC145-C距离、SC145CO进攻角 |
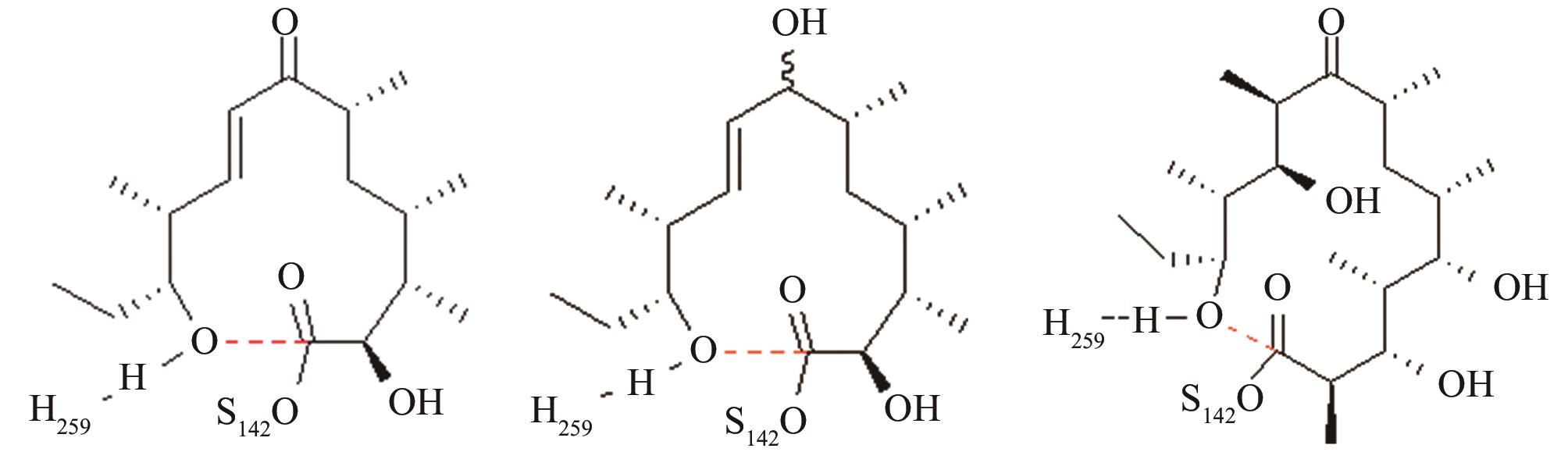
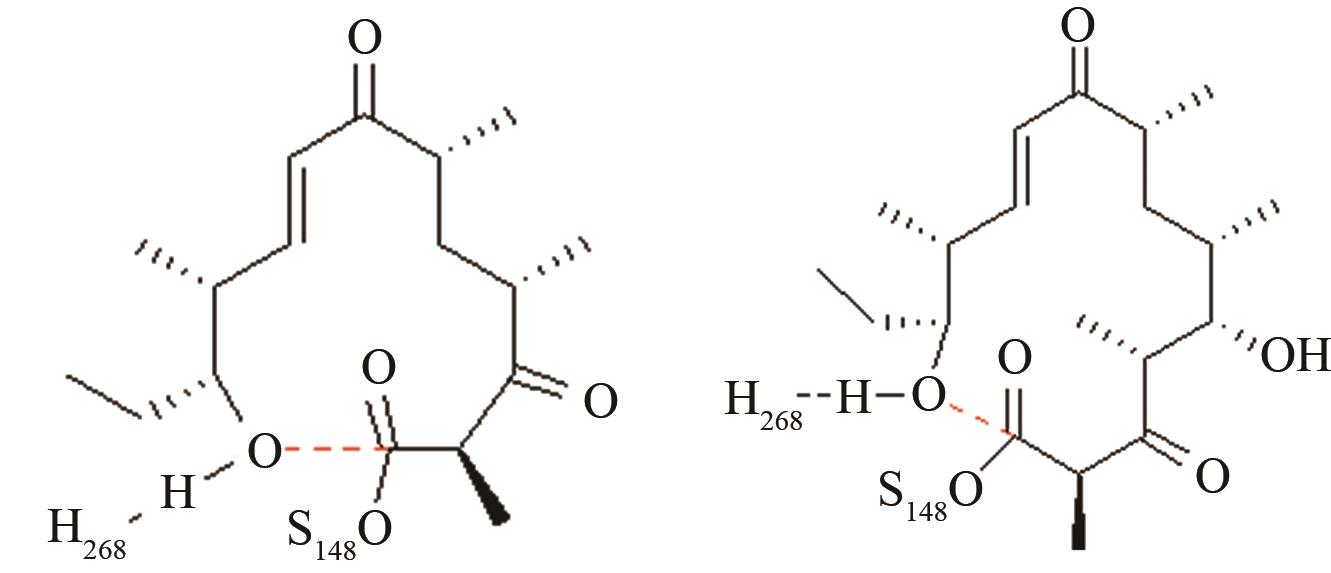
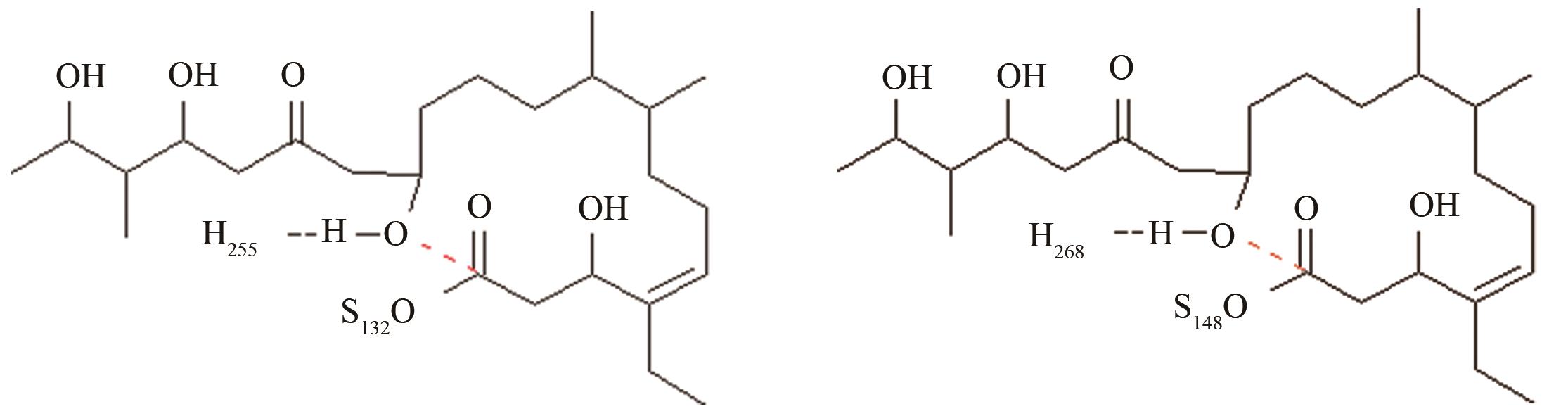
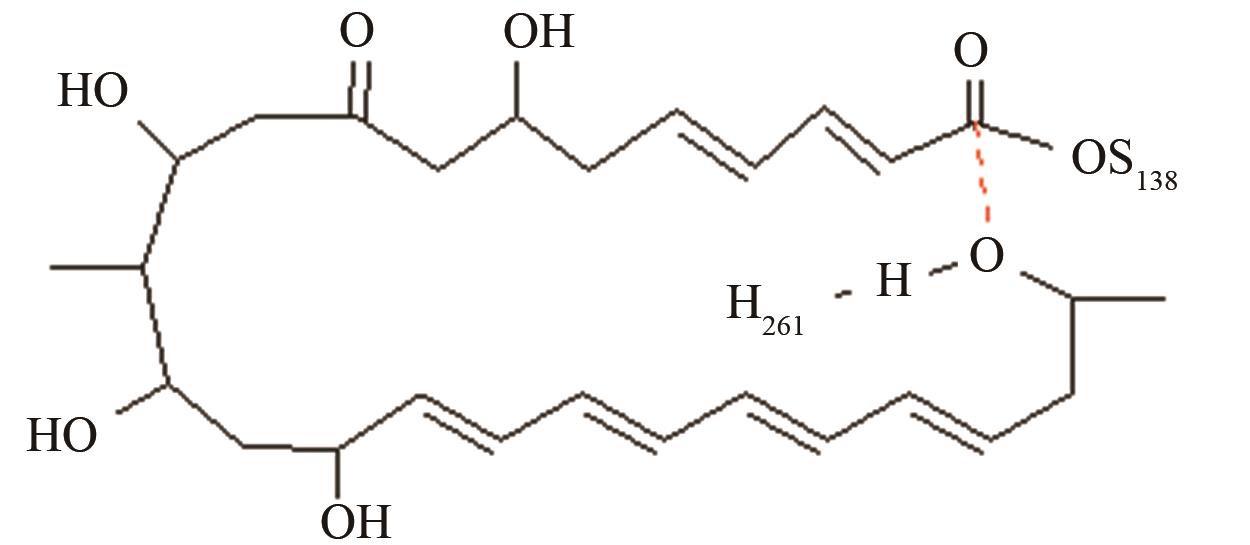
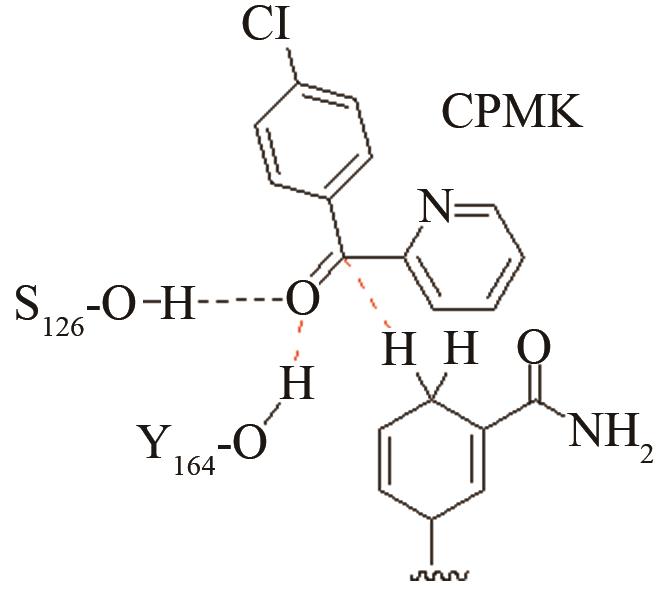
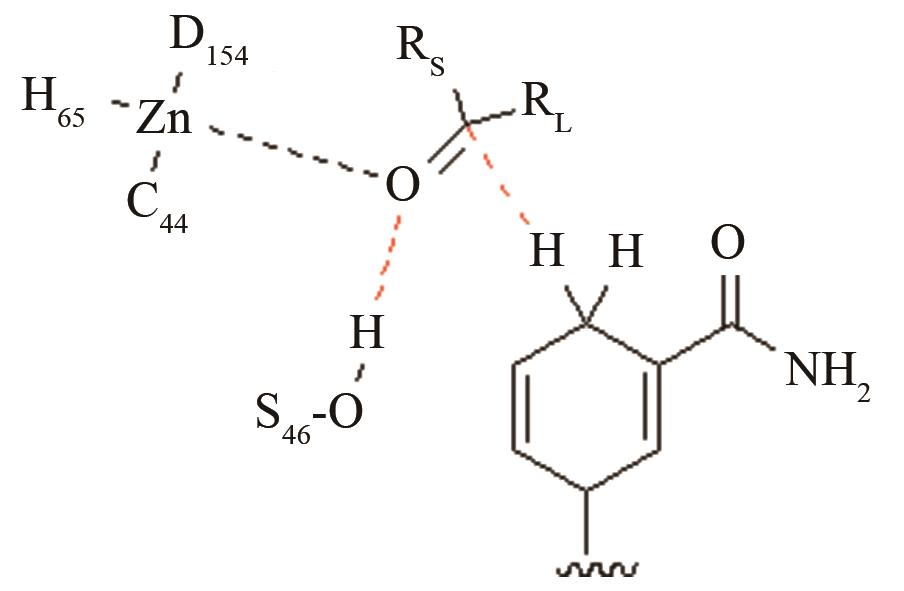
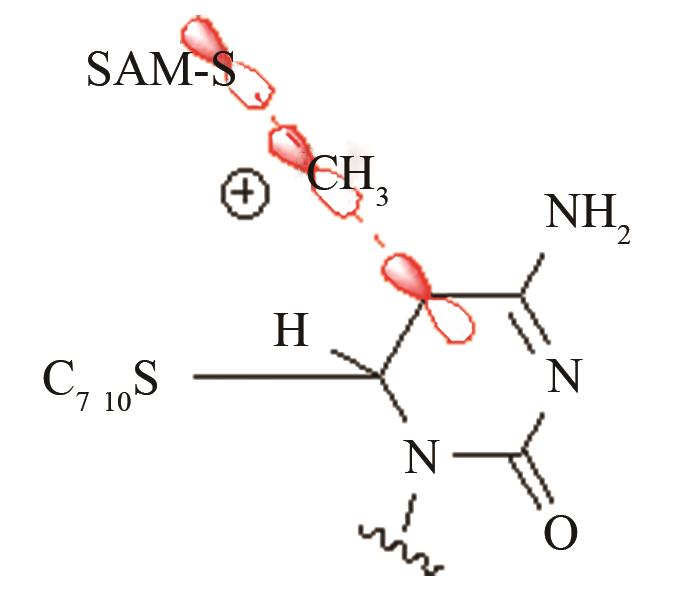
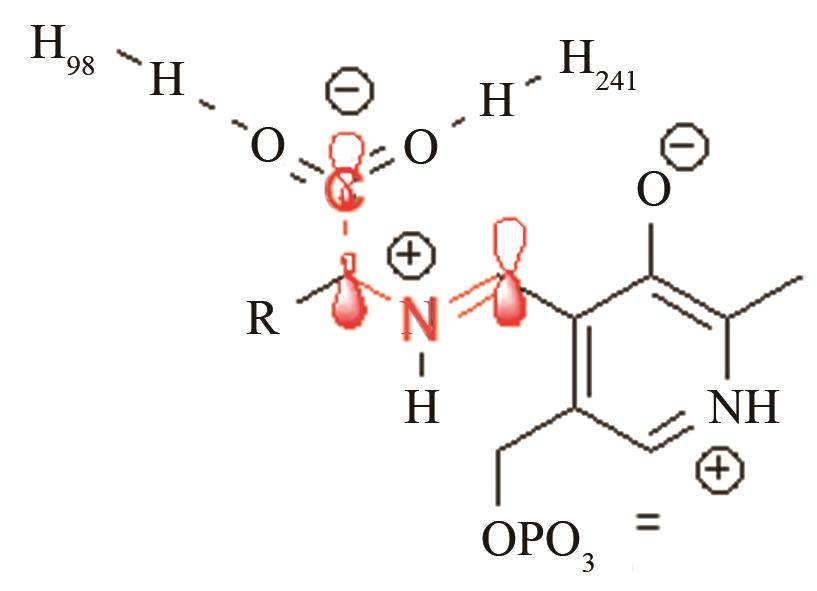
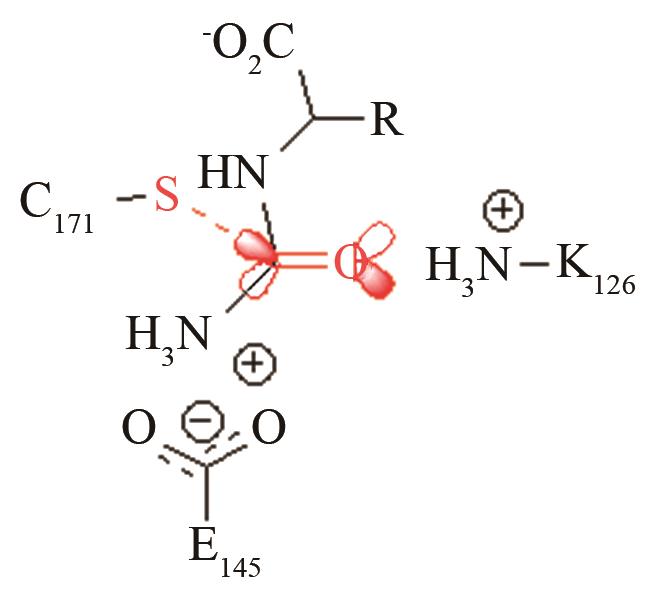
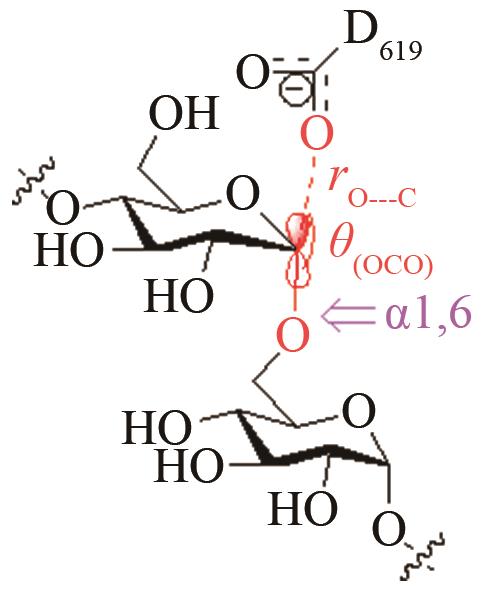
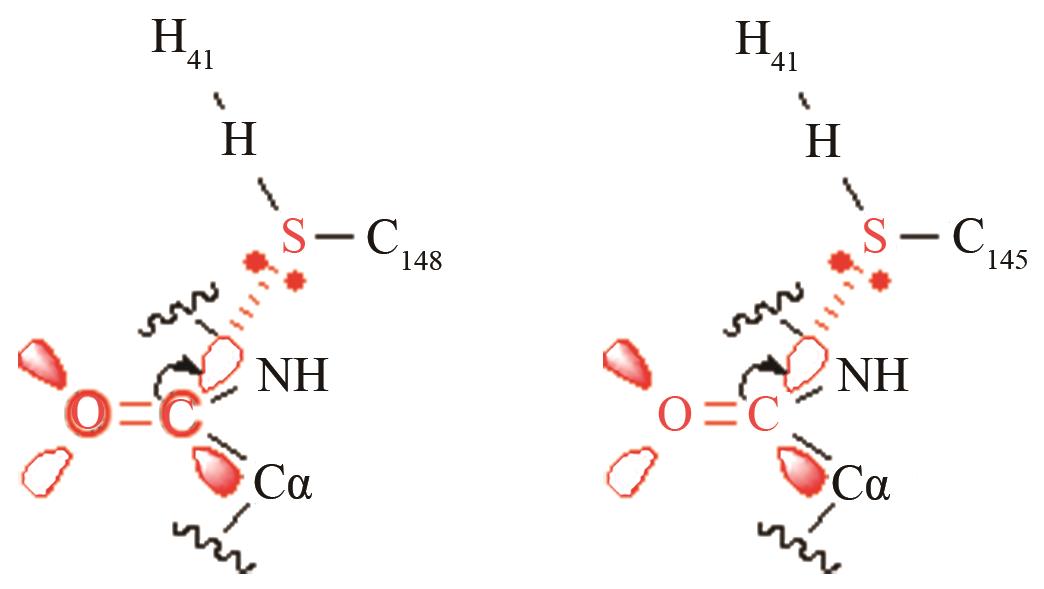
表1 预反应态模型的研究实例[95-98, 101,103,106,108,110,112,114]
Tab. 1 Pre-reaction states of the studied enzymes, control points, and geometric parameters[95-98, 101,103,106,108,110,112,114]
| 研究对象 | 预反应态所选择的控制点 | 实例 |
|---|---|---|
| 红霉素DEBS硫酯酶 | 底物上载后共价复合物水解和环化途径选择 |
与反应位点间隔4~5个化学键的碳原子对反应中心环化和水解活性结构的远程作用[ PRS结构参数:NH259-OH距离、OH-C距离 |
| 苦霉素硫酯酶 | 底物上载后共价复合物水解和环化途径选择 |
12元和14元大环成环对反应中心环化和水解活性结构的影响[ PRS结构参数:NH268-OH距离、OH-C距离 |
| 变构霉素硫酯酶/苦霉素硫酯酶 | 底物上载后共价复合物水解和环化途径选择 |
变构霉素生物合成时环化释放受阻,利用异源硫酯酶调整环化水解选择性,适配性分析[ 变构霉素硫酯酶PRS结构参数:NH255-OH距离、OH-C距离 苦霉素硫酯酶PRS结构参数:NH268-OH距离、OH-C距离 |
| 匹马霉素硫酯酶 | 底物上载后共价复合物水解和环化途径选择 |
与反应位点间隔11~13个化学键的碳原子修饰对反应中心环化和水解活性结构的远程作用[ PRS结构参数:NH261-OH距离、OH-C距离 |
| 醇脱氢酶KpADH | 质子偶合的负氢原子转移关键过渡态 |
高效突变体与野生型反应中心的活性构象分布变化[ PRS结构参数:HY164-O距离、HNADPH-C距离 |
| 醇脱氢酶CpRCR | 质子偶合的负氢原子转移关键过渡态 |
高效突变体与野生型反应中心的活性构象分布变化[ PRS结构参数:HS46-O距离、HNADPH-C距离 |
| DNA甲基化酶3A | SAM甲基转移到DNA-酶共价结合中间体 |
白血病高发的R882H远端突变对活性中心SAM甲基转移的影响[ PRS结构参数:CSAM-C距离 |
| 脱羧酶TyDC | PLP辅因子与底物的共价加合物C-C键断裂 |
通过H98、H251控制共价加合物的反应前线轨道布局[ PRS结构参数:CCαNSB角度,CCαNSBC4’二面角 |
| 酰胺水解酶NiHyuC | 碳四面体氧负离子关键过渡态 |
高效突变体对活化后巯基与被进攻的羧基反应前线轨道布局的影响[ PRS结构参数:SC171-C距离、SC171CO进攻角 |
| 普鲁兰多糖酶 | 取代反应SN2过渡态(碳氧键断裂) |
高效突变体对[O-C-O]取代反应前线轨道布局的影响[ PRS结构参数:OD619-C距离、OD619CO夹角 |
| MERS和SARS冠状病毒主蛋白水解酶 | 碳四面体氧负离子关键过渡态 |
两种病毒主蛋白水解酶的关键反应前线轨道布局[ MERS病毒PRS结构参数:SC148-C距离、SC148CO进攻角 SARS病毒PRS结构参数:SC145-C距离、SC145CO进攻角 |
| 20 | ZHOU Huaxiang, WLODEK S T, MCCAMMON J A. Conformation gating as a mechanism for enzyme specificity[J]. Proceedings of the National Academy of Sciences of the United States of America, 1998, 95(16): 9280-9283. |
| 21 | JOHNSON K A. Role of induced fit in enzyme specificity: a molecular forward/reverse switch[J]. Journal of Biological Chemistry, 2008, 283(39): 26297-26301. |
| 22 | BENKOVIC S J, HAMMES G G, HAMMES-SCHIFFER S. Free-energy landscape of enzyme catalysis[J]. Biochemistry, 2008, 47(11):3317-3321. |
| 23 | WEI Guanghong, XI Wenhui, NUSSINOV R, et al. Protein ensembles: how does nature harness thermodynamic fluctuations for life? The diverse functional roles of conformational ensembles in the cell[J]. Chemical Reviews, 2016, 116(11): 6516-6551. |
| 24 | BUCHHOLZ K, KASCHE V, BORNSCHEUER U T. Biocatalysts and enzyme technology[M]. Wiley-VCH, Weihheim, Germany, 2005. |
| 25 | ARNOLD F H. Combinatorial and computational challenges for biocatalyst design[J]. Nature, 2001, 409(6817): 253-257. |
| 26 | BURTON S G, COWAN D A, WOODLEY J M. The search for the ideal biocatalyst[J]. Nature Biotechnology, 2002, 20(1): 37-45. |
| 27 | KRAUT D A, CARROLL K S, HERSCHLAG D. Challenges in enzyme mechanism and energetics[J]. Annual Review of Biochemistry, 2003, 72: 517-571. |
