合成生物学 ›› 2022, Vol. 3 ›› Issue (3): 429-444.DOI: 10.12211/2096-8280.2021-032
人工智能辅助的蛋白质工程
卞佳豪, 杨广宇
- 上海交通大学 生命科学技术学院,微生物代谢国家重点实验室,上海 200240
-
收稿日期:2021-03-16修回日期:2021-05-24出版日期:2022-06-30发布日期:2022-07-13 -
通讯作者:杨广宇 -
作者简介:卞佳豪 (1997—),男,硕士研究生。研究方向为人工智能辅助定向进化的组合方法。 E-mail:bjh2170@sjtu.edu.cn杨广宇 (1980—),男,研究员,博士生导师。研究方向为酶定向进化、酶高通量筛选、酶技术应用、体外合成生物学等。E-mail:yanggy@sjtu.edu.cn -
基金资助:国家自然科学基金(32030063)
Artificial intelligence-assisted protein engineering
BIAN Jiahao, YANG Guangyu
- State Key Laboratory of Microbial Metabolism,School of Life Sciences and Biotechnology,Shanghai Jiao Tong University,Shanghai 200240,China
-
Received:2021-03-16Revised:2021-05-24Online:2022-06-30Published:2022-07-13 -
Contact:YANG Guangyu
摘要:
蛋白质工程是合成生物学领域的重要研究方向之一。但目前人类对于蛋白质折叠、酶天然进化机制等基础生物学问题的理解仍很有限,因此基于理性设计方法进行蛋白质的功能从头设计(de novo design)仍然是一个难题。定向进化(directed evolution)通过在实验室模拟自然进化的原理,可以在不依赖结构和机制信息的基础上对蛋白质的功能进行有效优化。但是定向进化高度依赖高通量筛选方法,也限制了其对缺少高通量筛选方法的蛋白质进行改造的能力。近年来,人工智能辅助的蛋白质工程逐渐发展成为一种高效的蛋白质分子设计新策略,在蛋白质的结构预测、功能预测、溶解度预测和指导智能文库设计等多个方面显现出独特的优势,成为理性设计和定向进化之后的又一次技术发展的浪潮。本文综述了近年来人工智能辅助的蛋白质工程的应用进展,对其中的代表性工作进行了重点阐述。在简单介绍了人工智能蛋白质工程策略的原理和流程之后,对数据、分子描述符和人工智能算法等三个影响预测模型性能的关键点进行了分析,总结了该策略中的主要数据库、分子描述符和算法的主流工具包及平台,介绍了它们的功能、用途和网址。我们还对人工智能策略目前仍面临的不足进行了探讨,如高质量数据不足、实验数据存在偏差、缺少通用模型等。随着自动基因功能注释技术、超高通量筛选技术和人工智能算法的不断发展,将会给人工智能辅助的蛋白质工程提供足够的高质量数据和更准确的算法,从而不断提升人工智能辅助的蛋白质工程预测准确度,为合成生物学研究提供更大的助力。
中图分类号:
引用本文
卞佳豪, 杨广宇. 人工智能辅助的蛋白质工程[J]. 合成生物学, 2022, 3(3): 429-444.
BIAN Jiahao, YANG Guangyu. Artificial intelligence-assisted protein engineering[J]. Synthetic Biology Journal, 2022, 3(3): 429-444.

图1 理性设计,定向进化和人工智能辅助的蛋白质工程策略示意图(理性设计依赖序列和结构信息,精准设计突变体文库,但难以应用于缺少结构功能信息的蛋白质。定向进化中对目标基因进行多轮突变和筛选实验,不受结构功能信息限制,但是需要进行高通量的筛选方法。人工智能辅助的蛋白质工程则需要大量的序列-功能数据,可以来源于实验、计算和数据库等多方面,通过构建的预测模型,能够更有效地探索蛋白质突变体序列空间)
Fig. 1 Schematic diagram for rational design, directed evolution and artificial intelligence-assisted protein engineering(Rational design relies on sequence and structural information to design mutant libraries accurately. However, it is difficult for being applied to proteins lacking structural and functional information. In the directed evolution strategy, multiple rounds of mutation and screening experiments are performed on target genes, which are not limited by structural and functional information, but high-throughput screening methods are required. Artificial intelligence-assisted protein engineering requires a large amount of sequence-function data, which can be derived from experiments, calculations, and databases. Through the predictive model, the sequence space of protein mutants can be explored more effectively)
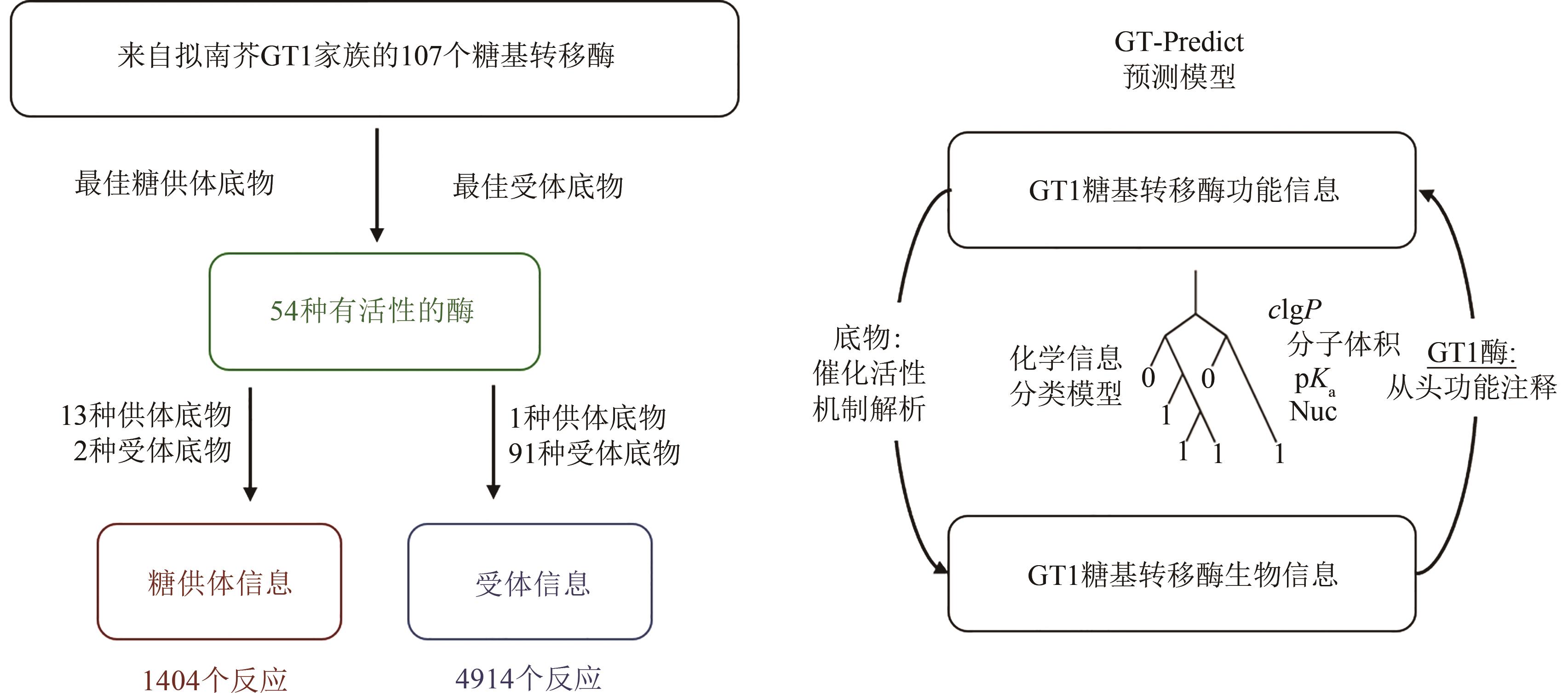
图2 GT1家族糖基转移酶预测模型(GT-Predict)的工作流程[19](基于功能的算法学习方法GT-Predict,使用来源于酶、亲电试剂和亲核试剂的多种训练集来创建基于物理化学和局部序列的分类器,从而预测GT1糖基转移酶的催化活性和功能信息。Nuc表示亲核基团的数量/类型)
Fig. 2 Workflow for predicting the GT1 glycosyltransferase model (GT-Predict)[19](The function-based algorithmic learning approach, GT-Predict, uses a diverse training set of enzymes, electrophiles, and nucleophiles to create a physicochemical and local-sequence-based classifier for predicting the novel transformations and functional annotation of GT group-transfer enzymes.)
| 名称 | 发表日期 | 分子描述符 | 程序/算法 | 软件/工具包地址 |
|---|---|---|---|---|
| PRIAM | 2003年 11月15日 | 序列比对构建的同源模块 | PSI-BLAST序列比对程序 | http://genopole.toulouse.inra.fr/bioinfo/priam/ |
| CatFam | 2008年 07月17日 | 序列比对和分层聚类 | ClustalW和PSI-BLAST 序列比对程序 | http://www.bhsai.org/downloads/catfam.tar.gz |
| EFICAz2.5 | 2012年 08月24日 | 序列比对和分层聚类 | 支持向量机(SVM)和 分类树(classification trees) | http://cssb.biology.gatech.edu/EFICAz2.5 |
| SVM-Prot | 2016年 08月15日 | 多种分子描述符(多种氨基酸 残基特性描述符和整体描述符) | 支持向量机(SVM), K最近邻(KNN)和概率 神经网络(sPNN) | http://bidd2.nus.edu.sg/cgi-bin/svmprot/svmprot.cgi |
| COFACTOR | 2017年 05月02日 | 来自BioLiP文库的结构信息 | 基于TM分数的蛋白质结构比对算法 | http://zhanglab.ccmb.med.umich.edu/COFACTOR/。 |
| DEEPre | 2017年 10月23日 | 序列独热编码,位置特异性得分矩阵,溶剂可及性,二级结构独热编码和功能结构域 | 卷积神经网络(CNN) 和循环神经网络(RNN) | http://www.cbrc.kaust.edu.sa/DEEPre |
| DETECT v2 | 2018年 05月02日 | 酶EC编号的正负密度分布图 | 贝叶斯框架(Bayesian framework) | https://github.com/ParkinsonLab/DETECT-v2 |
| ECPred | 2018年 09月21日 | 三种基于子序列,序列相似性和氨基酸物理化学特征的分子描述符:SPMap,BLAST-kNN和Pepstats-SVM | 二进制分类算法 | https://ecpred.kansil.org/ |
| DeepEC | 2019年 06月20日 | 独热编码 | 卷积神经网络(CNN) | https://bitbucket.org/kaistsystemsbiology/deepec |
表1 EC编号预测工具汇总表
Tab. 1 Forecast tools for EC numbers
| 名称 | 发表日期 | 分子描述符 | 程序/算法 | 软件/工具包地址 |
|---|---|---|---|---|
| PRIAM | 2003年 11月15日 | 序列比对构建的同源模块 | PSI-BLAST序列比对程序 | http://genopole.toulouse.inra.fr/bioinfo/priam/ |
| CatFam | 2008年 07月17日 | 序列比对和分层聚类 | ClustalW和PSI-BLAST 序列比对程序 | http://www.bhsai.org/downloads/catfam.tar.gz |
| EFICAz2.5 | 2012年 08月24日 | 序列比对和分层聚类 | 支持向量机(SVM)和 分类树(classification trees) | http://cssb.biology.gatech.edu/EFICAz2.5 |
| SVM-Prot | 2016年 08月15日 | 多种分子描述符(多种氨基酸 残基特性描述符和整体描述符) | 支持向量机(SVM), K最近邻(KNN)和概率 神经网络(sPNN) | http://bidd2.nus.edu.sg/cgi-bin/svmprot/svmprot.cgi |
| COFACTOR | 2017年 05月02日 | 来自BioLiP文库的结构信息 | 基于TM分数的蛋白质结构比对算法 | http://zhanglab.ccmb.med.umich.edu/COFACTOR/。 |
| DEEPre | 2017年 10月23日 | 序列独热编码,位置特异性得分矩阵,溶剂可及性,二级结构独热编码和功能结构域 | 卷积神经网络(CNN) 和循环神经网络(RNN) | http://www.cbrc.kaust.edu.sa/DEEPre |
| DETECT v2 | 2018年 05月02日 | 酶EC编号的正负密度分布图 | 贝叶斯框架(Bayesian framework) | https://github.com/ParkinsonLab/DETECT-v2 |
| ECPred | 2018年 09月21日 | 三种基于子序列,序列相似性和氨基酸物理化学特征的分子描述符:SPMap,BLAST-kNN和Pepstats-SVM | 二进制分类算法 | https://ecpred.kansil.org/ |
| DeepEC | 2019年 06月20日 | 独热编码 | 卷积神经网络(CNN) | https://bitbucket.org/kaistsystemsbiology/deepec |
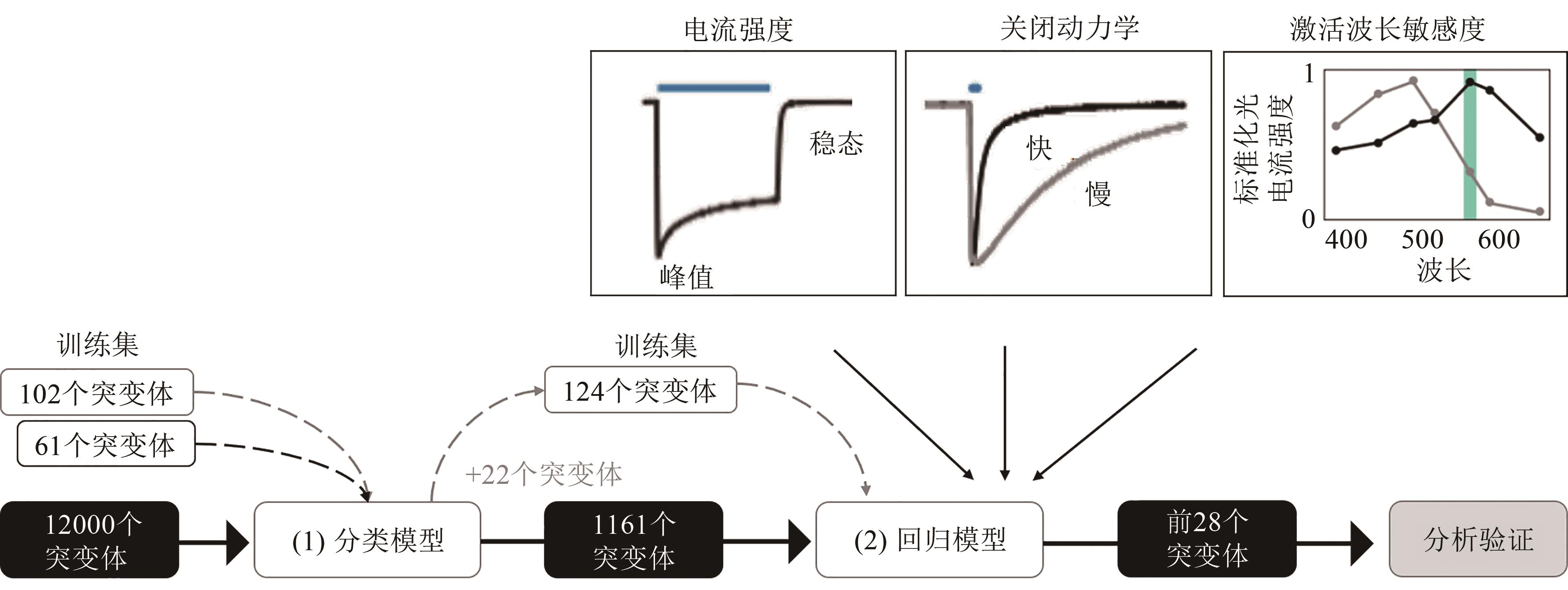
图3 人工智能辅助的视紫红质通道蛋白改造的工作流程[26][在重组文库中表征的102种ChR蛋白和文献中报道的61种变体,共同构成了(1)分类模型的训练集。然后,使用经过训练的分类模型来预测12000个未表征的ChR序列变体是否具有功能。接下来,构建了三个(2)回归模型,分别针对不同的ChR光电流特性:光电流强度,关闭动力学和光电流的波长敏感性]
Fig. 3 Workflow for machine learning-guided channelrhodopsin engineering[26][102 ChR proteins characterized in the recombinant library, together with 61 variants reported in the literature, constitute the training set of the classification model (1). Then the trained classification model was used to predict whether 12000 uncharacterized ChR sequence variants are functional, and three regression models (2) were trained, one for each of the ChR photocurrent properties of interest: photocurrent strength, off-kinetics and wavelength sensitivity of the photocurrents.]