| 28 | RADZICKA A, WOLFENDEN R. A proficient enzyme[J]. Science, 1995, 267(5194): 90-93. |
| 29 | ZHANG Xiyun, HOUK K N. Why enzymes are proficient catalysts: beyond the Pauling paradigm[J]. Accounts of Chemical Research, 2005, 38(5): 379-385. |
| 30 | TANTILLO D J, CHEN Jiangang, HOUK K N. Theozymes and compuzymes: theoretical models for biological catalysis[J]. Current Opinion in Chemical Biology, 1998, 2(6): 743-750. |
| 31 | KISS G, ÇELEBI-ÖLÇÜM N, MORETTI R, et al. Computational enzyme design[J]. Angewandte Chemie International Edition, 2013, 52(22): 5700-5725. |
| 32 | FRUSHICHEVA M P, MILLS M J, SCHOPF P, et al. Computer aided enzyme design and catalytic concepts[J]. Current Opinion in Chemical Biology, 2014, 21: 56-62. |
| 33 | ANDERSON V E. Quantifying energetic contributions to ground state destabilization[J]. Archives of Biochemistry and Biophysics, 2005, 433(1): 27-33. |
| 34 | SCHRAMM V L. Enzymatic transition states and transition state analog design[J]. Annual Review of Biochemistry, 1998, 67: 693-720. |
| 35 | KOLESNIKOVÁ L, LEÓN I, ALONSO E R, et al. Laser ablation assists cyclization reactions of hydantoic acid: a proof for the near-attack conformation theory?[J]. The Journal of Physical Chemistry Letters, 2019, 10(6): 1325-1330. |
| 36 | CROSS P J, ALLISON T M, DOBSON R C J, et al. Engineering allosteric control to an unregulated enzyme by transfer of a regulatory domain[J]. Proceedings of the National Academy of Sciences of the United States of America, 2013, 110(6): 2111-2116. |
| 37 | ALHAMBRA C, GAO Jiali, CORCHADO J C, et al. Quantum mechanical dynamical effects in an enzyme-catalyzed proton transfer reaction[J]. Journal of the American Chemical Society, 1999, 121(10): 2253-2258. |
| 38 | RIBEIRO J M L, TSAI Sui-ting, PRAMANIK D, et al. Kinetics of ligand-protein dissociation from all-atom simulations: are we there yet?[J]. Biochemistry, 2019, 58(3): 156-165. |
| 39 | BARON R, MCCAMMON J A. Molecular recognition and ligand association[J]. Annual Review of Physical Chemistry, 2013, 64: 151-175. |
| 40 | AGARWAL V, LIN S, LUKK T, et al. Structure of the enzyme-acyl carrier protein (ACP) substrate gatekeeper complex required for biotin synthesis[J]. Proceedings of the National Academy of Sciences of the United States of America, 2012, 109(43): 17406-17411. |
| 41 | YING Hanxiao, WANG Jing, SHI Ting, et al. Engineering of lysine cyclodeaminase conformational dynamics for relieving substrate and product inhibitions in the biosynthesis of L-pipecolic acid[J]. Catalysis Science & Technology, 2019, 9(2): 398-405. |
| 42 | BUTTON D K. Biochemical basis for whole-cell uptake kinetics: specific affinity, oligotrophic capacity, and the meaning of the Michaelis constant[J]. Applied and Environmental Microbiology, 1991, 57(7): 2033-2038. |
| 43 | LEVINE R D. Molecular reaction dynamics[M]. Cambridge, U.K: Cambridge University Press, 2005. |
| 44 | GRUEBELE M, ZEWAIL A H. Ultrafast reaction dynamics[J]. Berichte der Bunsengesellschaft für Physikalische Chemie, 1990, 94(11): 1210-1218. |