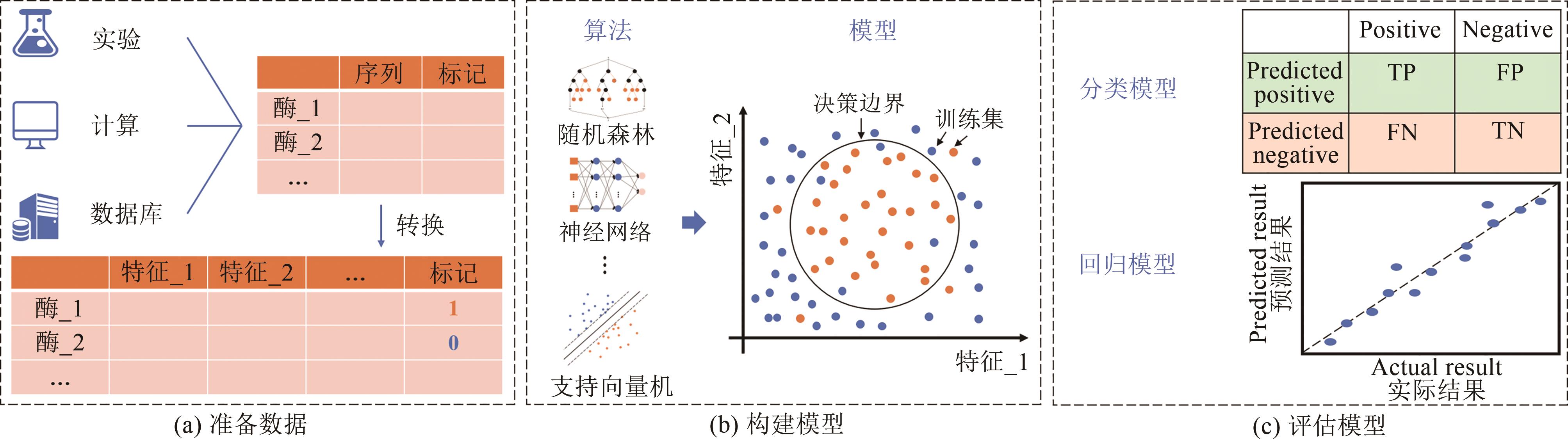
图4 监督学习的流程示意图[27](a)准备数据:来源于实验,计算或数据库的数据通常会转换成计算机可以识别的格式,并拆分为训练集和测试集;(b)构建预测模型:利用训练集训练不同的算法以找到决策边界,构建预测模型,例如随机森林,神经网络和支持向量机;(c)验证模型:对于分类问题或者回归问题,应选择合适的评估方法
Fig. 4 Schematic diagram of the supervised learning process[27]Step (a): Preparing data. The data from experiments, calculations or databases are usually converted to a format that the computer can recognize and split into the training and test parts. Step(b): Constructing a predictive model. Using the training set to train different algorithms to find decision boundaries, such as random forests, neural networks and support vector machines, so as to build predictive models. Step (c): Validating the model. An appropriate evaluation method should be selected for tasks with classification or regression.
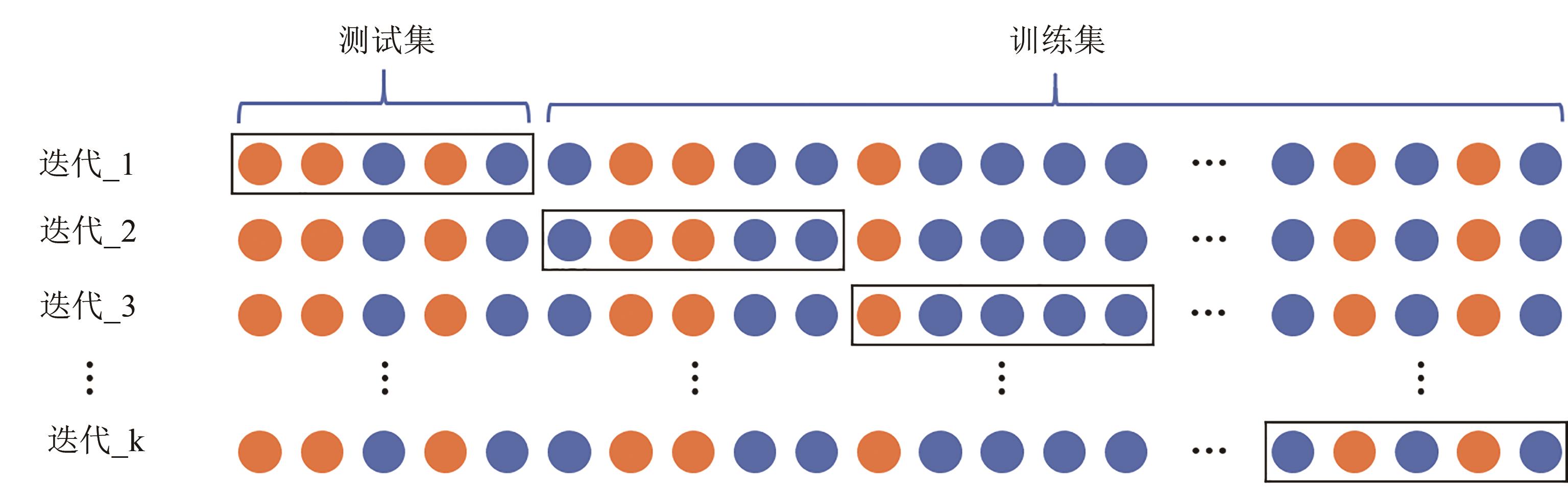
图5 k折交叉验证示意图(将训练数据进一步细分为k个子集,并且将训练工作流程重复k次,同时保留k个子集中的一个用于评估,其余k-1个子集用于训练)
Fig. 5 Schematic diagram for k-fold cross-validation(The training data is further subsplit into k subsets, and the training workflow is repeated k times with each of the k subsets holding for evaluation and the remaining k-1 subsets used for training)
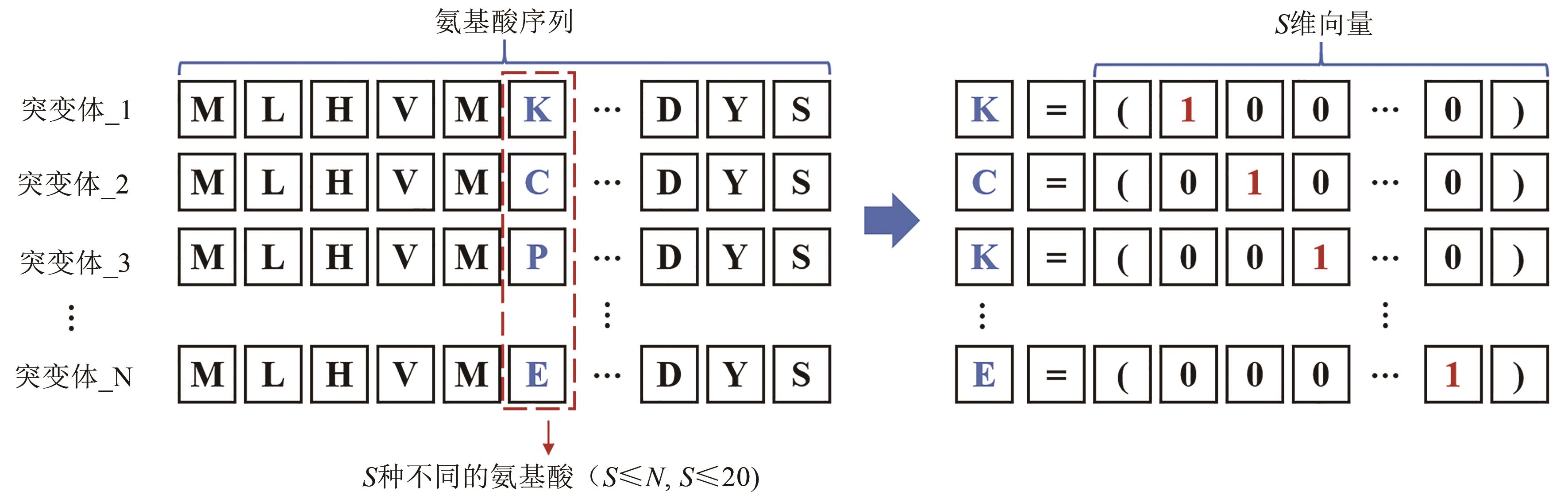
图6 独热编码示意图(N个蛋白质突变体序列中L个氨基酸中某一相同位置包含S种不同的氨基酸,独热编码将这S个氨基酸都表示为包括S-1个0和一个1的S维向量,其中1的位置表示该位置的氨基酸的种类)
Fig. 6 Schematic diagram for one-hot encoding(A certain position of the L amino acids in the N protein mutant sequence contains S different amino acids. The one-hot encoding represents all S amino acids as an S-dimensional vector including S-1 zeros and one 1. The position of 1 indicates the type of amino acid at that position.)
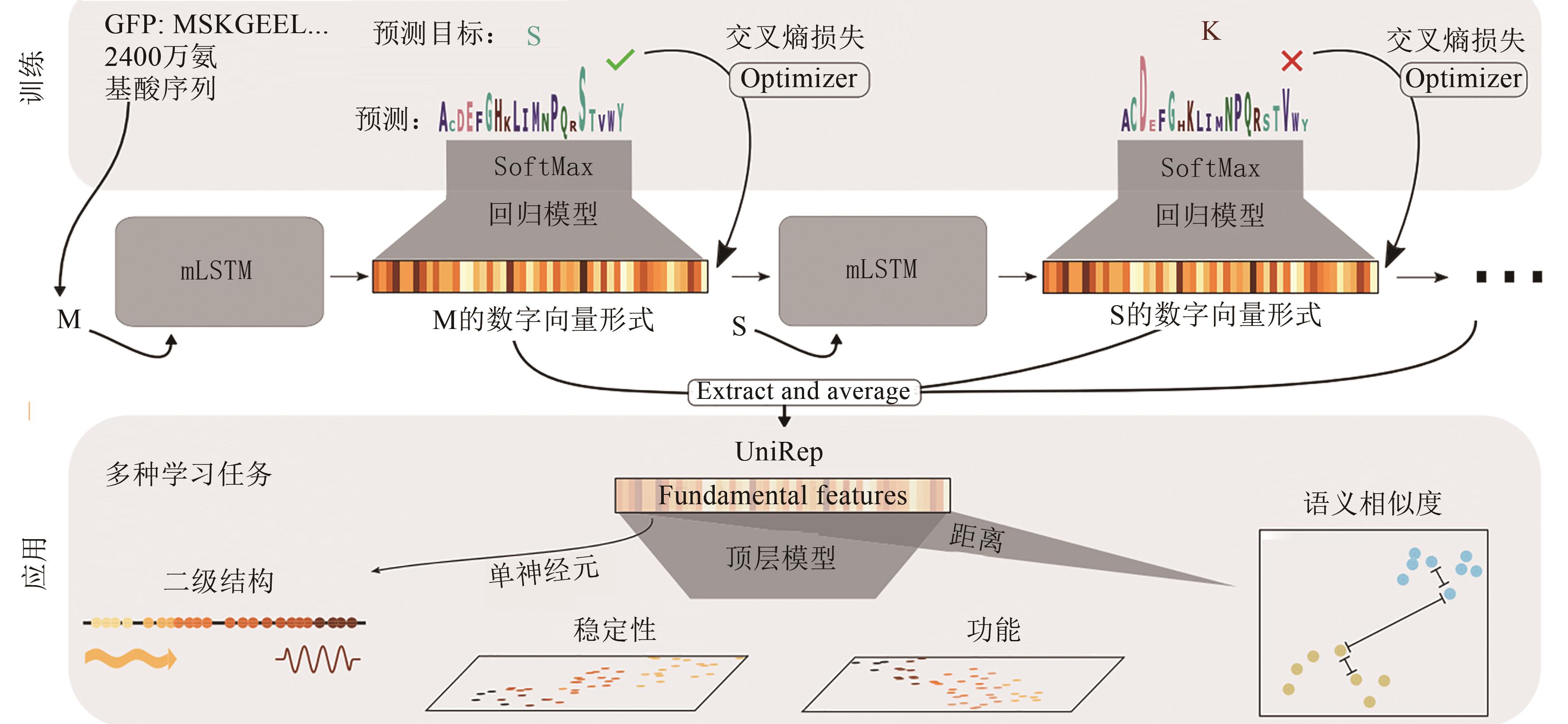
图7 UniRep模型的工作流程[67][在训练部分,UniRep模型使用了2400万个氨基酸序列作为训练集。然后使用训练好的模型来预测下一个氨基酸(使交叉熵损失最小化),从而学会如何正确表示氨基酸。在应用部分中,训练后的模型通过提取和平均各个氨基酸的数字向量,从而生成输入序列的单个固定长度矢量表示。这些向量可以用于训练顶级模型,从而应用于多种序列-功能预测任务]
Fig. 7 Workflow for the UniRep model[67][In the training part, 24 million amino acid sequences are used to train the UniRep model. Then the trained model is used to predict the next amino acid (minimizing the cross-entropy loss), so as to learn how to correctly represent the amino acid. In the application part, by extracting and assessing the numerical vector associated with the amino acid, the trained model is used to generate a single fixed-length vector representing the input sequence. Next, these vectors can be used to train top models, which can be applied to various sequence-function prediction tasks.]