| 45 | LAIDLER K J, KING M C. Development of transition-state theory[J]. The Journal of Physical Chemistry, 1983, 87(15): 2657-2664. |
| 46 | OSUNA S, JIMÉNEZ-OSÉS G, NOEY E L, et al. Molecular dynamics explorations of active site structure in designed and evolved enzymes[J]. Accounts of Chemical Research, 2015, 48(4): 1080-1089. |
| 47 | LIGHTSTONE F C, BRUICE T C. Ground state conformations and entropic and enthalpic factors in the efficiency of intramolecular and enzymatic reactions (I): Cyclic anhydride formation by substituted glutarates, succinate, and 3,6-endoxo-Δ 4-tetrahydrophthalate monophenyl esters[J]. Journal of the American Chemical Society, 1996, 118(11): 2595-2605. |
| 48 | HUR S, BRUICE T C. The near attack conformation approach to the study of the chorismate to prephenate reaction[J]. Proceedings of the National Academy of Sciences of the United States of America, 2003, 100(21): 12015-12020. |
| 49 | BRUICE T C. A view at the millennium: the efficiency of enzymatic catalysis[J]. Accounts of Chemical Research, 2002, 35(3): 139-148. |
| 50 | LAU E Y, BRUICE T C. Importance of correlated motions in forming highly reactive near attack conformations in catechol O-methyltransferase[J]. Journal of the American Chemical Society, 1998, 120(48): 12387-12394. |
| 51 | LAU E Y, BRUICE T C. Active site dynamics of the HhaI methyltransferase: insights from computer simulation[J]. Journal of Molecular Biology, 1999, 293(1): 9-18. |
| 52 | TORRES R A, SCHIØTT B, BRUICE T C. Molecular dynamics simulations of ground and transition states for the hydride transfer from formate to NAD+ in the active site of formate dehydrogenase[J]. Journal of the American Chemical Society, 1999, 121(36): 8164-8173. |
| 53 | RADKIEWICZ J L, BROOKS C L. Protein dynamics in enzymatic catalysis: exploration of dihydrofolate reductase[J]. Journal of the American Chemical Society, 2000, 122(2): 225-231. |
| 54 | LAITINEN T, ROUVINEN J, PERÄKYLÄ M. Inversion of the roles of the nucleophile and acid/base catalysts in the covalent binding of epoxyalkyl xyloside inhibitor to the catalytic glutamates of endo-1, 4-β-xylanase (XYNII): A molecular dynamics study[J]. Protein Engineering, Design and Selection, 2000, 13(4): 247-252. |
| 55 | LUO Jia, BRUICE T C. Dynamic structures of horse liver alcohol dehydrogenase (HLADH): results of molecular dynamics simulations of HLADH-NAD+-PhCH2OH, HLADH-NAD+-PhCH2O-, and HLADH-NADH-PhCHO[J]. Journal of the American Chemical Society, 2001, 123(48): 11952-11959. |
| 56 | REDDY S Y, KAHN K, ZHENG Yajun, et al. Protein engineering of nitrile hydratase activity of papain: molecular dynamics study of a mutant and wild-type enzyme[J]. Journal of the American Chemical Society, 2002, 124(44): 12979-12990. |
| 57 | SCHIØTT B, BRUICE T C. Reaction mechanism of soluble epoxide hydrolase: insights from molecular dynamics simulations[J]. Journal of the American Chemical Society, 2002, 124(49): 14558-14570. |
| 58 | MAZUMDER-SHIVAKUMAR D, BRUICE T C. Molecular dynamics studies of ground state and intermediate of the hyperthermophilic indole-3-glycerol phosphate synthase[J]. Proceedings of the National Academy of Sciences of the United States of America, 2004, 101(40): 14379-14384. |