| 名称 | 类型 | 数目/大小 | 参考文献 | 特点 |
|---|---|---|---|---|
| UniProtKB | 蛋白质序列、功能信息、研究论文索引的蛋白质数据库 | UniProtKB/Swiss-Prot包括560 000多条手动注释的蛋白序列,UniProtKB/TrEMBL则包括了2亿多条自动注释的蛋白序列 | [ | 已有学者利用该数据库提供的大量蛋白质序列信息,利用自然语言处理技术成功构建预测模型[ |
| Protein Data Bank | 生物大分子三维结构的数据库 | 145 000多个来源于X射线单晶衍射、核磁共振、电子衍射等实验手段确定的蛋白质、DNA、RNA等生物大分子结构 | [ | 该数据库为蛋白质结构预测模型的构建提供了大量的初始数据 |
| ProThermDB | 蛋白质信息、结构信息、实验条件、文献信息和实验热力学数据库 | 32 000多条数据 | [ | 突变体数据中突变类型包括野生型、单点突变和多点突变 |
| FireProtDB | 蛋白质稳定性数据的数据库 | 242个蛋白质的6715个变体数据 | [ | 手动管理,仅包含单点突变体蛋白质数据 |
| SoluProtMut DB | 突变体蛋白质溶解度数据库 | 917条突变数据 | [ | 手动管理,数据已经针对机器学习应用进行了整理 |
| ProtaBank | 蛋白质工程数据的数据库 | 700多种蛋白质的1 800 000多个突变体 | [ | 手动输入,不仅仅储存各种类型的突变信息,还提供完整的序列信息 |
表2 常见数据库汇总表
Tab. 2 Commonly used database
| 名称 | 类型 | 数目/大小 | 参考文献 | 特点 |
|---|---|---|---|---|
| UniProtKB | 蛋白质序列、功能信息、研究论文索引的蛋白质数据库 | UniProtKB/Swiss-Prot包括560 000多条手动注释的蛋白序列,UniProtKB/TrEMBL则包括了2亿多条自动注释的蛋白序列 | [ | 已有学者利用该数据库提供的大量蛋白质序列信息,利用自然语言处理技术成功构建预测模型[ |
| Protein Data Bank | 生物大分子三维结构的数据库 | 145 000多个来源于X射线单晶衍射、核磁共振、电子衍射等实验手段确定的蛋白质、DNA、RNA等生物大分子结构 | [ | 该数据库为蛋白质结构预测模型的构建提供了大量的初始数据 |
| ProThermDB | 蛋白质信息、结构信息、实验条件、文献信息和实验热力学数据库 | 32 000多条数据 | [ | 突变体数据中突变类型包括野生型、单点突变和多点突变 |
| FireProtDB | 蛋白质稳定性数据的数据库 | 242个蛋白质的6715个变体数据 | [ | 手动管理,仅包含单点突变体蛋白质数据 |
| SoluProtMut DB | 突变体蛋白质溶解度数据库 | 917条突变数据 | [ | 手动管理,数据已经针对机器学习应用进行了整理 |
| ProtaBank | 蛋白质工程数据的数据库 | 700多种蛋白质的1 800 000多个突变体 | [ | 手动输入,不仅仅储存各种类型的突变信息,还提供完整的序列信息 |
| 名称 | 主要功能 | 类型 | 参考文献 |
|---|---|---|---|
| PseAAC | 从蛋白质序列生成氨基酸的疏水性、亲水性、侧链质量、α-COOH的pK值、α-NH3+的pK值以及25 °C时的pI值6种特征 | 网页平台 | [ |
| PROFEAT | 从蛋白质序列生成多种氨基酸分子的结构和物理化学特征,包含11种不同类型分子描述符 | 网页平台 | [ |
| propy | 从蛋白质序列生成多种氨基酸分子的结构和物理化学特征,包含13种不同类型分子描述符 | Python工具包 | [ |
| PseAAC-General | 从蛋白质序列生成多种氨基酸分子的结构和物理化学特征,包含13种基于PseAAC的分子描述符 | Linux/Windows软件 | [ |
| protr/ProtrWeb | 从蛋白质序列生成多种氨基酸分子的结构和物理化学特征,包含22种分子描述符 | R工具包/网页平台 | [ |
| Rcpi | 从蛋白质序列生成多种氨基酸分子的结构和物理化学特征,包含10种分子描述符 | R/Bioconductor工具包 | [ |
| Pse-in-One | 从蛋白质序列生成多种氨基酸分子的结构和物理化学特征,包含8种分子描述符 | 网页平台 | [ |
| ProFET | 从蛋白质序列生成多种氨基酸分子的结构和物理化学特征,不支持PSSM矩阵和GO注释等非基于序列的特征 | Python工具包 | [ |
| PseKRAAC | 从蛋白质序列生成多种基于PseAAC的特征,并且利用氨基酸簇的概念,降低了特征向量的维度 | 网页平台 | [ |
| POSSUM | 从蛋白质序列生成21种基于PSSM矩阵的特征 | 网页平台 | [ |
| iFeature | 从蛋白质序列生成多种氨基酸分子的结构和物理化学特征,包含18种分子描述符,并且提供12种常用的特征聚类,选择和降维算法 | Python工具包/ 网页平台 | [ |
| BioSeq-Analysis2.0 | 从蛋白质序列生成多种氨基酸分子的结构和物理化学特征;提供多种人工智能算法构建预测模型;提供特征选择算法和模型验证方法 | Windows/Linux/ Unix软件/网页平台 | [ |
| iLearn | 从蛋白质序列生成多种氨基酸分子的结构和物理化学特征;提供多种人工智能算法构建预测模型;提供特征选择算法和模型验证方法 | Python工具包/ 网页平台 | [ |
| SOLart | 支持从特征提取、预测模型构建到性能评估的完整流程。但是用户不能获得特征信息,不能选择算法和评估方式 | 网页平台 | [ |
表3 基于蛋白质序列的特征生成工具汇总表
Tab. 3 Feature generation tools based on protein sequences
| 名称 | 主要功能 | 类型 | 参考文献 |
|---|---|---|---|
| PseAAC | 从蛋白质序列生成氨基酸的疏水性、亲水性、侧链质量、α-COOH的pK值、α-NH3+的pK值以及25 °C时的pI值6种特征 | 网页平台 | [ |
| PROFEAT | 从蛋白质序列生成多种氨基酸分子的结构和物理化学特征,包含11种不同类型分子描述符 | 网页平台 | [ |
| propy | 从蛋白质序列生成多种氨基酸分子的结构和物理化学特征,包含13种不同类型分子描述符 | Python工具包 | [ |
| PseAAC-General | 从蛋白质序列生成多种氨基酸分子的结构和物理化学特征,包含13种基于PseAAC的分子描述符 | Linux/Windows软件 | [ |
| protr/ProtrWeb | 从蛋白质序列生成多种氨基酸分子的结构和物理化学特征,包含22种分子描述符 | R工具包/网页平台 | [ |
| Rcpi | 从蛋白质序列生成多种氨基酸分子的结构和物理化学特征,包含10种分子描述符 | R/Bioconductor工具包 | [ |
| Pse-in-One | 从蛋白质序列生成多种氨基酸分子的结构和物理化学特征,包含8种分子描述符 | 网页平台 | [ |
| ProFET | 从蛋白质序列生成多种氨基酸分子的结构和物理化学特征,不支持PSSM矩阵和GO注释等非基于序列的特征 | Python工具包 | [ |
| PseKRAAC | 从蛋白质序列生成多种基于PseAAC的特征,并且利用氨基酸簇的概念,降低了特征向量的维度 | 网页平台 | [ |
| POSSUM | 从蛋白质序列生成21种基于PSSM矩阵的特征 | 网页平台 | [ |
| iFeature | 从蛋白质序列生成多种氨基酸分子的结构和物理化学特征,包含18种分子描述符,并且提供12种常用的特征聚类,选择和降维算法 | Python工具包/ 网页平台 | [ |
| BioSeq-Analysis2.0 | 从蛋白质序列生成多种氨基酸分子的结构和物理化学特征;提供多种人工智能算法构建预测模型;提供特征选择算法和模型验证方法 | Windows/Linux/ Unix软件/网页平台 | [ |
| iLearn | 从蛋白质序列生成多种氨基酸分子的结构和物理化学特征;提供多种人工智能算法构建预测模型;提供特征选择算法和模型验证方法 | Python工具包/ 网页平台 | [ |
| SOLart | 支持从特征提取、预测模型构建到性能评估的完整流程。但是用户不能获得特征信息,不能选择算法和评估方式 | 网页平台 | [ |
| 1 | WAY J C, COLLINS J J, KEASLING J D, et al. Integrating biological redesign: Where synthetic biology came from and where it needs to go[J]. Cell, 2014, 157(1): 151-161. |
| 2 | XIE M Q, HAELLMAN V, FUSSENEGGER M. Synthetic biology—application-oriented cell engineering[J]. Current Opinion in Biotechnology, 2016, 40: 139-148. |
| 3 | BOYLE P M, SILVER P A. Parts plus pipes: Synthetic biology approaches to metabolic engineering[J]. Metabolic Engineering, 2012, 14(3): 223-232. |
| 4 | FOO J L, CHING C B, CHANG M W, et al. The imminent role of protein engineering in synthetic biology[J]. Biotechnology Advances, 2012, 30(3): 541-549. |
| 5 | ERB T J, JONES P R, BAR-EVEN A. Synthetic metabolism: Metabolic engineering meets enzyme design[J]. Current Opinion in Chemical Biology, 2017, 37: 56-62. |
| 6 | PLEISS J. Protein design in metabolic engineering and synthetic biology[J]. Current Opinion in Biotechnology, 2011, 22(5): 611-617. |
| 7 | CHEN R P, GAYNOR A S, CHEN W. Synthetic biology approaches for targeted protein degradation[J]. Biotechnology Advances, 2019, 37(8): 107446. |
| 8 | GAINZA-CIRAUQUI P, CORREIA B E. Computational protein design—the next generation tool to expand synthetic biology applications[J]. Current Opinion in Biotechnology, 2018, 52: 145-152. |
| 9 | BADENHORST C P S, BORNSCHEUER U T. Getting momentum: From biocatalysis to advanced synthetic biology[J]. Trends in Biochemical Sciences, 2018, 43(3): 180-198. |
| 10 | EASON M G, DAMRY A M, CHICA R A. Structure-guided rational design of red fluorescent proteins: Towards designer genetically-encoded fluorophores[J]. Current Opinion in Structural Biology, 2017, 45: 91-99. |
| 11 | ZEYMER C, HILVERT D. Directed evolution of protein catalysts[J]. Annual Review of Biochemistry, 2018, 87: 131-157. |
| 12 | RIBEIRO L F, AMARELLE V, ALVES L F, et al. Genetically engineered proteins to improve biomass conversion: New advances and challenges for tailoring biocatalysts[J]. Molecules, 2019, 24(16): 2879. |
| 13 | MARKEL U, ESSANI K D, BESIRLIOGLU V, et al. Advances in ultrahigh-throughput screening for directed enzyme evolution[J]. Chemical Society Reviews, 2020, 49(1): 233-262. |
| 14 | LIU Q, XUN G H, FENG Y. The state-of-the-art strategies of protein engineering for enzyme stabilization[J]. Biotechnology Advances, 2019, 37(4): 530-537. |
| 15 | LIAO J, WARMUTH M K, GOVINDARAJAN S, et al. Engineering proteinase K using machine learning and synthetic genes[J]. BMC Biotechnology, 2007, 7: 16. |
| 16 | YANG K K, WU Z, ARNOLD F H. Machine-learning-guided directed evolution for protein engineering[J]. Nature Methods, 2019, 16(8): 687-694. |
| 17 | SENIOR A W, EVANS R, JUMPER J, et al. Improved protein structure prediction using potentials from deep learning[J]. Nature, 2020, 577(7792): 706-710. |
| 18 | YANG J Y, ANISHCHENKO I, PARK H, et al. Improved protein structure prediction using predicted interresidue orientations[J]. Proceedings of the National Academy of Sciences of the United States of America, 2020, 117(3): 1496-1503. |
| 19 | YANG M, FEHL C, LEES K V, et al. Functional and informatics analysis enables glycosyltransferase activity prediction[J]. Nature Chemical Biology, 2018, 14(12): 1109-1117. |
| 20 | RYU J Y, KIM H U, LEE S Y. Deep learning enables high-quality and high-throughput prediction of enzyme commission numbers[J]. Proceedings of the National Academy of Sciences of the United States of America, 2019, 116(28): 13996-14001. |
| 21 | HAN X, WANG X N, ZHOU K. Develop machine learning-based regression predictive models for engineering protein solubility[J]. Bioinformatics, 2019, 35(22): 4640-4646. |
| 22 | CHEN J W, ZHENG S J, ZHAO H Y, et al. Structure-aware protein solubility prediction from sequence through graph convolutional network and predicted contact map. Journal of Cheminformatics, 2021, 13: 7. |
| 23 | SAITO Y, OIKAWA M, NAKAZAWA H, et al. Machine-learning-guided mutagenesis for directed evolution of fluorescent proteins[J]. ACS Synthetic Biology, 2018, 7(9): 2014-2022. |
| 24 | WU Z, KAN S B J, LEWIS R D, et al. Machine learning-assisted directed protein evolution with combinatorial libraries[J]. Proceedings of the National Academy of Sciences of the United States of America, 2019, 116(18): 8852-8858. |
| 25 | CADET F, FONTAINE N, LI G Y, et al. A machine learning approach for reliable prediction of amino acid interactions and its application in the directed evolution of enantioselective enzymes[J]. Scientific Reports, 2018, 8: 16757. |
| 26 | BEDBROOK C N, YANG K K, ROBINSON J E, et al. Machine learning-guided channelrhodopsin engineering enables minimally invasive optogenetics[J]. Nature Methods, 2019, 16(11): 1176-1184. |
| 27 | MAZURENKO S, PROKOP Z, DAMBORSKY J. Machine learning in enzyme engineering[J]. ACS Catalysis, 2020, 10(2): 1210-1223. |
| 28 | YANG X, WANG Y F, BYRNE R, et al. Concepts of artificial intelligence for computer-assisted drug discovery[J]. Chemical Reviews, 2019, 119(18): 10520-10594. |
| 29 | BADILLO S, BANFAI B, BIRZELE F, et al. An introduction to machine learning[J]. Clinical Pharmacology and Therapeutics, 2020, 107(4): 871-885. |
| 30 | 蒋迎迎, 曲戈, 孙周通. 机器学习助力酶定向进化[J]. 生物学杂志, 2020, 37(4): 1-11. |
| JIANG Y Y, QU G, SUN Z T. Machine learning-assisted enzyme directed evolution[J]. Journal of Biology, 2020, 37(4): 1-11. | |
| 31 | 胡如云, 张嵩亚, 蒙海林, 等. 面向合成生物学的机器学习方法及应用[J]. 科学通报, 2021, 66(3): 284-299. |
| HU R Y, ZHANG S Y, MENG H L, et al. Machine learning for synthetic biology: Methods and applications[J]. Chinese Science Bulletin, 2021, 66(3): 284-299. | |
| 32 | CONSORTIUM T U. UniProt: a worldwide hub of protein knowledge[J]. Nucleic Acids Research, 2018, 47(D1): D506-D515. |
| 33 | MUGGLETON S, KING R D, STENBERG M J E. Protein secondary structure prediction using logic-based machine learning[J]. Protein Engineering, Design and Selection, 1992, 5(7): 647-657. |
| 34 | ALQURAISHI M. AlphaFold at CASP13[J]. Bioinformatics, 2019, 35(22): 4862-4865. |
| 35 | KINCH L N, SHI S Y, CHENG H, et al. CASP9 target classification[J]. Proteins: Structure, Function, and Bioinformatics, 2011, 79(S10): 21-36. |
| 36 | LI Y, WANG S, UMAROV R, et al. DEEPre: sequence-based enzyme EC number prediction by deep learning[J]. Bioinformatics, 2017, 34(5): 760-769. |
| 37 | BOUTET E, LIEBERHERR D, TOGNOLLI M, et al. UniProtKB/Swiss-prot, the manually annotated section of the UniProt KnowledgeBase: How to use the entry view[J]. Methods in Molecular Biology, 2016, 1374: 23-54. |
| 38 | CLAUDEL-RENARD C, CHEVALET C, FARAUT T, et al. Enzyme-specific profiles for genome annotation: PRIAM[J]. Nucleic Acids Research, 2003, 31(22): 6633-6639. |
| 39 | YU C G, ZAVALJEVSKI N, DESAI V, et al. Genome-wide enzyme annotation with precision control: Catalytic families (CatFam) databases[J]. Proteins, 2009, 74(2): 449-460. |
| 40 | KUMAR N, SKOLNICK J. EFICAz2.5: application of a high-precision enzyme function predictor to 396 proteomes[J]. Bioinformatics, 2012, 28(20): 2687-2688. |
| 41 | LI Y H, XU J Y, TAO L, et al. SVM-prot 2016: A web-server for machine learning prediction of protein functional families from sequence irrespective of similarity[J]. PLoS One, 2016, 11(8): e0155290. |
| 42 | ZHANG C X, FREDDOLINO P L, ZHANG Y. COFACTOR: improved protein function prediction by combining structure, sequence and protein-protein interaction information[J]. Nucleic Acids Research, 2017, 45(W1): W291-W299. |
| 43 | NURSIMULU N, XU L L, WASMUTH J D, et al. Improved enzyme annotation with EC-specific cutoffs using DETECT v2[J]. Bioinformatics, 2018, 34(19): 3393-3395. |
| 44 | DALKIRAN A, RIFAIOGLU A S, MARTIN M J, et al. ECPred: a tool for the prediction of the enzymatic functions of protein sequences based on the EC nomenclature[J]. BMC Bioinformatics, 2018, 19(1): 334. |