| 59 | BRUICE T C. Computational approaches: reaction trajectories, structures, and atomic motions. enzyme reactions and proficiency[J]. Chemical Reviews, 2006, 106(8): 3119-3139. |
| 60 | SENN H M, THIEL W. QM/MM methods for biomolecular systems[J]. Angewandte Chemie International Edition, 2009, 48(7): 1198-1229. |
| 61 | CLAEYSSENS F, RANAGHAN K E, MANBY F R, et al. Multiple high-level QM/MM reaction paths demonstrate transition-state stabilization in chorismate mutase: correlation of barrier height with transition-state stabilization[J]. Chemical Communications, 2005(40): 5068-5070. |
| 62 | ZHANG Xiaodong, Zhang Xiaohua, BRUICE T C. A definitive mechanism for chorismate mutase[J]. Biochemistry, 2005, 44(31): 10443-10448. |
| 63 | GIRALDO J, ROCHE D, ROVIRA X, et al. The catalytic power of enzymes: conformational selection or transition state stabilization?[J]. FEBS Letters, 2006, 580(9): 2170-2177. |
| 64 | RYDE U. How many conformations need to be sampled to obtain converged QM/MM energies? the curse of exponential averaging[J]. Journal of Chemical Theory and Computation, 2017, 13(11): 5745-5752. |
| 65 | COOPER A M, KÄSTNER J. Averaging techniques for reaction barriers in QM/MM simulations[J]. ChemPhysChem, 2014, 15(15): 3264-3269. |
| 66 | LI Yanwei, ZHANG Ruiming, DU Likai, et al. Catalytic mechanism of C-F bond cleavage: insights from QM/MM analysis of fluoroacetate dehalogenase[J]. Catalysis Science & Technology, 2016, 6(1): 73-80. |
| 67 | DIETERICH J M, WERNER H J, MATA R A, et al. Reductive half-reaction of aldehyde oxidoreductase toward acetaldehyde: ab initio and free energy quantum mechanical/molecular mechanical calculations[J]. Journal of Chemical Physics, 2010, 132(3):035101. |
| 1 | CANTON B, LABNO A, ENDY D. Refinement and standardization of synthetic biological parts and devices[J]. Nature Biotechnology, 2008, 26(7): 787-793. |
| 2 | 崔颖璐, 吴边. 符合工程化需求的生物元件设计[J]. 中国科学院院刊, 2018, 33(11): 1150-1157. |
| CUI Yinglu, WU Bian. Biological components design for engineering requirements[J]. Bulletin of Chinese Academy of Sciences, 2018, 33(11): 1150-1157. | |
| 3 | ZEYMER C, HILVERT D. Directed evolution of protein catalysts[J]. Annual Review of Biochemistry, 2018, 87: 131-157. |
| 4 | REETZ M T. KAHAKEAW D, LOHMER R. Addressing the numbers problem in directed evolution[J]. ChemBioChem, 2008, 9(11):1797-1804. |
| 5 | HUANG P S, BOYKEN S E, BAKER D. The coming of age of de novo protein design[J]. Nature, 2016, 537(7620): 320-327. |
| 6 | KLEPEIS J L, LINDORFF-LARSEN K, DROR R O, et al. Long-timescale molecular dynamics simulations of protein structure and function[J]. Current Opinion in Structural Biology, 2009, 19(2): 120-127. |
| 7 | AHMADI S, BARRIOS HERRERA L, CHEHELAMIRANI M, et al. Multiscale modeling of enzymes: QM-cluster, QM/MM, and QM/MM/MD: a tutorial review[J]. International Journal of Quantum Chemistry, 2018, 118(9): e25558. |
| 8 | SALOMON-FERRER R, CASE D A, WALKER R C. An overview of the Amber biomolecular simulation package[J]. Wiley Interdisciplinary Reviews: Computational Molecular Science, 2013, 3(2): 198-210. |
| 9 | HOUK K N, LIU Fang. Holy grails for computational organic chemistry and biochemistry[J]. Accounts of Chemical Research, 2017, 50(3): 539-543. |
| 10 | KRYLOV A, WINDUS T L, BARNES T, et al. Perspective: Computational chemistry software and its advancement as illustrated through three grand challenge cases for molecular science[J]. The Journal of Chemical Physics, 2018, 149(18): 180901. |
| 11 | GRIMME S, SCHREINER P R. Computational chemistry: The fate of current methods and future challenges[J]. Angewandte Chemie International Edition, 2018, 57(16): 4170-4176. |
| 68 | LONSDALE R, REETZ M T. Reduction of α, β-unsaturated ketones by old yellow enzymes: mechanistic insights from quantum mechanics/molecular mechanics calculations[J]. Journal of the American Chemical Society, 2015, 137(46): 14733-14742. |
| 69 | VASILEVSKAYA T, KHRENOVA M G, NEMUKHIN A V, et al. Methodological aspects of QM/MM calculations: a case study on matrix metalloproteinase-2[J]. Journal of Computational Chemistry, 2016, 37(19): 1801-1809. |
| 70 | ASTRUC D. Organometallic chemistry and catalysis[M]. New York: Springer, 2007. |
| 71 | ROONEY J J. The extended Eyring kinetic equation and the compensation effect in catalysis[J]. Journal of Molecular Catalysis A: Chemical, 1998, 129(2/3): 131-134. |
| 72 | KOZUCH S, SHAIK S. How to conceptualize catalytic cycles? The energetic span model[J]. Accounts of Chemical Research, 2011, 44(2): 101-110. |
| 73 | AMATORE C, JUTAND A. Mechanistic and kinetic studies of palladium catalytic systems[J]. Journal of Organometallic Chemistry, 1999, 576(1/2): 254-278. |
| 74 | CHRISTIANSEN J A. The elucidation of reaction mechanisms by the method of intermediates in quasi-stationary concentrations[J]. Advances in Catalysis, 1953, 5: 311-353. |
| 75 | KOZUCH S, SHAIK S. A combined kinetic-quantum mechanical model for assessment of catalytic cycles: application to cross-coupling and Heck reactions[J]. Journal of the American Chemical Society, 2006, 128(10): 3355-3365. |
| 76 | CAMPBELL C T. Finding the rate-determining step in a mechanism: comparing DeDonder relations with the "degree of rate control"[J]. Journal of Catalysis, 2001, 204(2): 520-524. |
| 77 | KOZUCH S, SHAIK S. Kinetic-quantum chemical model for catalytic cycles: The Haber-Bosch process and the effect of reagent concentration[J]. The Journal of Physical Chemistry A, 2008, 112(26): 6032-6041. |
| 78 | KOZUCH S, SHAIK S. Defining the optimal inductive and steric requirements for a cross-coupling catalyst using the energetic span model[J]. Journal of Molecular Catalysis A: Chemical, 2010, 324(1/2): 120-126. |
| 79 | STEGELMANN C, ANDREASEN A, CAMPBELL C T. Degree of rate control: how much the energies of intermediates and transition states control rates[J]. Journal of the American Chemical Society, 2009, 131(23): 8077-8082. |
| 12 | CHOWDHURY R, MARANAS C D. From directed evolution to computational enzyme engineering-a review[J]. AIChE Journal, 2020, 66(3): e16847. |
| 13 | OSUNA S. The challenge of predicting distal active site mutations in computational enzyme design[J]. WIREs Computational Molecular Science, 2020, e1502. |