| 45 | HOU Q Z, BOURGEAS R, PUCCI F, et al. Computational analysis of the amino acid interactions that promote or decrease protein solubility[J]. Scientific Reports, 2018, 8: 14661. |
| 46 | BHANDARI B K, GARDNER P P, LIM C S. Solubility-Weighted Index: Fast and accurate prediction of protein solubility[J]. Bioinformatics, 2020, 36(18): 4691-4698. |
| 47 | LI G Y, DONG Y J, REETZ M T. Can machine learning revolutionize directed evolution of selective enzymes? [J]. Advanced Synthesis and Catalysis, 2019, 361(11): 2377-2386. |
| 48 | SONG H, BREMER B J, HINDS E C, et al. Inferring protein sequence-function relationships with large-scale positive-unlabeled learning[J]. Cell Systems, 2021, 12(1): 92-101.e8. |
| 49 | YU L A, WANG S Y, LAI K K. An integrated data preparation scheme for neural network data analysis[J]. IEEE Transactions on Knowledge and Data Engineering, 2006, 18(2): 217-230. |
| 50 | YANG Y, UROLAGIN S, NIROULA A, et al. PON-tstab: Protein variant stability predictor. importance of training data quality[J]. International Journal of Molecular Sciences, 2018, 19(4): 1009. |
| 51 | SIEDHOFF N E, SCHWANEBERG U, DAVARI M D. Machine learning-assisted enzyme engineering[J]. Methods in Enzymology, 2020, 643: 281-315. |
| 52 | BASTIAN F B, CHIBUCOS M C, GAUDET P, et al. The Confidence Information Ontology: A step towards a standard for asserting confidence in annotations[J]. Database, 2015, 2015:bav043. |
| 53 | 周志华. 机器学习[M]. 北京: 清华大学出版社, 2016. |
| ZHOU Z H. Machine learning[M]. Beijing: Tsinghua University Press, 2016. | |
| 54 | KAWASHIMA S, POKAROWSKI P, POKAROWSKA M, et al. AAindex: amino acid index database, progress report 2008[J]. Nucleic Acids Research, 2007, 36(): D202-D205. |
| 55 | XU Y T, VERMA D, SHERIDAN R P, et al. Deep dive into machine learning models for protein engineering[J]. Journal of Chemical Information and Modeling, 2020, 60(6): 2773-2790. |
| 56 | LECUN Y, BENGIO Y, HINTON G. Deep learning[J]. Nature, 2015, 521(7553): 436-444. |
| 57 | ABDI H. Partial least squares regression and projection on latent structure regression (PLS Regression)[J]. WIREs Computational Statistics, 2010, 2(1): 97-106. |
| 58 | CORTES C, VAPNIK V. Support-vector networks[J]. Machine Learning, 1995, 20(3): 273-297. |
| 59 | QUINLAN J R. Induction of decision trees[J]. Machine Learning, 1986, 1(1): 81-106. |
| 60 | HECKERMAN D. A tutorial on learning with Bayesian networks [M]//HOLMES D E, JAIN L C. Innovations in Bayesian networks: theory and applications. Berlin, Heidelberg; Springer, 2008: 33-82. |
| 61 | SINAI S, KELSIC E, CHURCH G M, et al. Variational auto-encoding of protein sequences[EB/OL]. 2017: arXiv: 1712.03346[q-bio.QM]. |
| 62 | LECUN Y, BENGIO Y. Convolutional networks for images, speech, and time series[J]. The handbook of brain theory and neural networks, 1995, 3361(10): 1995. |
| 63 | LOHMANN R, SCHNEIDER G, BEHRENS D, et al. A neural network model for the prediction of membrane-spanning amino acid sequences[J]. Protein Science, 1994, 3(9): 1597-1601. |
| 64 | 曲戈, 朱彤, 蒋迎迎, 等. 蛋白质工程:从定向进化到计算设计[J]. 生物工程学报, 2019, 35(10): 1843-1856. |
| QU G, ZHU T, JIANG Y Y, et al. Protein engineering: From directed evolution to computational design[J]. Chinese Journal of Biotechnology, 2019, 35(10): 1843-1856. | |
| 65 | WOLPERT D H. The lack of A priori distinctions between learning algorithms[J]. Neural Computation, 1996, 8(7): 1341-1390. |
| 66 | YANG K K, WU Z, BEDBROOK C N, et al. Learned protein embeddings for machine learning[J]. Bioinformatics, 2018, 34(15): 2642-2648. |
| 67 | ALLEY E C, KHIMULYA G, BISWAS S, et al. Unified rational protein engineering with sequence-based deep representation learning[J]. Nature Methods, 2019, 16(12): 1315-1322. |
| 68 | CONSORTIUM T U, BATEMAN A, MARTIN M J, et al. UniProt: the universal protein knowledgebase in 2021[J]. Nucleic Acids Research, 2020, 49(D1): D480-D489. |
| 69 | RIVES A, MEIER J, SERCU T, et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences[EB/OL]. bioRxiv, 2020, DOI:10.1101/622803 . |
| 70 | BURLEY S K, BHIKADIYA C, BI C X, et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences[J]. Nucleic Acids Research, 2020, 49(D1): D437-D451. |
| 71 | NIKAM R, KULANDAISAMY A, HARINI K, et al. ProThermDB: thermodynamic database for proteins and mutants revisited after 15 years[J]. Nucleic Acids Research, 2020, 49(D1): D420-D424. |
| 72 | STOURAC J, DUBRAVA J, MUSIL M, et al. FireProtDB: database of manually curated protein stability data[J]. Nucleic Acids Research, 2020, 49(D1): D319-D324. |
| 73 | TIAN Y, DEUTSCH C, KRISHNAMOORTHY B. Scoring function to predict solubility mutagenesis[J]. Algorithms for Molecular Biology: AMB, 2010, 5: 33. |
| 74 | SORMANNI P, APRILE F A, VENDRUSCOLO M. The CamSol method of rational design of protein mutants with enhanced solubility[J]. Journal of Molecular Biology, 2015, 427(2): 478-490. |
| 75 | ZAMBRANO R, JAMROZ M, SZCZASIUK A, et al. AGGRESCAN3D (A3D): Server for prediction of aggregation properties of protein structures[J]. Nucleic Acids Research, 2015, 43(W1): W306-W313. |
| 76 | YANG Y, NIROULA A, SHEN B R, et al. PON-Sol: Prediction of effects of amino acid substitutions on protein solubility[J]. Bioinformatics, 2016, 32(13): 2032-2034. |
| 77 | PALADIN L, PIOVESAN D, TOSATTO S C E. SODA: prediction of protein solubility from disorder and aggregation propensity[J]. Nucleic Acids Research, 2017, 45(W1): W236-W240. |
| 78 | MAZURENKO S. Predicting protein stability and solubility changes upon mutations: data perspective[J]. Chem Cat Chem, 2020, 12,5590-5598. |
| 79 | WANG C Y, CHANG P M, ARY M L, et al. ProtaBank: A repository for protein design and engineering data[J]. Protein Science: a Publication of the Protein Society, 2018, 27(6): 1113-1124. |