| 14 | MAGALHÃES R P, FERNANDES H S, SOUSA S F. Modelling enzymatic mechanisms with QM/MM approaches: Current status and future challenges[J]. Israel Journal of Chemistry, 2020, 60(7): 655-666. |
| 15 | PAULING L. Nature of forces between large molecules of biological interest[J]. Nature, 1948, 161(4097): 707-709. |
| 16 | MICHAELIS L, MENTEN M L, JOHNSON K A, et al. The original Michaelis constant: translation of the 1913 Michaelis-Menten paper[J]. Biochemistry, 2011, 50(39): 8264-8269. |
| 17 | TOMCZAK J M, WĘGLARZ-TOMCZAK E. Estimating kinetic constants in the Michaelis-Menten model from one enzymatic assay using Approximate Bayesian Computation[J]. FEBS Letters, 2019, 593(19): 2742-2750. |
| 18 | HOUK K N, LEACH A G, KIM S P, et al. Binding affinities of host-guest, protein-ligand, and protein-transition-state complexes[J]. Angewandte Chemie International Edition, 2003, 42(40): 4872-4897. |
| 19 | SCHRAMM V L. Enzymatic transition states and transition state analogues[J]. Current Opinion in Structural Biology, 2005, 15(6): 604-613. |
| 80 | ROMERO P A, ARNOLD F H. Exploring protein fitness landscapes by directed evolution[J]. Nature Reviews Molecular Cell Biology, 2009, 10(12): 866-876. |
| 81 | VOIGT C A, MAYO S L, ARNOLD F H, et al. Computational method to reduce the search space for directed protein evolution[J]. Proceedings of the National Academy of Sciences of the United States of America, 2001, 98(7): 3778-3783. |
| 82 | TRACEWELL C A, ARNOLD F H. Directed enzyme evolution: climbing fitness peaks one amino acid at a time[J]. Current Opinion in Chemical Biology, 2009, 13(1): 3-9. |
| 83 | UHE A, KOZUCH S, SHAIK S. Automatic analysis of computed catalytic cycles[J]. Journal of Computational Chemistry, 2011, 32(5): 978-985. |
| 84 | SOLEL E, TARANNAM N, KOZUCH S. Catalysis: energy is the measure of all things[J]. Chemical Communications, 2019, 55(37): 5306-5322. |
| 85 | EINSIEDLER M, JAMIESON C S, MASKERI M A, et al. Fungal dioxygenase AsqJ is promiscuous and bimodal: substrate-directed formation of quinolones versus quinazolinones[J]. Angewandte Chemie International Edition, 2021, 60(15): 8297-8302. |
| 86 | LIU Fengjiao, YANG Zhongyue, YU Yanmin, et al. Bimodal Evans-Polanyi relationships in dioxirane oxidations of sp3 C—H: non-perfect synchronization in generation of delocalized radical intermediates[J]. Journal of the American Chemical Society, 2017, 139(46):16650-16656. |
| 87 | WILLIAMS G J. Engineering polyketide synthases and nonribosomal peptide synthetases[J]. Current Opinion in Structural Biology, 2013, 23(4): 603-612. |
| 88 | LARSEN E M, WILSON M R, TAYLOR R E. Conformation-activity relationships of polyketide natural products[J]. Natural Product Reports, 2015, 32(8): 1183-1206. |
| 89 | MEIER J L, BURKART M D. The chemical biology of modular biosynthetic enzymes[J]. Chemical Society Reviews, 2009, 38(7): 2012-2045. |
| 90 | SMITH S, TSAI S C. The type I fatty acid and polyketide synthases: a tale of two megasynthases[J]. Natural Product Reports, 2007, 24(5): 1041-1072. |
| 91 | KEATINGE-CLAY A T. The structures of type I polyketide synthases[J]. Natural Product Reports, 2012, 29(10): 1050-1073. |
| 92 | KHOSLA C, TANG Yingyuan, CHEN A Y, et al. Structure and mechanism of the 6-deoxyerythronolide B synthase[J]. Annual Reviews of Biochemistry, 2007, 76: 195-221. |
| 93 | ALDRICH C C, VENKATRAMAN L, SHERMAN D H, et al. Chemoenzymatic synthesis of the polyketide macrolactone 10-deoxymethynolide[J]. Journal of the American Chemical Society, 2005, 127(25): 8910-8911. |
| 94 | HE Weiguo, WU Jiaquan, KHOSLA C, et al. Macrolactonization to 10-deoxymethynolide catalyzed by the recombinant thioesterase of the picromycin/methymycin polyketide synthase[J]. Bioorganic & Medicinal Chemistry Letters, 2006, 16(2): 391-394. |
| 95 | CHEN Xiongping, SHI Ting, WANG Xiaolei, et al. Theoretical studies on the mechanism of thioesterase-catalyzed macrocyclization in erythromycin biosynthesis[J]. ACS Catalysis, 2016, 6(7):4369-4378. |
| 96 | SHI Ting, LIU Lanxuan, TAO Wentao, et al. Theoretical studies on the catalytic mechanism and substrate diversity for macrocyclization of pikromycin thioesterase[J]. ACS Catalysis, 2018, 8(5): 4323-4332. |
| 97 | LIU Lei, TAO Wentao, BAI Linquan, et al. Why does tautomycetin thioesterase prefer hydrolysis to macrocyclization? Theoretical study on its catalytic mechanism[J]. Catalysis Science & Technology, 2019, 9(22): 6391-6403. |
| 98 | FAN Shuobing, WANG Rufan, LI Chen, et al. Insight into structural characteristics of protein-substrate interaction in pimaricin thioesterase[J]. International Journal of Molecular Sciences, 2019, 20(4):877. |
| 99 | HALL M, BOMMARIUS A S. Enantioenriched compounds via enzyme-catalyzed redox reactions[J]. Chemical Reviews, 2011, 111(7): 4088-4110. |
| 100 | KULIG J, SIMON R C, ROSE C A, et al. Stereoselective synthesis of bulky 1,2-diols with alcohol dehydrogenases[J]. Catalysis Science & Technology, 2012, 2(8): 1580. |
| 101 | ZHOU Jieyu, WANG Yue, XU Guochao, et al. Structural insight into enantioselective inversion of an alcohol dehydrogenase reveals a "polar gate" in stereorecognition of diaryl ketones[J]. Journal of the American Chemical Society, 2018, 140(39): 12645-12654. |
| 102 | NOEY E L, TIBREWAL N, JIMÉNEZ-OSÉS G, et al. Origins of stereoselectivity in evolved ketoreductases[J]. Proceedings of the National Academy of Sciences of the United States of America, 2015, 112(51): E7065-E7072. |
| 103 | NIE Yao, WANG Shanshan, XU Yan, et al. Enzyme engineering based on X-ray structures and kinetic profiling of substrate libraries: alcohol dehydrogenases for stereospecific synthesis of a broad range of chiral alcohols[J]. ACS Catalysis, 2018, 8(6): 5145-5152. |
| 104 | MESSERSCHMIDT D M, KNOWLES B B, SOLTER D. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos[J]. Genes & Development, 2014, 28(8): 812-828. |
| 105 | YANG Li, LIU Yanan, ZHANG Na, et al. Novel impact of the DNMT3A R882H mutation on GSH metabolism in a K562 cell model established by TALENs[J]. Oncotarget, 2017, 8(18): 30395-30409. |