| 80 | SHEN H B, CHOU K C. PseAAC: A flexible web server for generating various kinds of protein pseudo amino acid composition[J]. Analytical Biochemistry, 2008, 373(2): 386-388. |
| 81 | RAO H B, ZHU F, YANG G B, et al. Update of PROFEAT: A web server for computing structural and physicochemical features of proteins and peptides from amino acid sequence[J]. Nucleic Acids Research, 2011, 39(): W385-W390. |
| 82 | CAO D S, XU Q S, LIANG Y Z. Propy: a tool to generate various modes of Chou's PseAAC[J]. Bioinformatics, 2013, 29(7): 960-962. |
| 83 | DU P F, GU S W, JIAO Y S. PseAAC-General: Fast building various modes of general form of Chou's pseudo-amino acid composition for large-scale protein datasets[J]. International Journal of Molecular Sciences, 2014, 15(3): 3495-3506. |
| 84 | XIAO N, CAO D S, ZHU M F, et al. Protr/ProtrWeb: R package and web server for generating various numerical representation schemes of protein sequences[J]. Bioinformatics, 2015, 31(11): 1857-1859. |
| 85 | CAO D S, XIAO N, XU Q S, et al. Rcpi: R/Bioconductor package to generate various descriptors of proteins, compounds and their interactions[J]. Bioinformatics, 2014, 31(2): 279-281. |
| 86 | LIU B, LIU F L, WANG X L, et al. Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences[J]. Nucleic Acids Research, 2015, 43(W1): W65-W71. |
| 87 | OFER D, LINIAL M. ProFET: Feature engineering captures high-level protein functions[J]. Bioinformatics, 2015, 31(21): 3429-3436. |
| 88 | ZUO Y C, LI Y, CHEN Y L, et al. PseKRAAC: a flexible web server for generating pseudo K-tuple reduced amino acids composition[J]. Bioinformatics, 2016, 33(1): 122-124. |
| 89 | WANG J W, YANG B J, REVOTE J, et al. POSSUM: a bioinformatics toolkit for generating numerical sequence feature descriptors based on PSSM profiles[J]. Bioinformatics, 2017, 33(17): 2756-2758. |
| 90 | CHEN Z, ZHAO P, LI F Y, et al. iFeature: a Python package and web server for features extraction and selection from protein and peptide sequences[J]. Bioinformatics, 2018, 34(14): 2499-2502. |
| 91 | LIU B, GAO X, ZHANG H Y. BioSeq-Analysis2.0: An updated platform for analyzing DNA, RNA and protein sequences at sequence level and residue level based on machine learning approaches[J]. Nucleic Acids Research, 2019, 47(20): e127. |
| 92 | CHEN Z, ZHAO P, LI F Y, et al. iLearn: an integrated platform and meta-learner for feature engineering, machine-learning analysis and modeling of DNA, RNA and protein sequence data[J]. Briefings in Bioinformatics, 2019, 21(3): 1047-1057. |
| 93 | HOU Q Z, KWASIGROCH J M, ROOMAN M, et al. SOLart: a structure-based method to predict protein solubility and aggregation[J]. Bioinformatics, 2019, 36(5): 1445-1452. |
| 94 | LIU Y M, WANG X L, LIU B. IDP-CRF: intrinsically disordered protein/region identification based on conditional random fields[J]. International Journal of Molecular Sciences, 2018, 19(9): 2483. |
| 95 | CHEN Z, HE N N, HUANG Y, et al. Integration of A deep learning classifier with A random forest approach for predicting malonylation sites[J]. Genomics, Proteomics & Bioinformatics, 2018, 16(6): 451-459. |
| 96 | SCHNOES A M, REAM D C, THORMAN A W, et al. Biases in the experimental annotations of protein function and their effect on our understanding of protein function space[J]. PLoS Computational Biology, 2013, 9(5): e1003063. |
| 97 | ZHOU N H, JIANG Y X, BERGQUIST T R, et al. The CAFA challenge reports improved protein function prediction and new functional annotations for hundreds of genes through experimental screens[J]. Genome Biology, 2019, 20(1): 244. |
| 98 | GERLT J A, ALLEN K N, ALMO S C, et al. The enzyme function initiative[J]. Biochemistry, 2011, 50(46): 9950-9962. |
| 99 | ROBERTS R J, CHANG Y C, HU Z J, et al. COMBREX: a project to accelerate the functional annotation of prokaryotic genomes[J]. Nucleic Acids Research, 2010, 39(S1): D11-D14. |
| 100 | FOWLER D M, FIELDS S. Deep mutational scanning: A new style of protein science[J]. Nature Methods, 2014, 11(8): 801-807. |
| 101 | FOWLER D M, STEPHANY J J, FIELDS S. Measuring the activity of protein variants on a large scale using deep mutational scanning[J]. Nature Protocols, 2014, 9(9): 2267-2284. |
| 102 | NEWBERRY R W, LEONG J T, CHOW E D, et al. Deep mutational scanning reveals the structural basis for α-synuclein activity[J]. Nature Chemical Biology, 2020, 16(6): 653-659. |
| 103 | Riesselman A J, Ingraham J B, Marks D S. Deep generative models of genetic variation capture the effects of mutations[J]. Nature Methods, 2018, 15(10): 816-822. |
| [1] | 高歌, 边旗, 王宝俊. 合成基因线路的工程化设计研究进展与展望[J]. 合成生物学, 2025, 6(1): 45-64. |
| [2] | 李冀渊, 吴国盛. 合成生物学视域下有机体的两种隐喻[J]. 合成生物学, 2025, 6(1): 190-202. |
| [3] | 焦洪涛, 齐蒙, 邵滨, 蒋劲松. DNA数据存储技术的法律治理议题[J]. 合成生物学, 2025, 6(1): 177-189. |
| [4] | 唐兴华, 陆钱能, 胡翌霖. 人类世中对合成生物学的哲学反思[J]. 合成生物学, 2025, 6(1): 203-212. |
| [5] | 徐怀胜, 石晓龙, 刘晓光, 徐苗苗. DNA存储的关键技术:编码、纠错、随机访问与安全性[J]. 合成生物学, 2025, 6(1): 157-176. |
| [6] | 温艳华, 刘合栋, 曹春来, 巫瑞波. 蛋白质工程在医药产业中的应用[J]. 合成生物学, 2025, 6(1): 65-86. |
| [7] | 石婷, 宋展, 宋世怡, 张以恒. 体外生物转化(ivBT):生物制造的新前沿[J]. 合成生物学, 2024, 5(6): 1437-1460. |
| [8] | 柴猛, 王风清, 魏东芝. 综合利用木质纤维素生物转化合成有机酸[J]. 合成生物学, 2024, 5(6): 1242-1263. |
| [9] | 邵明威, 孙思勉, 杨时茂, 陈国强. 基于极端微生物的生物制造[J]. 合成生物学, 2024, 5(6): 1419-1436. |
| [10] | 王子渊, 杨立荣, 吴坚平, 郑文隆. 酶促合成手性氨基酸的研究进展[J]. 合成生物学, 2024, 5(6): 1319-1349. |
| [11] | 陈雨, 张康, 邱以婧, 程彩云, 殷晶晶, 宋天顺, 谢婧婧. 微生物电合成技术转化二氧化碳研究进展[J]. 合成生物学, 2024, 5(5): 1142-1168. |
| [12] | 郑皓天, 李朝风, 刘良叙, 王嘉伟, 李恒润, 倪俊. 负碳人工光合群落的设计、优化与应用[J]. 合成生物学, 2024, 5(5): 1189-1210. |
| [13] | 夏孔晨, 徐维华, 吴起. 光酶催化混乱性反应的研究进展[J]. 合成生物学, 2024, 5(5): 997-1020. |
| [14] | 陈子苓, 向阳飞. 类器官技术与合成生物学协同研究进展[J]. 合成生物学, 2024, 5(4): 795-812. |
| [15] | 蔡冰玉, 谭象天, 李伟. 合成生物学在干细胞工程化改造中的研究进展[J]. 合成生物学, 2024, 5(4): 782-794. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