| 106 | LIU Lanxuan, SHI Ting, HOUK K N, et al. Understanding the R882H mutation effects of DNA methyltransferase DNMT3A: a combination of molecular dynamics simulations and QM/MM calculations[J]. RSC Advance, 2019, 9:31425-31434. |
| 107 | DUNATHAN H C. Conformation and reaction specificity in pyridoxal phosphate enzymes[J]. Proceedings of the National Academy of Sciences of the United States, 1966, 55(4): 712-716. |
| 108 | NI Jie, XU Guochao, DAI Wei, et al. Hyperconjugation promoted by hydrogen bonding between His98/His241 and a carboxyl group contributes to tyrosine decarboxylase catalysis[J]. Catalysis Science & Technology, 2019, 9(22): 6222-6226. |
| 109 | HERAS-YAZQUEZ F, CLEMENTE-JIMENEZ J, MARTINEZ-RODRIGUEZ S, et al. Optically pure α-amino acids production by the "hydantoinase process"[J]. Recent Patents Biotechnology, 2008, 2(1): 35-46. |
| 110 | LIU Yafei, XU Guocao, ZHOU Jieyu, et al. Structure-guided engineering of D-carbamoylase reveals a key loop at substrate entrance tunnel[J]. ACS Catalysis, 2020, 10(21): 12393-12402. |
| 111 | BERTOLDO C, ANTRANIKIAN G. Starch-hydrolyzing enzymes from thermophilic archaea and bacteria[J]. Current Opinion in Chemical Biology, 2002, 6(2): 151-160. |
| 112 | WANG Xinye, JING Xiaoran, Deng Yi, et al. Evolutionary coupling saturation mutagenesis: coevolution‐guided identification of distant sites influencing Bacillus naganoensis pullulanase activity[J]. FEBS Letters, 2020, 594(5): 799-812. |
| 113 | TARANTO A G, CARVALHO P, AVERY M A. QM/QM studies for Michael reaction in coronavirus main protease (3CLPro)[J]. Journal of Molecular Graphics and Modelling, 2008, 27(3): 275-285. |
| 114 | WANG Hao, He Shuai, DENG Weilong, et al. Comprehensive insights into the catalytic mechanism of middle east respiratory syndrome 3C-like protease and severe acute respiratory syndrome 3C-like protease[J]. ACS Catalysis, 2020, 10: 5871-5890. |
| [1] | 张成辛. 基于文本数据挖掘的蛋白功能预测的机遇与挑战[J]. 合成生物学, 2025, (): 1-14. |
| [2] | 李倩, James E. Ferrell, 陈于平. 细胞质浓度:细胞生物学的老问题、新参数[J]. 合成生物学, 2025, (): 1-18. |
| [3] | 姜百翼, 钱珑. 活细胞记录器在细胞谱系追踪中的应用和前景[J]. 合成生物学, 2025, (): 1-17. |
| [4] | 温艳华, 刘合栋, 曹春来, 巫瑞波. 蛋白质工程在医药产业中的应用[J]. 合成生物学, 2025, 6(1): 65-86. |
| [5] | 董颖, 马孟丹, 黄卫人. CRISPR-Cas系统的小型化研究进展[J]. 合成生物学, 2025, 6(1): 105-117. |
| [6] | 张日新, 田晓军. 合成基因回路面临的细胞‘经济学窘境’[J]. 合成生物学, 2025, (): 1-14. |
| [7] | 谭骁天, 李睿涵, 杨慧. 生物分子传感中的抗体探针:由碳基计算走向硅基计算[J]. 合成生物学, 2025, (): 1-9. |
| [8] | 邵明威, 孙思勉, 杨时茂, 陈国强. 基于极端微生物的生物制造[J]. 合成生物学, 2024, 5(6): 1419-1436. |
| [9] | 刘建明, 张炽坚, 张冰, 曾安平. 巴氏梭菌作为工业底盘细胞高效生产1,3-丙二醇——从代谢工程和菌种进化到过程工程和产品分离[J]. 合成生物学, 2024, 5(6): 1386-1403. |
| [10] | 刘益宁, 蒲伟, 杨金星, 王钰. ω-氨基酸与内酰胺的生物合成研究进展[J]. 合成生物学, 2024, 5(6): 1350-1366. |
| [11] | 赵亮, 李振帅, 付丽平, 吕明, 王士安, 张全, 刘立成, 李福利, 刘自勇. 生物转化一碳化合物原料产油脂与单细胞蛋白研究进展[J]. 合成生物学, 2024, 5(6): 1300-1318. |
| [12] | 宋开南, 张礼文, 王超, 田平芳, 李广悦, 潘国辉, 徐玉泉. 小分子生物农药及其生物合成研究进展[J]. 合成生物学, 2024, (): 1-21. |
| [13] | 伊进行, 唐宇琳, 李春雨, 吴鹤云, 马倩, 谢希贤. 氨基酸衍生物在化妆品中的应用及其生物合成研究进展[J]. 合成生物学, 2024, (): 1-36. |
| [14] | 瞿旭东, 唐功利, 丁奎岭. 打破学科界限——合成科学的新发展[J]. 合成生物学, 2024, 5(5): 909-912. |
| [15] | 张守祺, 王涛, 孔尧, 邹家胜, 刘元宁, 徐正仁. 天然产物的化学-酶法合成:方法与策略的演进[J]. 合成生物学, 2024, 5(5): 913-940. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
